


Uncovering the Gene Regulatory Network of Endothelial Cells in Mouse Duchenne Muscular Dystrophy: Insights from Single-Nuclei RNA Sequencing Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. SnRNA-Seq Datasets
2.2. SnRNA-Seq Data Analysis
2.3. Gene Ontology (GO) and GSEA Enrichment Analysis
2.4. Single-Cell Metabolic Analysis
2.5. Histology
2.6. Statistical Analysis
3. Results
3.1. Single Cell Transcriptomics Reveals Nine EC Clusters
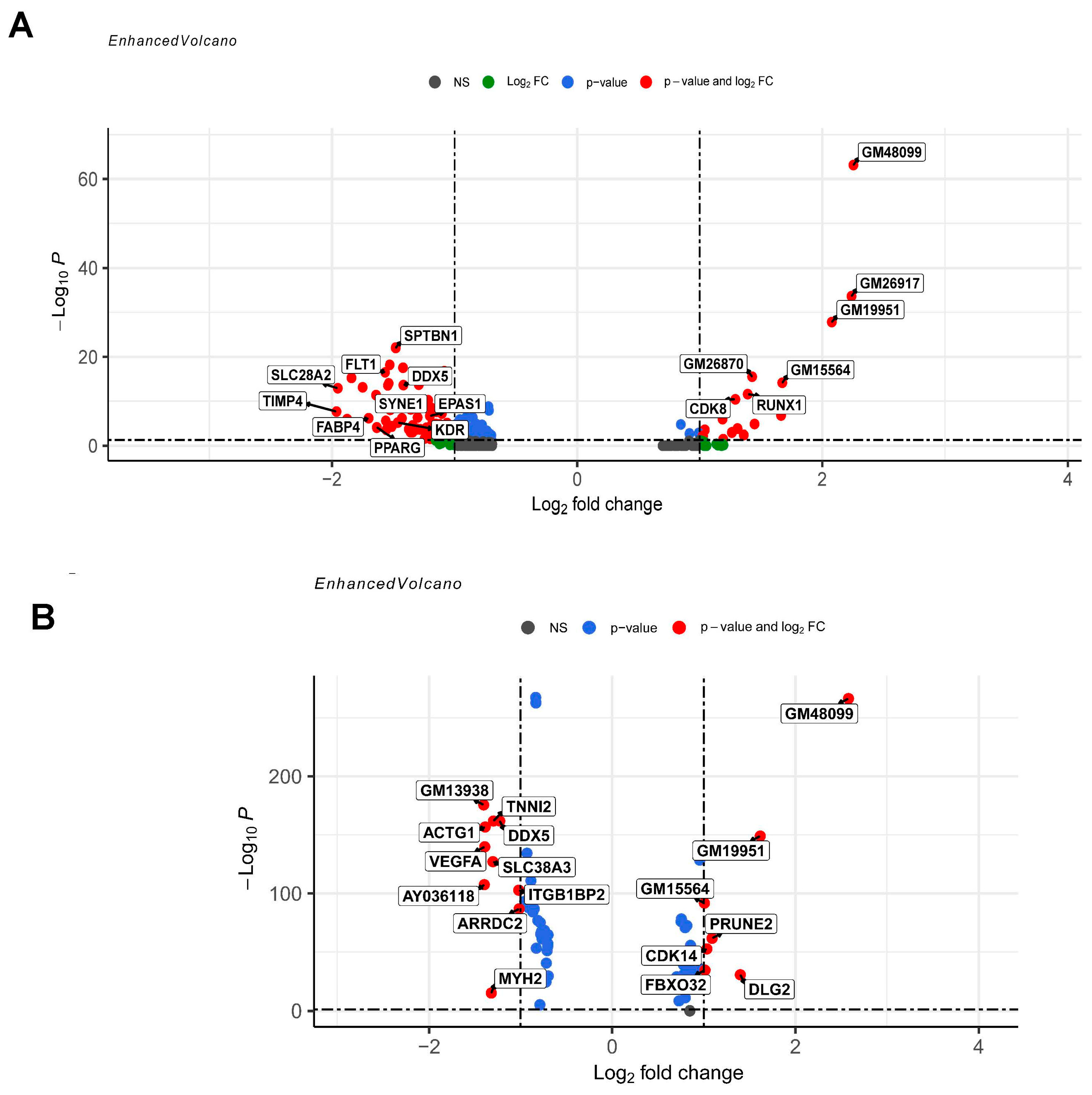
3.2. Differential Gene Expression and Functional Enrichment Analysis of ECs in DMDmut versus Control Muscles
3.3. Differential Metabolic Pathways of ECs within DMDmut and Control Muscles
- (i)
- ECs are more glycolytic and less dependent on oxidative phosphorylation (OXPHOS), despite having punctate mitochondria [18]. Recent studies demonstrated that both OXPHOS and glycolysis are essential for the angiogenic response of vascular ECs [19]. “scMetabolism” analysis revealed that ECs from DMDmut showed lower activities of oxidative citric acid cycle (TCA cycle), OXPHOS, fatty acid degradation, glycolysis and pyruvate metabolism compared to ECs from control muscles (Figure 4);
- (ii)
- Although the role of fatty acid synthesis in ECs remains incompletely described, recent study reported that fatty acid synthase knockdown in ECs impedes vascular sprouting by reducing proliferation. Loss of fatty acid synthase in ECs also impairs angiogenesis in vivo [20]. Our scRNA-seq analysis revealed that ECs from DMDmut showed high activity of fatty acid biosynthesis, which has been linked to vascular sprouting and angiogenesis (Figure 4);
- (iii)
- Trophic effects of purine as well as pyrimidine nucleosides and nucleotides promote migration and proliferation of ECs via P1 and P2Y receptors during angiogenesis, vascular remodeling, and atherosclerosis during restenosis after angioplasty [21,22]. One-carbon folate pool is needed for the synthesis of purines and thymidylate in nucleic acids [23]. “scMetabolism” analysis revealed that ECs from DMDmut muscles showed increased activities of purine metabolism, pyrimidine metabolism, and one carbon pool by folate, which play a role in promoting migration and proliferation of ECs during angiogenesis and restenosis (Figure 4);
- (iv)
- Recent studies demonstrated the role of the endothelial oxidized pentose phosphate pathway (oxPPP) in promoting cell coverage and a maturation of the vascular wall by controlling vascular matrix composition and vascular mural cell recruitment during early development. These findings establish a key role for oxPPP in the formation of the vascular system [24,25]. “scMetabolism” analysis shows that ECs from DMDmut muscle showed high pentose phosphate pathway activity, which plays a role in the formation of the vascular system (Figure 4);
- (v)
- Metabolism of serine, glycine, and threonine provides energy for the TCA cycle via pyruvate and acetyl CoA [26]. Glutathione (GSH) plays an important role in the cellular defense against peroxidative stress. In amino acid metabolic pathways, homocysteine is an intermediate amino acid that is metabolized from methionine to cysteine and has been shown to be associated with oxidative stress and endothelial damage [27]. Glutathione (GSH), superoxide dismutase (SOD), and glutathione peroxidase (GPX) are major antioxidant molecules in ECs [28]. A recent report showed that brain ECs directly absorb and metabolize glutamate and utilize the resulting α-ketoglutarate in the tricarboxylic acid cycle, ultimately producing ATP in the absence of glucose [29]. Our “scMetabolism” analysis shows that ECs from DMDmut showed high activities of glycine, serine, and threonine metabolism, glutathione metabolism, and alanine, aspartate, and glutamate metabolism, but low activity of cysteine and methionine metabolism compared to ECs from control muscle (Figure 4).

3.4. Impact of DMD Mutation on EC Density and Proliferation in Skeletal Muscles
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fayssoil, A.; Nardi, O.; Orlikowski, D.; Annane, D. Cardiomyopathy in Duchenne muscular dystrophy: Pathogenesis and therapeutics. Heart Fail. Rev. 2010, 15, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Gumerson, J.D.; Michele, D.E. The dystrophin-glycoprotein complex in the prevention of muscle damage. J. Biomed. Biotechnol. 2011, 2011, 210797. [Google Scholar] [CrossRef] [Green Version]
- D’Amario, D.; Amodeo, A.; Adorisio, R.; Tiziano, F.D.; Leone, A.M.; Perri, G.; Bruno, P.; Massetti, M.; Ferlini, A.; Pane, M.; et al. A current approach to heart failure in Duchenne muscular dystrophy. Heart 2017, 103, 1770–1779. [Google Scholar] [CrossRef] [PubMed]
- Kamdar, F.; Garry, D.J. Dystrophin-Deficient Cardiomyopathy. J. Am. Coll. Cardiol. 2016, 67, 2533–2546. [Google Scholar] [CrossRef] [PubMed]
- Loufrani, L.; Matrougui, K.; Gorny, D.; Duriez, M.; Blanc, I.; Lévy, B.I.; Henrion, D. Flow (shear stress)–Induced endothelium-dependent dilation is altered in mice lacking the gene encoding for dystrophin. Circulation 2001, 103, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Chemello, F.; Wang, Z.; Li, H.; McAnally, J.R.; Liu, N.; Bassel-Duby, R.; Olson, E.N. Degenerative and regenerative pathways underlying Duchenne muscular dystrophy revealed by single-nucleus RNA sequencing. Proc. Natl. Acad. Sci. USA 2020, 117, 29691–29701. [Google Scholar] [CrossRef]
- Galbraith, M.D.; Donner, A.J.; Espinosa, J.M. CDK8: A positive regulator of transcription. Transcription 1 2010, 1, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Okuda, T.; van Deursen, J.; Hiebert, S.W.; Grosveld, G.; Downing, J.R. AML1, the target of multiple chromosomal translocations in human leukemia, is essential for normal fetal liver hematopoiesis. Cell 1996, 84, 321–330. [Google Scholar] [CrossRef] [Green Version]
- Sugimura, R.; Jha, D.K.; Han, A.; Soria-Valles, C.; da Rocha, E.L.; Lu, Y.-F.; Goettel, J.A.; Serrao, E.; Rowe, R.G.; Malleshaiah, M.; et al. Haematopoietic stem and progenitor cells from human pluripotent stem cells. Nature 2017, 545, 432–438. [Google Scholar] [CrossRef] [Green Version]
- Kanno, S.; Oda, N.; Abe, M.; Terai, Y.; Ito, M.; Shitara, K.; Tabayashi, K.; Shibuya, M.; Sato, Y. Roles of two VEGF receptors, Flt-1 and KDR, in the signal transduction of VEGF effects in human vascular endothelial cells. Oncogene 2000, 19, 2138–2146. [Google Scholar] [CrossRef] [Green Version]
- Tian, H.; McKnight, S.L.; Russell, D.W. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 1997, 11, 72–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández, C.A.; Moses, M.A. Modulation of angiogenesis by tissue inhibitor of metalloproteinase-4. Biochem. Biophys. Res. Commun. 2006, 345, 523–529. [Google Scholar] [CrossRef] [PubMed]
- Redell, M.S.; Tsimelzon, A.; Hilsenbeck, S.G.; Tweardy, D.J. Conditional overexpression of Stat3alpha in differentiating myeloid cells results in neutrophil expansion and induces a distinct, antiapoptotic and pro-oncogenic gene expression pattern. J. Leukoc. Biol. 2007, 82, 975–985. [Google Scholar] [CrossRef] [PubMed]
- Saint-Geniez, M.; Ghelfi, E.; Liang, X.; Yu, D.; Spencer, C.; Abend, S.; Hotamisligil, G.; Cataltepe, S. Fatty acid binding protein 4 deficiency protects against oxygen-induced retinopathy in mice. PLoS ONE 2014, 9, e96253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Fountzilas, C.; Puzanov, I.; Attwood, K.M.; Morrison, C.; Ling, X. Multiple functions of the DEAD-box RNA helicase, DDX5 (p68), make DDX5 a superior oncogenic biomarker and target for targeted cancer therapy. Am. J. Cancer Res. 2021, 11, 5190–5213. [Google Scholar] [PubMed]
- Lin, S.; Tian, L.; Shen, H.; Gu, Y.; Li, J.-L.; Chen, Z.; Sun, X.; You, M.J.; Wu, L. DDX5 is a positive regulator of oncogenic NOTCH1 signaling in T cell acute lymphoblastic leukemia. Oncogene 2013, 32, 4845–4853. [Google Scholar] [CrossRef] [PubMed]
- Shi, H.; Zhao, W.; Luo, J.; Yao, D.; Sun, Y.; Li, J.; Loor, J. Peroxisome proliferator-activated receptor γ1 and γ2 isoforms alter lipogenic gene networks in goat mammary epithelial cells to different extents. J. Dairy Sci. 2014, 97, 5437–5447. [Google Scholar] [CrossRef] [Green Version]
- Garcia, D.; Carr, J.F.; Chan, F.; Peterson, A.L.; Ellis, K.A.; Scaffa, A.; Ghio, A.J.; Yao, H.; Dennery, P.A. Short exposure to hyperoxia causes cultured lung epithelial cell mitochondrial dysregulation and alveolar simplification in mice. Pediatr. Res. 2021, 90, 58–65. [Google Scholar] [CrossRef]
- Lapel, M.; Weston, P.; Strassheim, D.; Karoor, V.; Burns, N.; Lyubchenko, T.; Paucek, P.; Stenmark, K.R.; Gerasimovskaya, E.V. Glycolysis and oxidative phosphorylation are essential for purinergic receptor-mediated angiogenic responses in vasa vasorum endothelial cells. Am. J. Physiol. Physiol. 2017, 312, C56–C70. [Google Scholar] [CrossRef] [Green Version]
- Bruning, U.; Morales-Rodriguez, F.; Kalucka, J.; Goveia, J.; Taverna, F.; Queiroz, K.C.; Dubois, C.; Cantelmo, A.R.; Chen, R.; Loroch, S.; et al. Impairment of Angiogenesis by Fatty Acid Synthase Inhibition Involves mTOR Malonylation. Cell Metab. 2018, 28, 866–880.e15. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G. Purinergic Signaling in the Cardiovascular System. Circ. Res. 2017, 120, 207–228. [Google Scholar] [CrossRef] [Green Version]
- Burnstock, G.; Ralevic, V. Purinergic signaling and blood vessels in health and disease. Pharmacol. Rev. 2014, 66, 102–192. [Google Scholar] [CrossRef] [PubMed]
- Filipp, F.V.; Ratnikov, B.; De Ingeniis, J.; Smith, J.W.; Osterman, A.L.; Scott, D. Glutamine-fueled mitochondrial metabolism is decoupled from glycolysis in melanoma. Pigment. Cell Melanoma Res. 2012, 25, 732–739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cherepanova, O.A.; Byzova, T.V. Pentose phosphate pathway drives vascular maturation. Nat. Metab. 2022, 4, 15–16. [Google Scholar] [CrossRef] [PubMed]
- Facchinello, N.; Astone, M.; Audano, M.; Oberkersch, R.E.; Spizzotin, M.; Calura, E.; Marques, M.; Crisan, M.; Mitro, N.; Santoro, M.M. Oxidative pentose phosphate pathway controls vascular mural cell coverage by regulating extracellular matrix composition. Nat. Metab. 2022, 4, 123–140. [Google Scholar] [CrossRef]
- Hong, J.-L.; Wu, L.; Lu, J.-Q.; Zhou, W.-B.; Cao, Y.-J.; Lv, W.-L.; Liu, B.; Rao, P.-F.; Ni, L.; Lv, X.-C. Comparative transcriptomic analysis reveals the regulatory effects of inorganic nitrogen on the biosynthesis of Monascus pigments and citrinin. RSC Adv. 2020, 10, 5268–5282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdalla, B.M.Z.; de Oliveira, R.T.G.; Afonso, R.I.; Criado, P.R. A late diagnosis of hyperhomocysteinemia with probable evolution to verrucous elephantiasis nostra and leg ulcers. An. Bras. Dermatol. 2021, 96, 253–255. [Google Scholar] [CrossRef]
- Wu, D.; Ji, H.; Du, W.; Ren, L.; Qian, G. Mitophagy alleviates ischemia/reperfusion-induced microvascular damage through improving mitochondrial quality control. Bioengineered 2022, 13, 3596–3607. [Google Scholar] [CrossRef]
- Hinca, S.B.; Salcedo, C.; Wagner, A.; Goldeman, C.; Sadat, E.; Aibar, M.M.; Maechler, P.; Brodin, B.; Aldana, B.I.; Helms, H.C. Brain endothelial cells metabolize glutamate via glutamate dehydrogenase to replenish TCA-intermediates and produce ATP under hypoglycemic conditions. J. Neurochem. 2021, 157, 1861–1875. [Google Scholar] [CrossRef]
- Chen, M.; Zeng, J.; Chen, S.; Li, J.; Wu, H.; Dong, X.; Lei, Y.; Zhi, X.; Yao, L. SPTBN1 suppresses the progression of epithelial ovarian cancer via SOCS3-mediated blockade of the JAK/STAT3 signaling pathway. Aging 2020, 12, 10896–10911. [Google Scholar] [CrossRef]
- Duan, R.; Kim, J.H.; Shilagardi, K.; Schiffhauer, E.S.; Lee, D.M.; Son, S.; Li, S.; Thomas, C.; Luo, T.; Fletcher, D.A.; et al. Spectrin is a mechanoresponsive protein shaping fusogenic synapse architecture during myoblast fusion. Nature 2018, 20, 688–698. [Google Scholar] [CrossRef] [PubMed]
- McCourt, J.L.; Stearns-Reider, K.M.; Mamsa, H.; Kannan, P.; Afsharinia, M.H.; Shu, C.; Gibbs, E.M.; Shin, K.M.; Kurmangaliyev, Y.Z.; Schmitt, L.R.; et al. Multi-omics analysis of sarcospan overexpression in mdx skeletal muscle reveals compensatory remodeling of cytoskeleton-matrix interactions that promote mechanotransduction pathways. Skelet. Muscle 2023, 13, 1. [Google Scholar] [CrossRef] [PubMed]
- Loufrani, L.; Dubroca, C.; You, D.; Li, Z.; Levy, B.; Paulin, D.; Henrion, D. Absence of dystrophin in mice reduces NO-dependent vascular function and vascular density: Total recovery after a treatment with the aminoglycoside gentamicin. Arter. Thromb. Vasc. Biol. 2004, 24, 671–676. [Google Scholar] [CrossRef] [Green Version]
- Sato, K.; Yokota, T.; Ichioka, S.; Shibata, M.; Takeda, S. Vasodilation of intramuscular arterioles under shear stress in dystrophin-deficient skeletal muscle is impaired through decreased nNOS expression. Acta Myol. 2008, 27, 30–36. [Google Scholar] [PubMed]
- Kunert-Keil, C.; Gredes, T.; Lucke, S.; Morgenstern, S.; Mielczarek, A.; Sporniak-Tutak, K.; Gedrange, T.; Spassov, A. Caveolin-1, caveolin-3 and VEGF expression in the masticatory muscles of mdx mice. Folia Histochem. Cytobiol. 2011, 49, 291–298. [Google Scholar] [CrossRef] [Green Version]
- Nico, B.; Corsi, P.; Vacca, A.; Roncali, L.; Ribatti, D. Vascular endothelial growth factor and vascular endothelial growth factor receptor-2 expression in mdx mouse brain. Brain Res. 2002, 953, 12–16. [Google Scholar] [CrossRef]
- Nico, B.; Marzullo, A.; Corsi, P.; Vacca, A.; Roncali, L.; Ribatti, D. A possible role of tryptase in angiogenesis in the brain of mdx mouse, a model of Duchenne muscular dystrophy. Neuroscience 2004, 123, 585–588. [Google Scholar] [CrossRef]
- Nico, B.; Mangieri, D.; Crivellato, E.; Longo, V.; De Giorgis, M.; Capobianco, C.; Corsi, P.; Benagiano, V.; Roncali, L.; Ribatti, M. HIF activation and VEGF overexpression are coupled with ZO-1 up-phosphorylation in the brain of dystrophic mdx mouse. Brain Pathol. 2007, 17, 399–406. [Google Scholar] [CrossRef]
- Podkalicka, P.; Mucha, O.; Kaziród, K.; Bronisz-Budzyńska, I.; Ostrowska-Paton, S.; Tomczyk, M.; Andrysiak, K.; Stępniewski, J.; Dulak, J.; Łoboda, A. Age-Dependent Dysregulation of Muscle Vasculature and Blood Flow Recovery after Hindlimb Ischemia in the mdx Model of Duchenne Muscular Dystrophy. Biomedicines 2021, 9, 481. [Google Scholar] [CrossRef]
- Frankel, K.A.; Rosser, R.J. The pathology of the heart in progressive muscular dystrophy: Epimyocardial fibrosis. Hum. Pathol. 1976, 7, 375–386. [Google Scholar] [CrossRef]
- Moriuchi, T.; Kagawa, N.; Mukoyama, M.; Hizawa, K. Autopsy analyses of the muscular dystrophies. Tokushima J. Exp. Med. 1993, 40, 83–93. [Google Scholar] [PubMed]
- Starc, J.J.; Moore, R.A.; Rattan, M.S.; Villa, C.R.; Gao, Z.; Mazur, W.; Jefferies, J.L.; Taylor, M.D. Elevated Myocardial Extracellular Volume Fraction in Duchenne Muscular Dystrophy. Pediatr. Cardiol. 2017, 38, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Steplewski, A.; Fertala, A. Inhibition of collagen fibril formation. Fibrogenesis Tissue Repair 2012, 5, S29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guttman, M.; Rinn, J.L. Modular regulatory principles of large non-coding RNAs. Nature 2012, 482, 339–346. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Liang, Z.; Ge, M.; Qi, W.; Zhang, T.; Lin, F.; Peng, Z.; Zhao, H. Genome-wide identification and functional prediction of nitrogen-responsive intergenic and intronic long non-coding RNAs in maize (Zea mays L.). BMC Genom. 2016, 17, 350. [Google Scholar] [CrossRef] [Green Version]
- Bovolenta, M.; Erriquez, D.; Valli, E.; Brioschi, S.; Scotton, C.; Neri, M.; Falzarano, M.S.; Gherardi, S.; Fabris, M.; Rimessi, P.; et al. The DMD locus harbours multiple long non-coding RNAs which orchestrate and control transcription of muscle dystrophin mRNA isoforms. PLoS ONE 2012, 7, e45328. [Google Scholar] [CrossRef]
- Zhao, Q.; Sheng, M.-F.; Wang, Y.-Y.; Wang, X.-Y.; Liu, W.-Y.; Zhang, Y.-Y.; Ke, T.-Y.; Chen, S.; Pang, G.-Z.; Yong, L.; et al. LncRNA Gm26917 regulates inflammatory response in macrophages by enhancing Annexin A1 ubiquitination in LPS-induced acute liver injury. Front. Pharmacol. 2022, 13, 975250. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shen, Y.; Kim, I.-m.; Hamrick, M.; Tang, Y. Uncovering the Gene Regulatory Network of Endothelial Cells in Mouse Duchenne Muscular Dystrophy: Insights from Single-Nuclei RNA Sequencing Analysis. Biology 2023, 12, 422. https://doi.org/10.3390/biology12030422
Shen Y, Kim I-m, Hamrick M, Tang Y. Uncovering the Gene Regulatory Network of Endothelial Cells in Mouse Duchenne Muscular Dystrophy: Insights from Single-Nuclei RNA Sequencing Analysis. Biology. 2023; 12(3):422. https://doi.org/10.3390/biology12030422
Chicago/Turabian StyleShen, Yan, Il-man Kim, Mark Hamrick, and Yaoliang Tang. 2023. "Uncovering the Gene Regulatory Network of Endothelial Cells in Mouse Duchenne Muscular Dystrophy: Insights from Single-Nuclei RNA Sequencing Analysis" Biology 12, no. 3: 422. https://doi.org/10.3390/biology12030422
APA StyleShen, Y., Kim, I. -m., Hamrick, M., & Tang, Y. (2023). Uncovering the Gene Regulatory Network of Endothelial Cells in Mouse Duchenne Muscular Dystrophy: Insights from Single-Nuclei RNA Sequencing Analysis. Biology, 12(3), 422. https://doi.org/10.3390/biology12030422