

Modeling the Circadian Control of the Cell Cycle and Its Consequences for Cancer Chronotherapy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
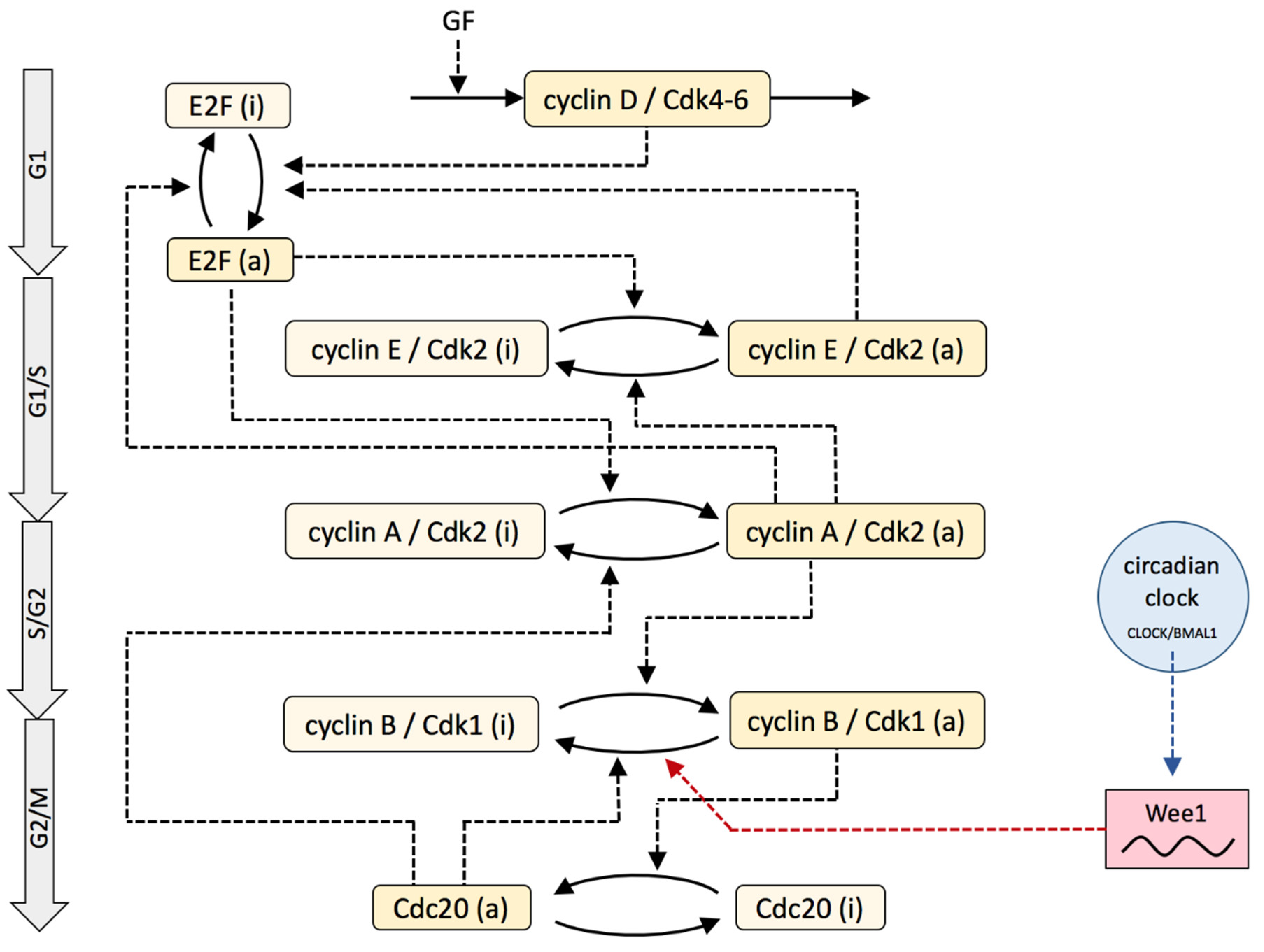
2. Model
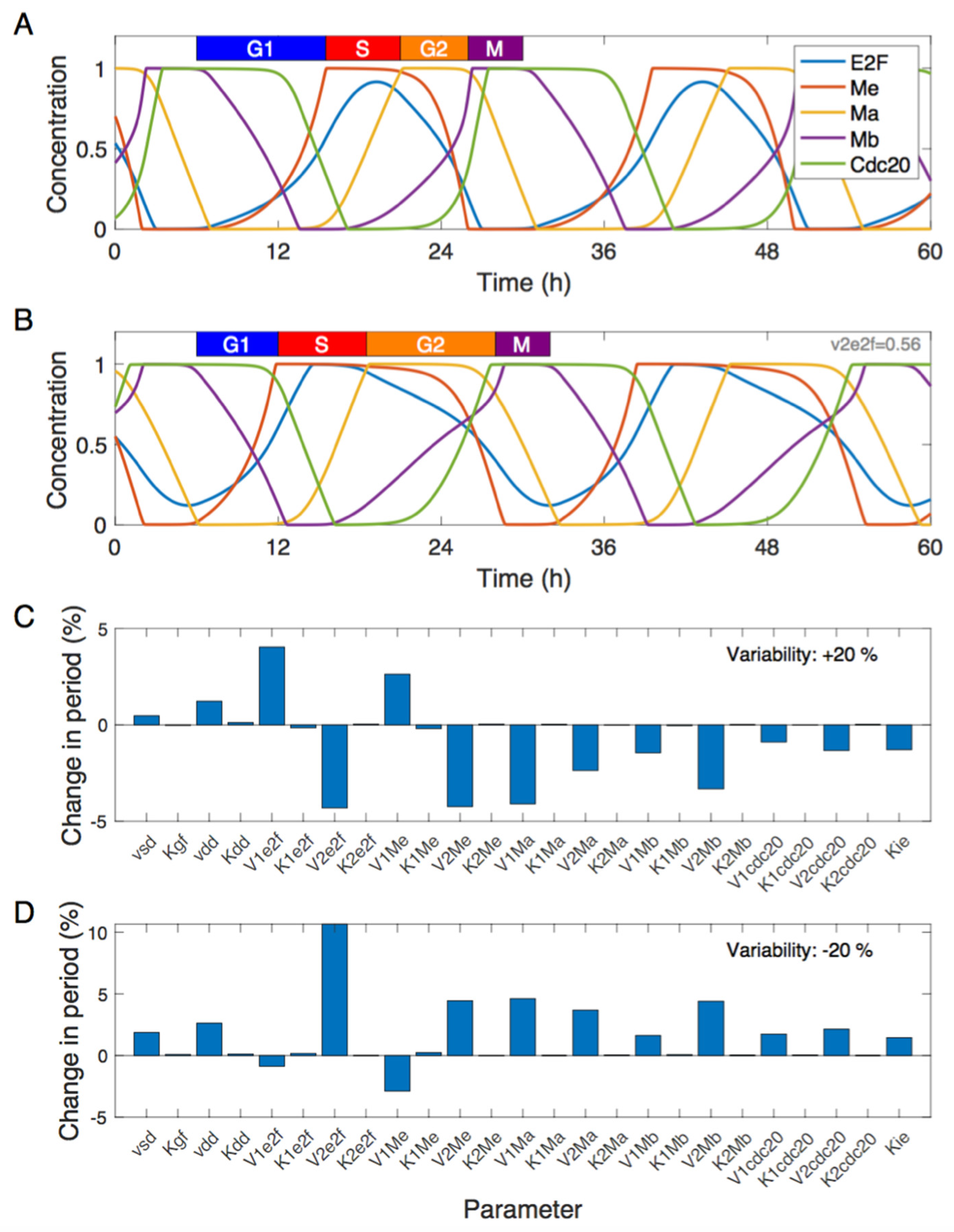
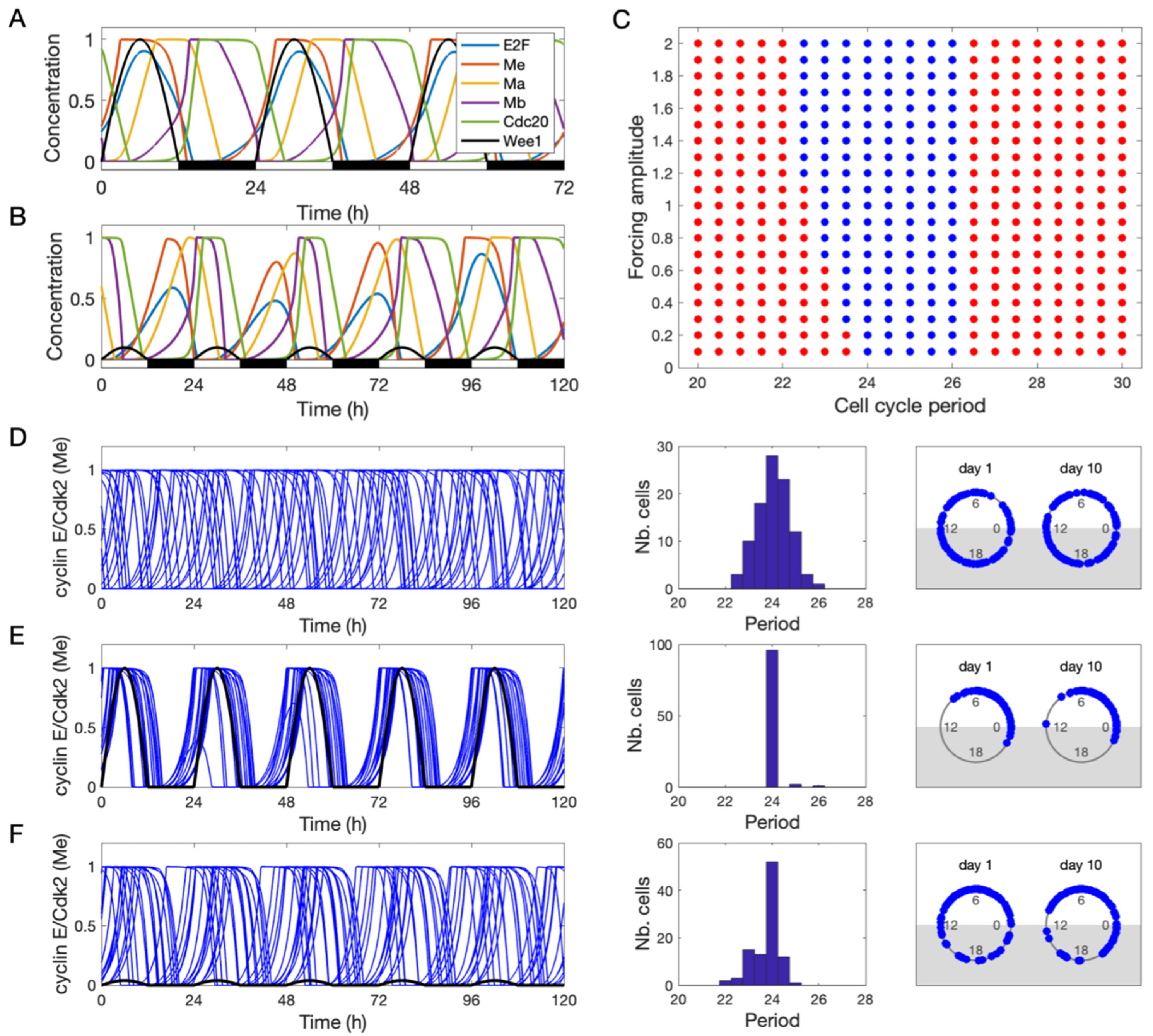
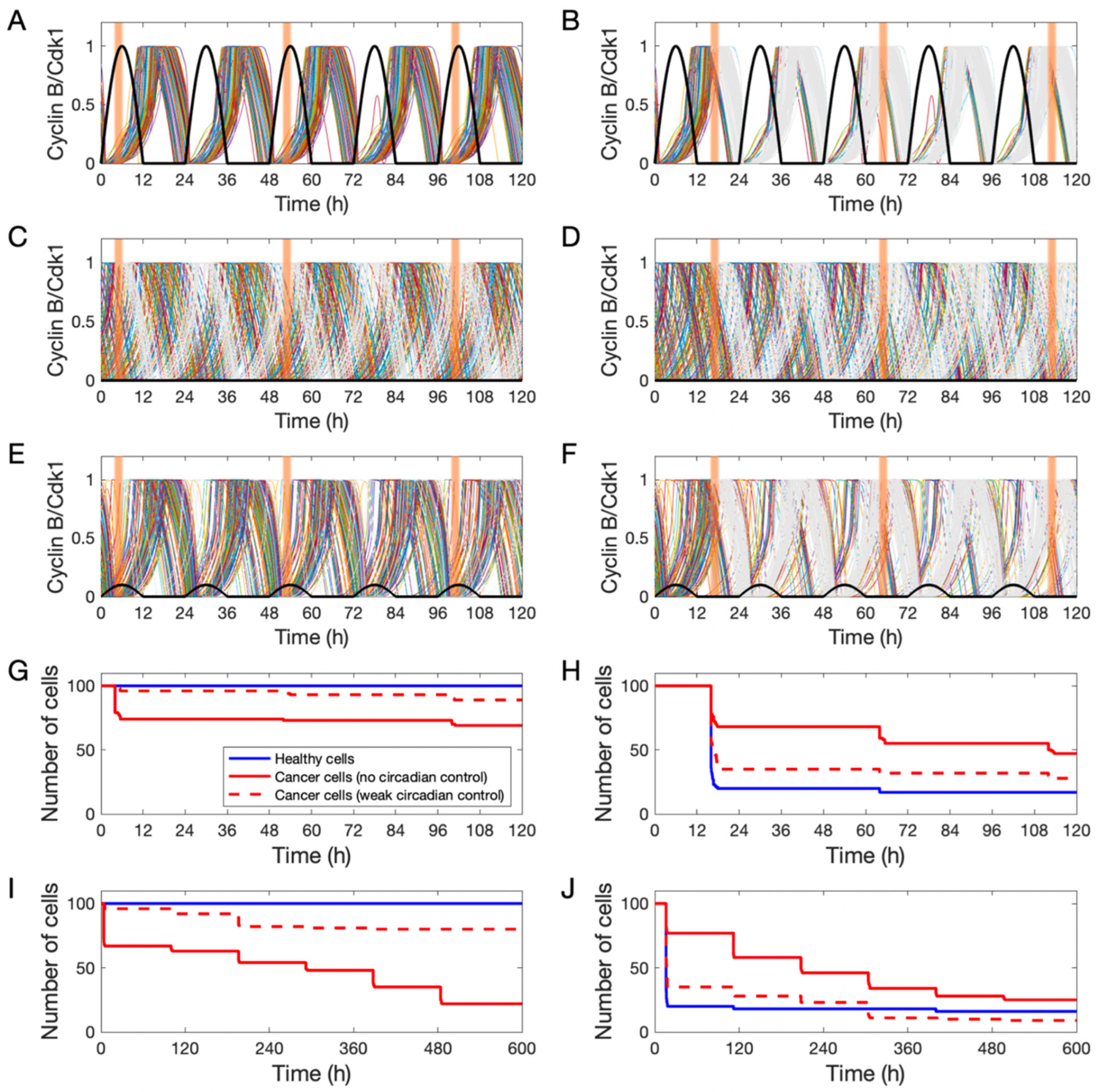
3. Results
3.1. Dynamics of the Cdk Network and Sensitivity Analysis
3.2. Entrainment of the Cell Cycle by the Circadian Clock
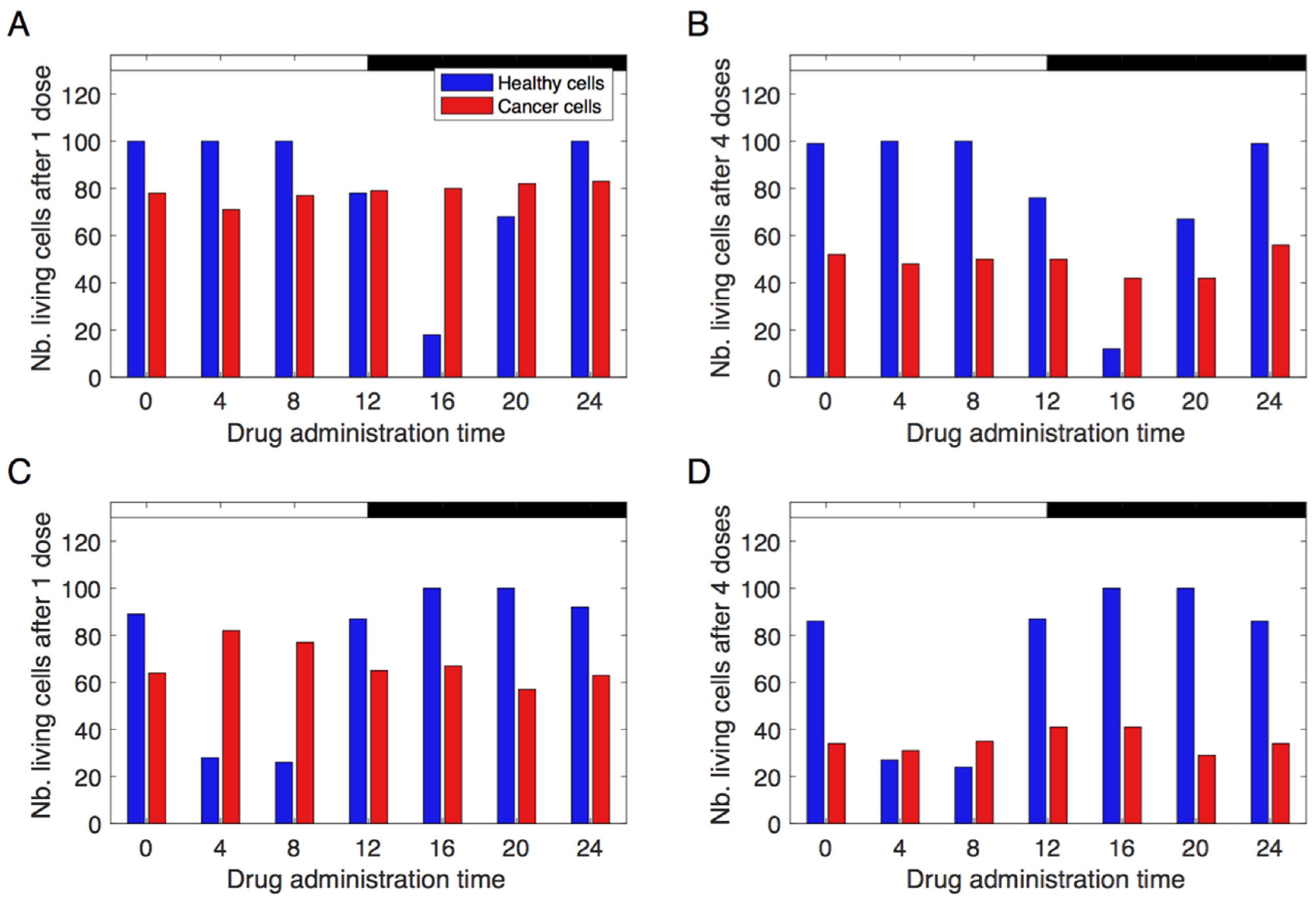
3.3. Simulating Chronotherapeutic Treatments
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cotter, T.G. Apoptosis and cancer: The genesis of a research field. Nat. Rev. Cancer 2009, 9, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Morgan, D.O. Principles of CDK regulation. Nature 1995, 374, 131–134. [Google Scholar] [CrossRef]
- Ballesta, A.; Innominato, P.F.; Dallmann, R.; Rand, D.A.; Lévi, F.A. Systems Chronotherapeutics. Pharmacol. Rev. 2017, 69, 161–199. [Google Scholar] [CrossRef] [PubMed]
- Amiama-Roig, A.; Verdugo-Sivianes, E.M.; Carnero, A.; Blanco, J.R. Cancers chronotherapy: Circadian rhythms and their influence in cancer therapy. Cancers 2022, 14, 5071. [Google Scholar] [CrossRef] [PubMed]
- Reppert, S.M.; Weaver, D.R. Coordination of circadian timing in mammals. Nature 2002, 418, 935–941. [Google Scholar] [CrossRef]
- Takahashi, J.S. Transcriptional architecture of the mammalian circadian clock. Nat. Rev. Genet. 2017, 18, 164–179. [Google Scholar] [CrossRef]
- Matsuo, T.; Yamaguchi, S.; Mitsui, S.; Emi, A.; Shimoda, F.; Okamura, H. Control mechanism of the circadian clock for timing of cell division in vivo. Science 2003, 302, 255–259. [Google Scholar] [CrossRef]
- Gréchez-Cassiau, A.; Rayet, B.; Guillaumond, F.; Teboul, M.; Delaunay, F. The circadian clock component Bmal1 is a critical regulator of p21WAF1/CIP1 expression and hepatocyte proliferation. J. Biol. Chem. 2008, 283, 4535–4542. [Google Scholar] [CrossRef]
- Fu, L.; Pelicano, H.; Liu, J.; Huang, P.; Lee, C.C. The circadian gene period2 plays an important role in tumor suppression and DNA damage response in vivo. Cell 2002, 111, 41–50. [Google Scholar] [CrossRef]
- Feillet, C.; Krusche, P.; Tamanini, F.; Janssens, R.C.; Downey, M.J.; Martin, P.; Teboul, M.; Saito, S.; Lévi, F.A.; Bretschneider, T.; et al. Phase locking and multiple oscillating attractors for the coupled mammalian clock and cell cycle. Proc. Natl. Acad. Sci. USA 2014, 111, 9828–9833. [Google Scholar] [CrossRef]
- Feillet, C.; van der Horst, G.T.; Levi, F.; Rand, D.A.; Delaunay, F. Coupling between the Circadian Clock and Cell Cycle Oscillators: Implication for Healthy Cells and Malignant Growth. Front. Neurol. 2015, 6, 96. [Google Scholar] [CrossRef] [PubMed]
- Shostak, A. Circadian clock, cell division, and cancer: From molecules to organism. Int. J. Mol. Sci. 2017, 18, 873. [Google Scholar] [CrossRef] [PubMed]
- Wood, P.; Yang, X.; Taber, A.; Oh, E.; Ansell, C.; Ayers, S.; Al-Assaad, Z.; Carnevale, K.; Berger, F.; Pena, M.; et al. Period 2 Mutation Accelerates ApcMin/+ Tumorigenesis. Mol. Cancer Res. 2008, 6, 1786–1793. [Google Scholar] [CrossRef] [PubMed]
- Davis, S.; Mirick, D.K.; Stevens, R.G. Night shift work, light at night, and risk of breast cancer. J. Natl. Cancer Inst. 2001, 93, 1557–1562. [Google Scholar] [CrossRef]
- Taniguchi, H.; Fernández, A.F.; Setién, F.; Ropero, S.; Ballestar, E.; Villanueva, A.; Yamamoto, H.; Imai, K.; Shinomura, Y.; Esteller, M. Epigenetic inactivation of the circadian clock gene BMAL1 in hematologic malignancies. Cancer Res. 2009, 69, 8447–8454. [Google Scholar] [CrossRef]
- Fu, L.; Kettner, N. The Circadian Clock in Cancer Development and Therapy. Prog. Mol. Biol. Transl. Sci. 2013, 119, 221–282. [Google Scholar] [PubMed]
- Amidi, A.; Wu, L.M. Circadian disruption and cancer- and treatment-related symptoms. Front. Oncol. 2022, 12, 1009064. [Google Scholar] [CrossRef]
- Pendergast, J.S.; Yeom, M.; Reyes, B.A.; Ohmiya, Y.; Yamazaki, S. Disconnected circadian and cell cycles in a tumor-driven cell line. Commun. Integr. Biol. 2010, 3, 536–539. [Google Scholar] [CrossRef]
- Lévi, F.; Altinok, A.; Clairambault, J.; Goldbeter, A. Implications of circadian clocks for the rhythmic delivery of cancer therapeutics. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2008, 366, 3575–3598. [Google Scholar] [CrossRef]
- Ozturk, N.; Ozturk, D.; Kavakli, I.H.; Okyar, A. Molecular aspects of circadian pharmacology and relevance for cancer chronotherapy. Int. J. Mol. Sci. 2017, 18, 2168. [Google Scholar] [CrossRef]
- Wang, T.H.; Wang, H.S.; Soong, Y.K. Paclitaxel-induced cell death: Where the cell cycle and apoptosis come together. Cancer 2000, 88, 2619–2628. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Cheng, B.; Xie, M.; Chen, Y.; Zhao, J.; Zhou, X.; Chen, L. Circadian clock gene Bmal1 inhibits tumorigenesis and increases paclitaxel sensitivity in tongue squamous cell carcinoma. Cancer Res. 2017, 77, 532–544. [Google Scholar] [CrossRef] [PubMed]
- Iurisci, I.; Filipski, E.; Reinhardt, J.; Bach, S.; Gianella-Borradori, A.; Iacobelli, S.; Meijer, L.; Lévi, F. Improved Tumor Control through Circadian Clock Induction by Seliciclib, a Cyclin-Dependent Kinase Inhibitor. Cancer Res. 2006, 66, 10720–10728. [Google Scholar] [CrossRef] [PubMed]
- Mizrahi, S.P.; Sandler, O.; Lande-Diner, L.; Balaban, N.Q.; Simon, I. Distinguishing between stochasticity and determinism: Examples from cell cycle duration variability. Bioessays 2015, 38, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Elowitz, M.B.; Levine, A.J.; Siggia, E.D.; Swain, P.S. Stochastic Gene Expression in a Single Cell. Science 2002, 297, 1183–1186. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; van Oudenaarden, A. Nature, nurture, or chance: Stochastic gene expression and its consequences. Cell 2008, 135, 216–226. [Google Scholar] [CrossRef] [PubMed]
- Govindaraj, V.; Sarma, S.; Karulkar, A.; Purwar, R.; Kar, S. Transcriptional Fluctuations Govern the Serum-Dependent Cell Cycle Duration Heterogeneities in Mammalian Cells. ACS Synth. Biol. 2022, 11, 3743–3758. [Google Scholar] [CrossRef]
- Huh, D.; Paulsson, J. Non-genetic heterogeneity from stochastic partitioning at cell division. Nat. Genet. 2011, 43, 95–100. [Google Scholar] [CrossRef]
- Sandler, O.; Mizrahi, S.P.; Weiss, N.; Agam, O.; Simon, I.; Balaban, N.Q. Lineage correlations of single cell division time as a probe of cell-cycle dynamics. Nature 2015, 519, 468–471. [Google Scholar] [CrossRef]
- Mura, M.; Feillet, C.; Bertolusso, R.; Delaunay, F.; Kimmel, M. Mathematical modelling reveals unexpected inheritance and variability patterns of cell cycle parameters in mammalian cells. PLoS Comput. Biol. 2019, 15, e1007054. [Google Scholar] [CrossRef]
- Gérard, C.; Goldbeter, A. Temporal self-organization of the cyclin/Cdk network driving the mammalian cell cycle. Proc. Natl. Acad. Sci. USA 2009, 106, 21643–21648. [Google Scholar] [CrossRef] [PubMed]
- Gérard, C.; Goldbeter, A. A skeleton model for the network of cyclin-dependent kinases driving the mammalian cell cycle. Interface Focus 2011, 1, 24–35. [Google Scholar] [CrossRef] [PubMed]
- Gérard, C.; Gonze, D.; Goldbeter, A. Effect of positive feedback loops on the robustness of oscillations in the network of cyclin-dependent kinases driving the mammalian cell cycle. FEBS J. 2012, 279, 3411–3431. [Google Scholar] [CrossRef] [PubMed]
- Gérard, C.; Goldbeter, A. Entrainment of the Mammalian Cell Cycle by the Circadian Clock: Modeling Two Coupled Cellular Rhythms. PLoS Comput. Biol. 2012, 8, e1002516. [Google Scholar] [CrossRef]
- Almeida, S.; Chaves, M.; Delaunay, F. Cell cycle period control through modulation of clock inputs. J. Bioinform. Comput. Biol. 2020, 18, 2040006. [Google Scholar] [CrossRef]
- Almeida, S.; Chaves, M.; Delaunay, F. Control of synchronization ratios in clock/cell cycle coupling by growth factors and glucocorticoids. R. Soc. Open Sci. 2020, 7, 192054. [Google Scholar] [CrossRef]
- Gérard, C.; Gonze, D.; Goldbeter, A. Revisiting a skeleton model for the mammalian cell cycle: From bistability to Cdk oscillations and cellular heterogeneity. J. Theor. Biol. 2019, 461, 276–290. [Google Scholar] [CrossRef]
- Ballesta, A.; Dulong, S.; Abbara, C.; Cohen, B.; Okyar, A.; Clairambault, J.; Lévi, F. A Combined Experimental and Mathematical Approach for Molecular-based Optimization of Irinotecan Circadian Delivery. PLoS Comput. Biol. 2011, 7, e1002143. [Google Scholar] [CrossRef]
- Dulong, S.; Ballesta, A.; Okyar, A.; Lévi, F. Identification of Circadian Determinants of Cancer Chronotherapy through In Vitro Chronopharmacology and Mathematical Modeling. Mol. Cancer Ther. 2015, 14, 2154–2164. [Google Scholar] [CrossRef]
- Hesse, J.; Martinelli, J.; Aboumanify, O.; Ballesta, A.; Relógio, A. A mathematical model of the circadian clock and drug pharmacology to optimize irinotecan administration timing in colorectal cancer. Comput. Struct. Biotechnol. J. 2021, 19, 5170–5183. [Google Scholar] [CrossRef]
- Martinelli, J.; Dulong, S.; Li, X.-M.; Teboul, M.; Soliman, S.; Lévi, F.; Fages, F.; Ballesta, A. Model learning to identify systemic regulators of the peripheral circadian clock. Bioinformatics 2021, 37, i401–i409. [Google Scholar] [CrossRef] [PubMed]
- Catozzi, S.; Hill, R.; Li, X.; Dulong, S.; Collard, E.; Ballesta, A. Interspecies and in vitro-in vivo scaling for quantitative modeling of whole-body drug pharmacokinetics in patients: Application to the anticancer drug oxaliplatin. CPT Pharmacomet. Syst. Pharmacol. 2023, 12, 221–235. [Google Scholar] [CrossRef] [PubMed]
- Altinok, A.; Gonze, D.; Lévi, F.; Goldbeter, A. An automaton model for the cell cycle. Interface Focus 2011, 1, 36–47. [Google Scholar] [CrossRef] [PubMed]
- Altinok, A.; Lévi, F.; Goldbeter, A. A cell cycle automaton model for probing circadian patterns of anticancer drug delivery. Adv. Drug Deliv. Rev. 2007, 59, 1036–1053. [Google Scholar] [CrossRef] [PubMed]
- Altinok, A.; Lévi, F.; Goldbeter, A. Identifying mechanisms of chronotolerance and chronoefficacy for the anticancer drugs 5-fluorouracil and oxaliplatin by computational modeling. Eur. J. Pharm. Sci. 2009, 36, 20–38. [Google Scholar] [CrossRef]
- Watanabe, N.; Broome, M.; Hunter, T. Regulation of the human WEE1Hu CDK tyrosine 15-kinase during the cell cycle. EMBO J. 1995, 14, 1878–1891. [Google Scholar] [CrossRef]
- Wu, C.L.; Kirley, S.D.; Xiao, H.; Chuang, Y.; Chung, D.C.; Zukerberg, L.R. Cables enhances cdk2 tyrosine 15 phosphorylation by Wee1, inhibits cell growth, and is lost in many human colon and squamous cancers. Cancer Res. 2001, 61, 7325–7332. [Google Scholar]
- Heldt, F.S.; Barr, A.R.; Cooper, S.; Bakal, C.; Novák, B. A comprehensive model for the proliferation-quiescence decision in response to endogenous DNA damage in human cells. Proc. Natl. Acad. Sci. USA 2018, 115, 2532–2537. [Google Scholar] [CrossRef]
- Spencer, S.L.; Cappell, S.D.; Tsai, F.-C.; Overton, K.W.; Wang, C.L.; Meyer, T. The proliferation-quiescence decision is controlled by a bifurcation in CDK2 activity at mitotic exit. Cell 2013, 155, 369–383. [Google Scholar] [CrossRef]
- Yan, J.; Goldbeter, A. Robust synchronization of the cell cycle and the circadian clock through bidirectional coupling. J. R. Soc. Interface 2019, 16, 20190376. [Google Scholar] [CrossRef]
- Hill, R.J.W.; Innominato, P.F.; Lévi, F.; Ballesta, A. Optimizing circadian drug infusion schedules towards personalized cancer chronotherapy. PLoS Comput. Biol. 2020, 16, e1007218. [Google Scholar] [CrossRef]
- Hesse, J.; Malhan, D.; Yalҫin, M.; Aboumanify, O.; Basti, A.; Relógio, A. An Optimal Time for Treatment—Predicting Circadian Time by Machine Learning and Mathematical Modelling. Cancers 2020, 12, 3103. [Google Scholar] [CrossRef] [PubMed]
- El Cheikh, R.; Bernard, S.; El Khatib, N. Modeling circadian clock–cell cycle interaction effects on cell population growth rates. J. Theor. Biol. 2014, 363, 318–331. [Google Scholar] [CrossRef] [PubMed]
- El Cheikh, R.; Bernard, S.; El Khatib, N. A multiscale modelling approach for the regulation of the cell cycle by the circadian clock. J. Theor. Biol. 2017, 426, 117–125. [Google Scholar] [CrossRef] [PubMed]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Leung, C.; Gérard, C.; Gonze, D. Modeling the Circadian Control of the Cell Cycle and Its Consequences for Cancer Chronotherapy. Biology 2023, 12, 612. https://doi.org/10.3390/biology12040612
Leung C, Gérard C, Gonze D. Modeling the Circadian Control of the Cell Cycle and Its Consequences for Cancer Chronotherapy. Biology. 2023; 12(4):612. https://doi.org/10.3390/biology12040612
Chicago/Turabian StyleLeung, Courtney, Claude Gérard, and Didier Gonze. 2023. "Modeling the Circadian Control of the Cell Cycle and Its Consequences for Cancer Chronotherapy" Biology 12, no. 4: 612. https://doi.org/10.3390/biology12040612
APA StyleLeung, C., Gérard, C., & Gonze, D. (2023). Modeling the Circadian Control of the Cell Cycle and Its Consequences for Cancer Chronotherapy. Biology, 12(4), 612. https://doi.org/10.3390/biology12040612