
The ACSL4 Network Regulates Cell Death and Autophagy in Diseases


Abstract
:Simple Summary
Abstract
1. Introduction
2. Discovery of ACSL4
3. Structure of ACSL4 Gene and Protein
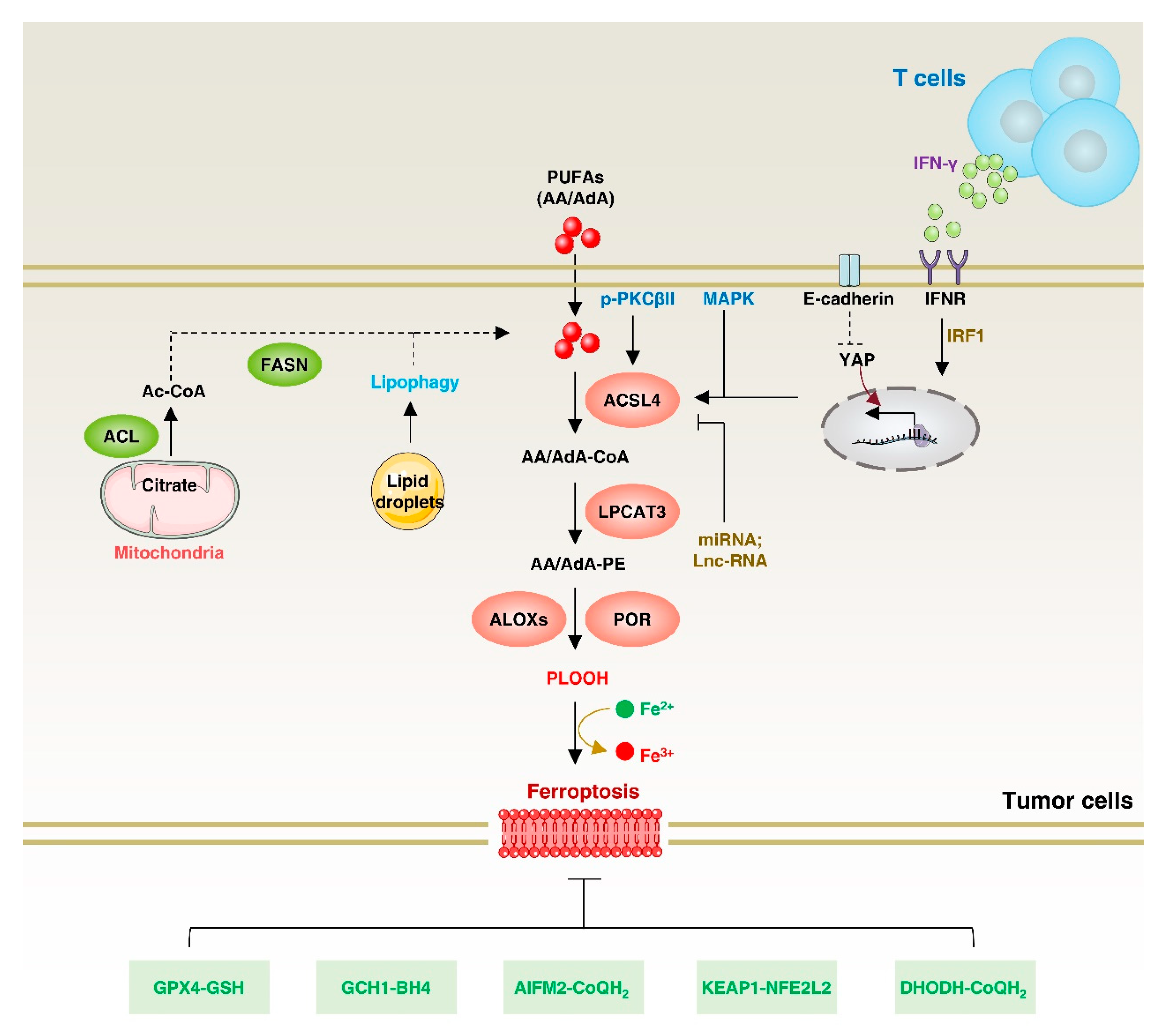
4. Function of ACSL4
5. Modulation of ACSL4
5.1. Transcriptional Regulation
5.2. Post-Translational Modifications
5.2.1. Phosphorylation
5.2.2. Methylation
5.2.3. Acetylation
5.2.4. O-GlcNAcylation
5.2.5. Ubiquitination
5.3. Protein–Protein Interaction
6. ACSL4 in Apoptosis
7. ACSL4 in Ferroptosis
8. ACSL4 in Autophagy
9. ACSL4 in Diseases
9.1. Obesity
9.2. Ischemia–Reperfusion Disease
9.3. Nonalcoholic Fatty Liver Disease (NAFLD)
9.4. Neurodegeneration
9.5. Exertional Heat Stroke
9.6. Acute Kidney Injury
9.7. Cancer
10. ACSL4-targeted Drugs
10.1. Inducers
10.2. Inhibitors
11. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Overmyer, K.; Brosché, M.; Kangasjärvi, J. Reactive oxygen species and hormonal control of cell death. Trends Plant Sci. 2003, 8, 335–342. [Google Scholar] [CrossRef] [PubMed]
- Livesey, K.M.; Kang, R.; Vernon, P.; Buchser, W.; Loughran, P.; Watkins, S.C.; Zhang, L.; Manfredi, J.J.; Zeh, H.J., 3rd; Li, L.; et al. p53/HMGB1 complexes regulate autophagy and apoptosis. Cancer Res. 2012, 72, 1996–2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.; Cao, L.; Kang, R.; Yang, M.; Wang, Z.; Peng, Y.; Tan, Y.; Liu, L.; Xie, M.; Zhao, Y.; et al. UV irradiation resistance-associated gene suppresses apoptosis by interfering with BAX activation. EMBO Rep. 2011, 12, 727–734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Kang, R.; Kroemer, G.; Tang, D. Broadening horizons: The role of ferroptosis in cancer. Nat. Rev. Clin. Oncol. 2021, 18, 280–296. [Google Scholar] [CrossRef]
- Yuan, H.; Li, X.; Zhang, X.; Kang, R.; Tang, D. Identification of ACSL4 as a biomarker and contributor of ferroptosis. Biochem. Biophys. Res. Commun. 2016, 478, 1338–1343. [Google Scholar] [CrossRef]
- Tang, D.; Kang, R.; Berghe, T.V.; Vandenabeele, P.; Kroemer, G. The molecular machinery of regulated cell death. Cell Res. 2019, 29, 347–364. [Google Scholar] [CrossRef] [Green Version]
- Chen, F.; Zhu, S.; Kang, R.; Tang, D.; Liu, J. ATP6V0D1 promotes alkaliptosis by blocking STAT3-mediated lysosomal pH homeostasis. Cell Rep. 2022, 42, 111911. [Google Scholar] [CrossRef]
- Kang, R.; Loux, T.; Tang, D.; Schapiro, N.E.; Vernon, P.; Livesey, K.M.; Krasinskas, A.; Lotze, M.T.; Zeh, H.J., 3rd. The expression of the receptor for advanced glycation endproducts (RAGE) is permissive for early pancreatic neoplasia. Proc. Natl. Acad. Sci. USA 2012, 109, 7031–7036. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, Y.; Wang, Y.; Li, C.; Xie, Y.; Klionsky, D.J.; Kang, R.; Tang, D. TMEM164 is a new determinant of autophagy-dependent ferroptosis. Autophagy 2022, 19, 945–956. [Google Scholar] [CrossRef]
- Liu, J.; Kuang, F.; Kroemer, G.; Klionsky, D.J.; Kang, R.; Tang, D. Autophagy-Dependent Ferroptosis: Machinery and Regulation. Cell Chem. Biol. 2020, 27, 420–435. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Cai, X.; Kang, R.; Liu, J.; Tang, D. Autophagy-dependent ferroptosis in cancer. Antioxid. Redox Signal. 2023. [Google Scholar] [CrossRef]
- Schütter, M.; Giavalisco, P.; Brodesser, S.; Graef, M. Local Fatty Acid Channeling into Phospholipid Synthesis Drives Phagophore Expansion during Autophagy. Cell 2020, 180, 135–149.e14. [Google Scholar] [CrossRef] [PubMed]
- Quan, J.; Bode, A.M.; Luo, X. ACSL family: The regulatory mechanisms and therapeutic implications in cancer. Eur. J. Pharmacol. 2021, 909, 174397. [Google Scholar] [CrossRef]
- Hatch, M.D.; Stumpf, P.K. Fat metabolism in higher plants. XVI. Acetyl coenzyme A carboxylase and acyl coenzyme A-malonyl coenzyme A transcarboxylase from wheat germ. J. Biol. Chem. 1961, 236, 2879–2885. [Google Scholar] [CrossRef] [PubMed]
- Brophy, P.J.; Vance, D.E. The synthesis and hydrolysis of long-chain fatty acyl-coenzyme A thioesters by soluble and microsomal fractions from the brain of the developing rat. Biochem. J. 1976, 160, 247–251. [Google Scholar] [CrossRef] [Green Version]
- Ockner, R.K.; Manning, J.A. Fatty acid binding protein. Role in esterification of absorbed long chain fatty acid in rat intestine. J. Clin. Investig. 1976, 58, 632–641. [Google Scholar] [CrossRef]
- Wilson, D.B.; Prescott, S.M.; Majerus, P.W. Discovery of an arachidonoyl coenzyme A synthetase in human platelets. J. Biol. Chem. 1982, 257, 3510–3515. [Google Scholar] [CrossRef]
- Nagamatsu, K.; Soeda, S.; Mori, M.; Kishimoto, Y. Lignoceroyl-coenzyme A synthetase from developing rat brain: Partial purification, characterization and comparison with palmitoyl-coenzyme A synthetase activity and liver enzyme. Biochim. Biophys. Acta 1985, 836, 80–88. [Google Scholar] [CrossRef]
- Abe, T.; Fujino, T.; Fukuyama, R.; Minoshima, S.; Shimizu, N.; Toh, H.; Suzuki, H.; Yamamoto, T. Human long-chain acyl-CoA synthetase: Structure and chromosomal location. J. Biochem. 1992, 111, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.J.; Fujino, T.; Sasano, H.; Minekura, H.; Yabuki, N.; Nagura, H.; Iijima, H.; Yamamoto, T.T. A novel arachidonate-preferring acyl-CoA synthetase is present in steroidogenic cells of the rat adrenal, ovary, and testis. Proc. Natl. Acad. Sci. USA 1997, 94, 2880–2884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccini, M.; Vitelli, F.; Bruttini, M.; Pober, B.R.; Jonsson, J.J.; Villanova, M.; Zollo, M.; Borsani, G.; Ballabio, A.; Renieri, A. FACL4, a new gene encoding long-chain acyl-CoA synthetase 4, is deleted in a family with Alport syndrome, elliptocytosis, and mental retardation. Genomics 1998, 47, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Traer, E.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Cloning, expression, and chromosomal localization of human long-chain fatty acid-CoA ligase 4 (FACL4). Genomics 1998, 49, 327–330. [Google Scholar] [CrossRef] [PubMed]
- Meloni, I.; Muscettola, M.; Raynaud, M.; Longo, I.; Bruttini, M.; Moizard, M.P.; Gomot, M.; Chelly, J.; des Portes, V.; Fryns, J.P.; et al. FACL4, encoding fatty acid-CoA ligase 4, is mutated in nonspecific X-linked mental retardation. Nat. Genet. 2002, 30, 436–440. [Google Scholar] [CrossRef]
- Cao, Y.; Dave, K.B.; Doan, T.P.; Prescott, S.M. Fatty acid CoA ligase 4 is up-regulated in colon adenocarcinoma. Cancer Res. 2001, 61, 8429–8434. [Google Scholar]
- Cao, Y.; Murphy, K.J.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Expression of fatty acid-CoA ligase 4 during development and in brain. FEBS Lett. 2000, 467, 263–267. [Google Scholar] [CrossRef]
- Sung, Y.K.; Park, M.K.; Hong, S.H.; Hwang, S.Y.; Kwack, M.H.; Kim, J.C.; Kim, M.K. Regulation of cell growth by fatty acid-CoA ligase 4 in human hepatocellular carcinoma cells. Exp. Mol. Med. 2007, 39, 477–482. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.; Zheng, Z.; An, C.; Gao, X.; Zhang, F. Genetic variations of FACL4 have no obvious influence on cognitive ability in young Chinese children. Psychiatry Res. 2010, 178, 202–204. [Google Scholar] [CrossRef]
- Minekura, H.; Kang, M.J.; Inagaki, Y.; Cho, Y.Y.; Suzuki, H.; Fujino, T.; Yamamoto, T.T. Exon/intron organization and transcription units of the human acyl-CoA synthetase 4 gene. Biochem. Biophys. Res. Commun. 2001, 286, 80–86. [Google Scholar] [CrossRef]
- Watkins, P.A.; Maiguel, D.; Jia, Z.; Pevsner, J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J. Lipid Res. 2007, 48, 2736–2750. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Shao, X.; Shen, J.; Lin, Q.; Zhu, X.; Li, S.; Li, J.; Zhou, W.; Qi, C.; Ni, Z. Downregulation of PPARα mediates FABP1 expression, contributing to IgA nephropathy by stimulating ferroptosis in human mesangial cells. Int. J. Biol. Sci. 2022, 18, 5438–5458. [Google Scholar] [CrossRef] [PubMed]
- Hoashi, S.; Hinenoya, T.; Tanaka, A.; Ohsaki, H.; Sasazaki, S.; Taniguchi, M.; Oyama, K.; Mukai, F.; Mannen, H. Association between fatty acid compositions and genotypes of FABP4 and LXR-alpha in Japanese black cattle. BMC Genet. 2008, 9, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Y.; Feng, D.; Wang, Z.; Zhao, Y.; Sun, R.; Tian, D.; Liu, D.; Zhang, F.; Ning, S.; Yao, J.; et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death Differ. 2019, 26, 2284–2299. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Li, X.; Ding, N.; Teng, J.; Zhang, S.; Zhang, Q.; Tang, H. miR-34a regulates adipogenesis in porcine intramuscular adipocytes by targeting ACSL4. BMC Genet. 2020, 21, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Zhao, R.; Lv, M.; Xu, X.; Liu, W.; Li, X.; Gao, Y.; Zhao, Z.; Zhang, Z.; Li, Y.; et al. ACSL4 promotes microglia-mediated neuroinflammation by regulating lipid metabolism and VGLL4 expression. Brain Behav. Immun. 2023, 109, 331–343. [Google Scholar] [CrossRef]
- Orlando, U.; Cooke, M.; Maciel, F.C.; Papadopoulos, V.; Podestá, E.J.; Maloberti, P. Characterization of the mouse promoter region of the acyl-CoA synthetase 4 gene: Role of Sp1 and CREB. Mol. Cell. Endocrinol. 2013, 369, 15–26. [Google Scholar] [CrossRef]
- Verot, L.; Alloisio, N.; Morlé, L.; Bozon, M.; Touraine, R.; Plauchu, H.; Edery, P. Localization of a non-syndromic X-linked mental retardation gene (MRX80) to Xq22-q24. Am. J. Med. Genet. Part A 2003, 122a, 37–41. [Google Scholar] [CrossRef]
- Ma, L.L.; Liang, L.; Zhou, D.; Wang, S.W. Tumor suppressor miR-424-5p abrogates ferroptosis in ovarian cancer through targeting ACSL4. Neoplasma 2021, 68, 165–173. [Google Scholar] [CrossRef]
- Xiao, F.J.; Zhang, D.; Wu, Y.; Jia, Q.H.; Zhang, L.; Li, Y.X.; Yang, Y.F.; Wang, H.; Wu, C.T.; Wang, L.S. miRNA-17-92 protects endothelial cells from erastin-induced ferroptosis through targeting the A20-ACSL4 axis. Biochem. Biophys. Res. Commun. 2019, 515, 448–454. [Google Scholar] [CrossRef]
- Cao, Y.; Pearman, A.T.; Zimmerman, G.A.; McIntyre, T.M.; Prescott, S.M. Intracellular unesterified arachidonic acid signals apoptosis. Proc. Natl. Acad. Sci. USA 2000, 97, 11280–11285. [Google Scholar] [CrossRef] [Green Version]
- Küch, E.M.; Vellaramkalayil, R.; Zhang, I.; Lehnen, D.; Brügger, B.; Stremmel, W.; Ehehalt, R.; Poppelreuther, M.; Füllekrug, J. Differentially localized acyl-CoA synthetase 4 isoenzymes mediate the metabolic channeling of fatty acids towards phosphatidylinositol. Biochim. Biophys. Acta 2014, 1841, 227–239. [Google Scholar] [CrossRef] [PubMed]
- Meloni, I.; Parri, V.; De Filippis, R.; Ariani, F.; Artuso, R.; Bruttini, M.; Katzaki, E.; Longo, I.; Mari, F.; Bellan, C.; et al. The XLMR gene ACSL4 plays a role in dendritic spine architecture. Neuroscience 2009, 159, 657–669. [Google Scholar] [CrossRef] [PubMed]
- Obermeyer, T.; Fraisl, P.; DiRusso, C.C.; Black, P.N. Topology of the yeast fatty acid transport protein Fat1p: Mechanistic implications for functional domains on the cytosolic surface of the plasma membrane. J. Lipid Res. 2007, 48, 2354–2364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimbara-Matsubayashi, S.; Kuwata, H.; Tanaka, N.; Kato, M.; Hara, S. Analysis on the Substrate Specificity of Recombinant Human Acyl-CoA Synthetase ACSL4 Variants. Biol. Pharm. Bull. 2019, 42, 850–855. [Google Scholar] [CrossRef] [Green Version]
- Soupene, E.; Kuypers, F.A. Multiple erythroid isoforms of human long-chain acyl-CoA synthetases are produced by switch of the fatty acid gate domains. BMC Mol. Biol. 2006, 7, 21. [Google Scholar] [CrossRef] [Green Version]
- Soupene, E.; Kuypers, F.A. Mammalian long-chain acyl-CoA synthetases. Exp. Biol. Med. 2008, 233, 507–521. [Google Scholar] [CrossRef] [Green Version]
- Nakahara, K.; Ohkuni, A.; Kitamura, T.; Abe, K.; Naganuma, T.; Ohno, Y.; Zoeller, R.A.; Kihara, A. The Sjögren-Larsson syndrome gene encodes a hexadecenal dehydrogenase of the sphingosine 1-phosphate degradation pathway. Mol. Cell 2012, 46, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Dhiman, H.; Gerstl, M.P.; Ruckerbauer, D.; Hanscho, M.; Himmelbauer, H.; Clarke, C.; Barron, N.; Zanghellini, J.; Borth, N. Genetic and Epigenetic Variation across Genes Involved in Energy Metabolism and Mitochondria of Chinese Hamster Ovary Cell Lines. Biotechnol. J. 2019, 14, e1800681. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Fu, C.Y.; Shen, G.; Wang, J.; Xu, L.; Li, H.; Cao, X.; Zheng, M.Z.; Shen, Y.L.; Zhong, J.; et al. Macrophages induce cardiomyocyte ferroptosis via mitochondrial transfer. Free Radic. Biol. Med. 2022, 190, 1–14. [Google Scholar] [CrossRef]
- Ohkuni, A.; Ohno, Y.; Kihara, A. Identification of acyl-CoA synthetases involved in the mammalian sphingosine 1-phosphate metabolic pathway. Biochem. Biophys. Res. Commun. 2013, 442, 195–201. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.B.; Kan, C.F.K.; Kraemer, F.B.; Sobel, R.A.; Liu, J. Liver-specific knockdown of long-chain acyl-CoA synthetase 4 reveals its key role in VLDL-TG metabolism and phospholipid synthesis in mice fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2019, 316, E880–E894. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Ding, C.; Chen, Y.; Hu, W.; Yu, C.; Peng, C.; Feng, X.; Cheng, Q.; Wu, W.; Lu, Y.; et al. ACSL4 reprograms fatty acid metabolism in hepatocellular carcinoma via c-Myc/SREBP1 pathway. Cancer Lett. 2021, 502, 154–165. [Google Scholar] [CrossRef] [PubMed]
- Cho, Y.Y. A novel role of brain-type ACS4 isotype in neuronal differentiation. Biochem. Biophys. Res. Commun. 2012, 419, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Shui, S.; Zhao, Z.; Wang, H.; Conrad, M.; Liu, G. Non-enzymatic lipid peroxidation initiated by photodynamic therapy drives a distinct ferroptosis-like cell death pathway. Redox Biol. 2021, 45, 102056. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Cao, D.; Qiu, S.; Li, Y.; Jiang, C.; Bian, R.; Yang, Y.; Li, L.; Li, X.; et al. SIRT3 inhibits gallbladder cancer by induction of AKT-dependent ferroptosis and blockade of epithelial-mesenchymal transition. Cancer Lett. 2021, 510, 93–104. [Google Scholar] [CrossRef]
- Wu, J.; Minikes, A.M.; Gao, M.; Bian, H.; Li, Y.; Stockwell, B.R.; Chen, Z.N.; Jiang, X. Intercellular interaction dictates cancer cell ferroptosis via NF2-YAP signalling. Nature 2019, 572, 402–406. [Google Scholar] [CrossRef]
- Chen, B.; Wang, H.; Lv, C.; Mao, C.; Cui, Y. Long non-coding RNA H19 protects against intracerebral hemorrhage injuries via regulating microRNA-106b-5p/acyl-CoA synthetase long chain family member 4 axis. Bioengineered 2021, 12, 4004–4015. [Google Scholar] [CrossRef]
- Lu, Y.; Chan, Y.T.; Tan, H.Y.; Zhang, C.; Guo, W.; Xu, Y.; Sharma, R.; Chen, Z.S.; Zheng, Y.C.; Wang, N.; et al. Epigenetic regulation of ferroptosis via ETS1/miR-23a-3p/ACSL4 axis mediates sorafenib resistance in human hepatocellular carcinoma. J. Exp. Clin. Cancer Res. CR 2022, 41, 3. [Google Scholar] [CrossRef]
- Li, Y.J.; Fahrmann, J.F.; Aftabizadeh, M.; Zhao, Q.; Tripathi, S.C.; Zhang, C.; Yuan, Y.; Ann, D.; Hanash, S.; Yu, H. Fatty acid oxidation protects cancer cells from apoptosis by increasing mitochondrial membrane lipids. Cell Rep. 2022, 39, 110870. [Google Scholar] [CrossRef]
- Shi, L.; Song, Z.; Li, Y.; Huang, J.; Zhao, F.; Luo, Y.; Wang, J.; Deng, F.; Shadekejiang, H.; Zhang, M.; et al. MiR-20a-5p alleviates kidney ischemia/reperfusion injury by targeting ACSL4-dependent ferroptosis. Am. J. Transplant. Off. J. Am. Soc. Transplant. Am. Soc. Transpl. Surg. 2023, 23, 11–25. [Google Scholar] [CrossRef]
- Smith, M.E.; Saraceno, G.E.; Capani, F.; Castilla, R. Long-chain acyl-CoA synthetase 4 is regulated by phosphorylation. Biochem. Biophys. Res. Commun. 2013, 430, 272–277. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.L.; Hu, B.X.; Li, Z.L.; Du, T.; Shan, J.L.; Ye, Z.P.; Peng, X.D.; Li, X.; Huang, Y.; Zhu, X.Y.; et al. PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nat. Cell Biol. 2022, 24, 88–98. [Google Scholar] [CrossRef]
- Zhao, Y.; Li, M.; Yao, X.; Fei, Y.; Lin, Z.; Li, Z.; Cai, K.; Zhao, Y.; Luo, Z. HCAR1/MCT1 Regulates Tumor Ferroptosis through the Lactate-Mediated AMPK-SCD1 Activity and Its Therapeutic Implications. Cell Rep. 2020, 33, 108487. [Google Scholar] [CrossRef] [PubMed]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar] [CrossRef]
- Dephoure, N.; Zhou, C.; Villén, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar] [CrossRef] [Green Version]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wang, G.; Qian, J. Transcription factors as readers and effectors of DNA methylation. Nat. Rev. Genet. 2016, 17, 551–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, K.; Liu, X.; Wen, B.; Liu, Y.; Zhang, W.; Hu, X.; Chen, L.; Hang, W.; Chen, J. GSK-J4, a Specific Histone Lysine Demethylase 6A Inhibitor, Ameliorates Lipotoxicity to Cardiomyocytes via Preserving H3K27 Methylation and Reducing Ferroptosis. Front. Cardiovasc. Med. 2022, 9, 907747. [Google Scholar] [CrossRef]
- Yang, H.; Hu, Y.; Weng, M.; Liu, X.; Wan, P.; Hu, Y.; Ma, M.; Zhang, Y.; Xia, H.; Lv, K. Hypoxia inducible lncRNA-CBSLR modulates ferroptosis through m6A-YTHDF2-dependent modulation of CBS in gastric cancer. J. Adv. Res. 2022, 37, 91–106. [Google Scholar] [CrossRef]
- Shvedunova, M.; Akhtar, A. Modulation of cellular processes by histone and non-histone protein acetylation. Nat. Rev. Mol. Cell Biol. 2022, 23, 329–349. [Google Scholar] [CrossRef]
- Li, H.; Song, J.; He, Y.; Liu, Y.; Liu, Z.; Sun, W.; Hu, W.; Lei, Q.Y.; Hu, X.; Chen, Z.; et al. CRISPR/Cas9 Screens Reveal that Hexokinase 2 Enhances Cancer Stemness and Tumorigenicity by Activating the ACSL4-Fatty Acid β-Oxidation Pathway. Adv. Sci. 2022, 9, e2105126. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Zhang, Y.; Yang, C.; Xiong, H.; Lin, Y.; Yao, J.; Li, H.; Xie, L.; Zhao, W.; Yao, Y.; et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 2010, 327, 1004–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Shen, Q.; Liu, Y.; Shi, Y.; Huang, W.; Wang, X.; Li, Z.; Chai, Y.; Wang, H.; Hu, X.; et al. Increased glucose metabolism in TAMs fuels O-GlcNAcylation of lysosomal Cathepsin B to promote cancer metastasis and chemoresistance. Cancer Cell 2022, 40, 1207–1222.e10. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, Z.; Yuan, J.; Wang, J.; Shen, X. The positive feedback between ACSL4 expression and O-GlcNAcylation contributes to the growth and survival of hepatocellular carcinoma. Aging 2020, 12, 7786–7800. [Google Scholar] [CrossRef]
- Xiang, J.; Chen, C.; Liu, R.; Gou, D.; Chang, L.; Deng, H.; Gao, Q.; Zhang, W.; Tuo, L.; Pan, X.; et al. Gluconeogenic enzyme PCK1 deficiency promotes CHK2 O-GlcNAcylation and hepatocellular carcinoma growth upon glucose deprivation. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Guo, B.; Liang, Q.; Li, L.; Hu, Z.; Wu, F.; Zhang, P.; Ma, Y.; Zhao, B.; Kovács, A.L.; Zhang, Z.; et al. O-GlcNAc-modification of SNAP-29 regulates autophagosome maturation. Nat. Cell Biol. 2014, 16, 1215–1226. [Google Scholar] [CrossRef]
- Wang, Y.; Liu, Y.; Liu, J.; Kang, R.; Tang, D. NEDD4L-mediated LTF protein degradation limits ferroptosis. Biochem. Biophys. Res. Commun. 2020, 531, 581–587. [Google Scholar] [CrossRef]
- Nguyen, K.T.; Mun, S.H.; Yang, J.; Lee, J.; Seok, O.H.; Kim, E.; Kim, D.; An, S.Y.; Seo, D.Y.; Suh, J.Y.; et al. The MARCHF6 E3 ubiquitin ligase acts as an NADPH sensor for the regulation of ferroptosis. Nat. Cell Biol. 2022, 24, 1239–1251. [Google Scholar] [CrossRef]
- Chen, J.; Ding, C.; Chen, Y.; Hu, W.; Lu, Y.; Wu, W.; Zhang, Y.; Yang, B.; Wu, H.; Peng, C.; et al. ACSL4 promotes hepatocellular carcinoma progression via c-Myc stability mediated by ERK/FBW7/c-Myc axis. Oncogenesis 2020, 9, 42. [Google Scholar] [CrossRef]
- Bao, Z.; Liu, Y.; Chen, B.; Miao, Z.; Tu, Y.; Li, C.; Chao, H.; Ye, Y.; Xu, X.; Sun, G.; et al. Prokineticin-2 prevents neuronal cell deaths in a model of traumatic brain injury. Nat. Commun. 2021, 12, 4220. [Google Scholar] [CrossRef]
- Sen, P.; Kan, C.F.K.; Singh, A.B.; Rius, M.; Kraemer, F.B.; Sztul, E.; Liu, J. Identification of p115 as a novel ACSL4 interacting protein and its role in regulating ACSL4 degradation. J. Proteom. 2020, 229, 103926. [Google Scholar] [CrossRef] [PubMed]
- Békés, M.; Langley, D.R.; Crews, C.M. PROTAC targeted protein degraders: The past is prologue. Nat. Rev. Drug Discov. 2022, 21, 181–200. [Google Scholar] [CrossRef]
- Liu, C.; Sun, W.; Zhu, T.; Shi, S.; Zhang, J.; Wang, J.; Gao, F.; Ou, Q.; Jin, C.; Li, J.; et al. Glia maturation factor-β induces ferroptosis by impairing chaperone-mediated autophagic degradation of ACSL4 in early diabetic retinopathy. Redox Biol. 2022, 52, 102292. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gao, Z.; He, W.; Wu, Y.; Liu, J.; Zhang, S.; Yan, L.; Mao, S.; Shi, X.; Fan, W.; et al. The gut microbiota metabolite glycochenodeoxycholate activates TFR-ACSL4-mediated ferroptosis to promote the development of environmental toxin-linked MAFLD. Free Radic. Biol. Med. 2022, 193, 213–226. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Li, A.; Zhang, X.; Xiao, H. A novel TIP30 protein complex regulates EGF receptor signaling and endocytic degradation. J. Biol. Chem. 2011, 286, 9373–9381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttlin, E.L.; Bruckner, R.J.; Paulo, J.A.; Cannon, J.R.; Ting, L.; Baltier, K.; Colby, G.; Gebreab, F.; Gygi, M.P.; Parzen, H.; et al. Architecture of the human interactome defines protein communities and disease networks. Nature 2017, 545, 505–509. [Google Scholar] [CrossRef] [Green Version]
- Hein, M.Y.; Hubner, N.C.; Poser, I.; Cox, J.; Nagaraj, N.; Toyoda, Y.; Gak, I.A.; Weisswange, I.; Mansfeld, J.; Buchholz, F.; et al. A human interactome in three quantitative dimensions organized by stoichiometries and abundances. Cell 2015, 163, 712–723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikari, H.; Counter, C.M. Interrogating the protein interactomes of RAS isoforms identifies PIP5K1A as a KRAS-specific vulnerability. Nat. Commun. 2018, 9, 3646. [Google Scholar] [CrossRef] [Green Version]
- Nitta, S.; Sakamoto, N.; Nakagawa, M.; Kakinuma, S.; Mishima, K.; Kusano-Kitazume, A.; Kiyohashi, K.; Murakawa, M.; Nishimura-Sakurai, Y.; Azuma, S.; et al. Hepatitis C virus NS4B protein targets STING and abrogates RIG-I-mediated type I interferon-dependent innate immunity. Hepatology 2013, 57, 46–58. [Google Scholar] [CrossRef]
- Li, X.; Wang, W.; Wang, J.; Malovannaya, A.; Xi, Y.; Li, W.; Guerra, R.; Hawke, D.H.; Qin, J.; Chen, J. Proteomic analyses reveal distinct chromatin-associated and soluble transcription factor complexes. Mol. Syst. Biol. 2015, 11, 775. [Google Scholar] [CrossRef]
- Livesey, K.M.; Kang, R.; Zeh, H.J., 3rd; Lotze, M.T.; Tang, D. Direct molecular interactions between HMGB1 and TP53 in colorectal cancer. Autophagy 2012, 8, 846–848. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Li, C.; Zhu, S.; Cao, L.; Kroemer, G.; Zeh, H.; Tang, D.; Kang, R. TFAM is a novel mediator of immunogenic cancer cell death. Oncoimmunology 2018, 7, e1431086. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Zhi, F.; Lun, W.; Deng, Q.; Zhang, W. Baicalin inhibits PDGF-BB-induced hepatic stellate cell proliferation, apoptosis, invasion, migration and activation via the miR-3595/ACSL4 axis. Int. J. Mol. Med. 2018, 41, 1992–2002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, C.; Zhuo, Y.; Liu, Y.; Chen, H. Itraconazole Inhibits the Growth of Cutaneous Squamous Cell Carcinoma by Targeting HMGCS1/ACSL4 Axis. Front. Pharmacol. 2022, 13, 828983. [Google Scholar] [CrossRef] [PubMed]
- Gubern, C.; Camós, S.; Ballesteros, I.; Rodríguez, R.; Romera, V.G.; Cañadas, R.; Lizasoain, I.; Moro, M.A.; Serena, J.; Mallolas, J.; et al. miRNA expression is modulated over time after focal ischaemia: Up-regulation of miR-347 promotes neuronal apoptosis. FEBS J. 2013, 280, 6233–6246. [Google Scholar] [CrossRef]
- Liu, X.; Hai, Y.; Dong, J.; Xu, L.; Hou, W.; Su, J.; Ren, W.; Liu, D. Realgar-induced KRAS mutation lung cancer cell death via KRAS/Raf/MAPK mediates ferroptosis. Int. J. Oncol. 2022, 61. [Google Scholar] [CrossRef] [PubMed]
- Tang, D.; Chen, X.; Kang, R.; Kroemer, G. Ferroptosis: Molecular mechanisms and health implications. Cell Res. 2021, 31, 107–125. [Google Scholar] [CrossRef]
- Lin, Z.; Liu, J.; Long, F.; Kang, R.; Kroemer, G.; Tang, D.; Yang, M. The lipid flippase SLC47A1 blocks metabolic vulnerability to ferroptosis. Nat. Commun. 2022, 13, 7965. [Google Scholar] [CrossRef] [PubMed]
- Kagan, V.E.; Mao, G.; Qu, F.; Angeli, J.P.; Doll, S.; Croix, C.S.; Dar, H.H.; Liu, B.; Tyurin, V.A.; Ritov, V.B.; et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat. Chem. Biol. 2017, 13, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Klett, E.L.; Chen, S.; Yechoor, A.; Lih, F.B.; Coleman, R.A. Long-chain acyl-CoA synthetase isoforms differ in preferences for eicosanoid species and long-chain fatty acids. J. Lipid Res. 2017, 58, 884–894. [Google Scholar] [CrossRef] [Green Version]
- Lei, G.; Zhang, Y.; Koppula, P.; Liu, X.; Zhang, J.; Lin, S.H.; Ajani, J.A.; Xiao, Q.; Liao, Z.; Wang, H.; et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020, 30, 146–162. [Google Scholar] [CrossRef]
- Grube, J.; Woitok, M.M.; Mohs, A.; Erschfeld, S.; Lynen, C.; Trautwein, C.; Otto, T. ACSL4-dependent ferroptosis does not represent a tumor-suppressive mechanism but ACSL4 rather promotes liver cancer progression. Cell Death Dis. 2022, 13, 704. [Google Scholar] [CrossRef]
- Kim, R.; Hashimoto, A.; Markosyan, N.; Tyurin, V.A.; Tyurina, Y.Y.; Kar, G.; Fu, S.; Sehgal, M.; Garcia-Gerique, L.; Kossenkov, A.; et al. Ferroptosis of tumour neutrophils causes immune suppression in cancer. Nature 2022, 612, 338–346. [Google Scholar] [CrossRef]
- Chu, B.; Kon, N.; Chen, D.; Li, T.; Liu, T.; Jiang, L.; Song, S.; Tavana, O.; Gu, W. ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat. Cell Biol. 2019, 21, 579–591. [Google Scholar] [CrossRef]
- Wang, X.; Chen, Y.; Yang, X.; Cheng, L.; He, Z.; Xin, Y.; Huang, S.; Meng, F.; Zhang, P.; Luo, L. Activation of ALOX12 by a multi-organelle-orienting photosensitizer drives ACSL4-independent cell ferroptosis. Cell Death Dis. 2022, 13, 1040. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Kang, R.; Sun, X.; Zhong, M.; Huang, J.; Klionsky, D.J.; Tang, D. Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy 2015, 11, 28–45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adhikari, S.; Moscatelli, J.; Puchner, E.M. Quantitative live-cell PALM reveals nanoscopic Faa4 redistributions and dynamics on lipid droplets during metabolic transitions of yeast. Mol. Biol. Cell 2021, 32, 1565–1578. [Google Scholar] [CrossRef]
- Helfenberger, K.E.; Argentino, G.F.; Benzo, Y.; Herrera, L.M.; Finocchietto, P.; Poderoso, C. Angiotensin II Regulates Mitochondrial mTOR Pathway Activity Dependent on Acyl-CoA Synthetase 4 in Adrenocortical Cells. Endocrinology 2022, 163, bqac170. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhu, X.; Liu, Y.; Zhu, K.; Lin, K.; Li, F. ACSL4 contributes to sevoflurane-induced ferroptotic neuronal death in SH-SY5Y cells via the 5′ AMP-activated protein kinase/mammalian target of rapamycin pathway. Ann. Transl. Med. 2021, 9, 1454. [Google Scholar] [CrossRef]
- Zhang, C.; Li, A.; Gao, S.; Zhang, X.; Xiao, H. The TIP30 protein complex, arachidonic acid and coenzyme A are required for vesicle membrane fusion. PLoS ONE 2011, 6, e21233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, B.; Liu, J.; Klionsky, D.J.; Tang, D.; Kang, R. Extracellular SQSTM1 as an inflammatory mediator. Autophagy 2020, 16, 2313–2315. [Google Scholar] [CrossRef]
- Killion, E.A.; Reeves, A.R.; El Azzouny, M.A.; Yan, Q.W.; Surujon, D.; Griffin, J.D.; Bowman, T.A.; Wang, C.; Matthan, N.R.; Klett, E.L.; et al. A role for long-chain acyl-CoA synthetase-4 (ACSL4) in diet-induced phospholipid remodeling and obesity-associated adipocyte dysfunction. Mol. Metab. 2018, 9, 43–56. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Guo, S.; Yang, C.; Liu, X.; Chen, X.; He, J.; Tong, C.; Ding, Y.; Peng, C.; Geng, Y.; et al. High-fat diet-induced obesity primes fatty acid β-oxidation impairment and consequent ovarian dysfunction during early pregnancy. Ann. Transl. Med. 2021, 9, 887. [Google Scholar] [CrossRef] [PubMed]
- Pei, Z.; Liu, Y.; Liu, S.; Jin, W.; Luo, Y.; Sun, M.; Duan, Y.; Ajoolabady, A.; Sowers, J.R.; Fang, Y.; et al. FUNDC1 insufficiency sensitizes high fat diet intake-induced cardiac remodeling and contractile anomaly through ACSL4-mediated ferroptosis. Metab. Clin. Exp. 2021, 122, 154840. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Hao, X.; Han, L.; Yan, Z.; Shen, W.J.; Dong, D.; Hasbargen, K.; Bittner, S.; Cortez, Y.; Greenberg, A.S.; et al. Tissue-Specific Ablation of ACSL4 Results in Disturbed Steroidogenesis. Endocrinology 2019, 160, 2517–2528. [Google Scholar] [CrossRef] [Green Version]
- Golej, D.L.; Askari, B.; Kramer, F.; Barnhart, S.; Vivekanandan-Giri, A.; Pennathur, S.; Bornfeldt, K.E. Long-chain acyl-CoA synthetase 4 modulates prostaglandin E2 release from human arterial smooth muscle cells. J. Lipid Res. 2011, 52, 782–793. [Google Scholar] [CrossRef] [Green Version]
- Fan, Z.; Cai, L.; Wang, S.; Wang, J.; Chen, B. Baicalin Prevents Myocardial Ischemia/Reperfusion Injury through Inhibiting ACSL4 Mediated Ferroptosis. Front. Pharmacol. 2021, 12, 628988. [Google Scholar] [CrossRef]
- Sun, W.; Wu, X.; Yu, P.; Zhang, Q.; Shen, L.; Chen, J.; Tong, H.; Fan, M.; Shi, H.; Chen, X. LncAABR07025387.1 Enhances Myocardial Ischemia/Reperfusion Injury via miR-205/ACSL4-Mediated Ferroptosis. Front. Cell Dev. Biol. 2022, 10, 672391. [Google Scholar] [CrossRef]
- Xu, Y.; Li, X.; Cheng, Y.; Yang, M.; Wang, R. Inhibition of ACSL4 attenuates ferroptotic damage after pulmonary ischemia-reperfusion. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2020, 34, 16262–16275. [Google Scholar] [CrossRef]
- Li, M.; Meng, Z.; Yu, S.; Li, J.; Wang, Y.; Yang, W.; Wu, H. Baicalein ameliorates cerebral ischemia-reperfusion injury by inhibiting ferroptosis via regulating GPX4/ACSL4/ACSL3 axis. Chem.-Biol. Interact. 2022, 366, 110137. [Google Scholar] [CrossRef]
- Tuo, Q.Z.; Liu, Y.; Xiang, Z.; Yan, H.F.; Zou, T.; Shu, Y.; Ding, X.L.; Zou, J.J.; Xu, S.; Tang, F.; et al. Thrombin induces ACSL4-dependent ferroptosis during cerebral ischemia/reperfusion. Signal Transduct. Target. Ther. 2022, 7, 59. [Google Scholar] [CrossRef]
- Tao, W.H.; Shan, X.S.; Zhang, J.X.; Liu, H.Y.; Wang, B.Y.; Wei, X.; Zhang, M.; Peng, K.; Ding, J.; Xu, S.X.; et al. Dexmedetomidine Attenuates Ferroptosis-Mediated Renal Ischemia/Reperfusion Injury and Inflammation by Inhibiting ACSL4 via α2-AR. Front. Pharmacol. 2022, 13, 782466. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Wu, J.; Xu, H.; Zhou, C.; Han, B.; Zhu, H.; Hu, Z.; Ma, Z.; Ming, Z.; Yao, Y.; et al. XJB-5-131 inhibited ferroptosis in tubular epithelial cells after ischemia-reperfusion injury. Cell Death Dis. 2020, 11, 629. [Google Scholar] [CrossRef]
- Sun, Z.; Wu, J.; Bi, Q.; Wang, W. Exosomal lncRNA TUG1 derived from human urine-derived stem cells attenuates renal ischemia/reperfusion injury by interacting with SRSF1 to regulate ASCL4-mediated ferroptosis. Stem Cell Res. Ther. 2022, 13, 297. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Li, G.; Wang, Y.; Li, Y.; Xu, H.; Liu, W.; Hao, W.; Yao, Y.; Zeng, R. Cytoplasmic HMGB1 induces renal tubular ferroptosis after ischemia/reperfusion. Int. Immunopharmacol. 2023, 116, 109757. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, M.; Bi, R.; Su, Y.; Quan, F.; Lin, Y.; Yue, C.; Cui, X.; Zhao, Q.; Liu, S.; et al. ACSL4 deficiency confers protection against ferroptosis-mediated acute kidney injury. Redox Biol. 2022, 51, 102262. [Google Scholar] [CrossRef]
- Zhongyin, Z.; Wei, W.; Juan, X.; Guohua, F. Isoliquiritin apioside relieves intestinal ischemia/reperfusion-induced acute lung injury by blocking Hif-1α-mediated ferroptosis. Int. Immunopharmacol. 2022, 108, 108852. [Google Scholar] [CrossRef]
- Duan, J.; Wang, Z.; Duan, R.; Yang, C.; Zhao, R.; Feng, Q.; Qin, Y.; Jiang, J.; Gu, S.; Lv, K.; et al. Therapeutic targeting of hepatic ACSL4 ameliorates NASH in mice. Hepatology 2022, 75, 140–153. [Google Scholar] [CrossRef] [PubMed]
- Wei, S.; Qiu, T.; Wang, N.; Yao, X.; Jiang, L.; Jia, X.; Tao, Y.; Zhang, J.; Zhu, Y.; Yang, G.; et al. Ferroptosis mediated by the interaction between Mfn2 and IREα promotes arsenic-induced nonalcoholic steatohepatitis. Environ. Res. 2020, 188, 109824. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.Y.; Liu, Y.D.; Gong, Y.; Jin, W.; Topchiy, E.; Turdi, S.; Gao, Y.F.; Culver, B.; Wang, S.Y.; Ge, W.; et al. Mitochondrial aldehyde dehydrogenase (ALDH2) rescues cardiac contractile dysfunction in an APP/PS1 murine model of Alzheimer’s disease via inhibition of ACSL4-dependent ferroptosis. Acta Pharmacol. Sin. 2022, 43, 39–49. [Google Scholar] [CrossRef]
- Thomas, M.H.; Paris, C.; Magnien, M.; Colin, J.; Pelleïeux, S.; Coste, F.; Escanyé, M.C.; Pillot, T.; Olivier, J.L. Dietary arachidonic acid increases deleterious effects of amyloid-β oligomers on learning abilities and expression of AMPA receptors: Putative role of the ACSL4-cPLA2 balance. Alzheimer’s Res. Ther. 2017, 9, 69. [Google Scholar] [CrossRef]
- Bouchaoui, H.; Mahoney-Sanchez, L.; Garçon, G.; Berdeaux, O.; Alleman, L.Y.; Devos, D.; Duce, J.A.; Devedjian, J.C. ACSL4 and the lipoxygenases 15/15B are pivotal for ferroptosis induced by iron and PUFA dyshomeostasis in dopaminergic neurons. Free Radic. Biol. Med. 2023, 195, 145–157. [Google Scholar] [CrossRef]
- Luoqian, J.; Yang, W.; Ding, X.; Tuo, Q.Z.; Xiang, Z.; Zheng, Z.; Guo, Y.J.; Li, L.; Guan, P.; Ayton, S.; et al. Ferroptosis promotes T-cell activation-induced neurodegeneration in multiple sclerosis. Cell. Mol. Immunol. 2022, 19, 913–924. [Google Scholar] [CrossRef]
- He, S.; Li, R.; Peng, Y.; Wang, Z.; Huang, J.; Meng, H.; Min, J.; Wang, F.; Ma, Q. ACSL4 contributes to ferroptosis-mediated rhabdomyolysis in exertional heat stroke. J. Cachexia Sarcopenia Muscle 2022, 13, 1717–1730. [Google Scholar] [CrossRef]
- Gazou, A.; Riess, A.; Grasshoff, U.; Schäferhoff, K.; Bonin, M.; Jauch, A.; Riess, O.; Tzschach, A. Xq22.3-q23 deletion including ACSL4 in a patient with intellectual disability. Am. J. Med. Genet. Part A 2013, 161a, 860–864. [Google Scholar] [CrossRef]
- Chao, A.M.; Quigley, K.M.; Wadden, T.A. Dietary interventions for obesity: Clinical and mechanistic findings. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Morigny, P.; Boucher, J.; Arner, P.; Langin, D. Lipid and glucose metabolism in white adipocytes: Pathways, dysfunction and therapeutics. Nat. Rev. Endocrinol. 2021, 17, 276–295. [Google Scholar] [CrossRef]
- Wei, H.; Lin, X.; Liu, L.; Peng, X. Flaxseed Polysaccharide Alters Colonic Gene Expression of Lipid Metabolism and Energy Metabolism in Obese Rats. Foods 2022, 11, 1991. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.Q.; Guo, Y.; Li, X.; Zhang, G.Q.; Li, P. Small molecules as modulators of regulated cell death against ischemia/reperfusion injury. Med. Res. Rev. 2022, 42, 2067–2101. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.H.; Yang, K.T.; Lee, W.S.; Ting, P.C.; Luo, Y.P.; Lin, D.J.; Wang, Y.S.; Chang, J.C. Xanthohumol Protects the Rat Myocardium against Ischemia/Reperfusion Injury-Induced Ferroptosis. Oxidative Med. Cell. Longev. 2022, 2022, 9523491. [Google Scholar] [CrossRef] [PubMed]
- Cui, Y.; Zhang, Y.; Zhao, X.; Shao, L.; Liu, G.; Sun, C.; Xu, R.; Zhang, Z. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav. Immun. 2021, 93, 312–321. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Yang, L.; Geng, L.; He, J.; Chen, L.; Sun, Q.; Zhao, J.; Wang, X. Inhibition of Acyl-CoA Synthetase Long-Chain Family Member 4 Facilitates Neurological Recovery After Stroke by Regulation Ferroptosis. Front. Cell. Neurosci. 2021, 15, 632354. [Google Scholar] [CrossRef]
- Ferronato, S.; Scuro, A.; Gomez-Lira, M.; Mazzucco, S.; Olivato, S.; Turco, A.; Elisa, O.; Malerba, G.; Romanelli, M.G. Correlations between gene expression highlight a different activation of ACE/TLR4/PTGS2 signaling in symptomatic and asymptomatic plaques in atherosclerotic patients. Mol. Biol. Rep. 2018, 45, 657–662. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, H.; Hua, L.; Hou, C.; Jia, Q.; Chen, J.; Zhang, S.; Wang, Y.; He, S.; Jia, E. Verification of ferroptosis and pyroptosis and identification of PTGS2 as the hub gene in human coronary artery atherosclerosis. Free Radic. Biol. Med. 2021, 171, 55–68. [Google Scholar] [CrossRef]
- Askari, B.; Kanter, J.E.; Sherrid, A.M.; Golej, D.L.; Bender, A.T.; Liu, J.; Hsueh, W.A.; Beavo, J.A.; Coleman, R.A.; Bornfeldt, K.E. Rosiglitazone inhibits acyl-CoA synthetase activity and fatty acid partitioning to diacylglycerol and triacylglycerol via a peroxisome proliferator-activated receptor-gamma-independent mechanism in human arterial smooth muscle cells and macrophages. Diabetes 2007, 56, 1143–1152. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Li, S.; Yin, M.; Li, Y.; Chen, C.; Zhang, J.; Sun, K.; Kong, X.; Chen, Z.; Qian, J. Isorhapontigenin Attenuates Cardiac Microvascular Injury in Diabetes via the Inhibition of Mitochondria-Associated Ferroptosis through PRDX2-MFN2-ACSL4 Pathways. Diabetes 2023, 72, 389–404. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD development and therapeutic strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Chen, J.; Jin, Z.; Zhang, S.; Zhang, X.; Li, P.; Yang, H.; Ma, Y. Arsenic trioxide elicits prophylactic and therapeutic immune responses against solid tumors by inducing necroptosis and ferroptosis. Cell. Mol. Immunol. 2023, 20, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Shou, Y.; Yang, L.; Yang, Y.; Xu, J. Inhibition of keratinocyte ferroptosis suppresses psoriatic inflammation. Cell Death Dis. 2021, 12, 1009. [Google Scholar] [CrossRef] [PubMed]
- Shimano, H.; Sato, R. SREBP-regulated lipid metabolism: Convergent physiology—divergent pathophysiology. Nat. Rev. Endocrinol. 2017, 13, 710–730. [Google Scholar] [CrossRef]
- Qu, X.F.; Liang, T.Y.; Wu, D.G.; Lai, N.S.; Deng, R.M.; Ma, C.; Li, X.; Li, H.Y.; Liu, Y.Z.; Shen, H.T.; et al. Acyl-CoA synthetase long chain family member 4 plays detrimental role in early brain injury after subarachnoid hemorrhage in rats by inducing ferroptosis. CNS Neurosci. Ther. 2021, 27, 449–463. [Google Scholar] [CrossRef]
- Gao, Y.; Li, J.; Wu, Q.; Wang, S.; Yang, S.; Li, X.; Chen, N.; Li, L.; Zhang, L. Tetrahydroxy stilbene glycoside ameliorates Alzheimer’s disease in APP/PS1 mice via glutathione peroxidase related ferroptosis. Int. Immunopharmacol. 2021, 99, 108002. [Google Scholar] [CrossRef]
- Bouchama, A.; Abuyassin, B.; Lehe, C.; Laitano, O.; Jay, O.; O’Connor, F.G.; Leon, L.R. Classic and exertional heatstroke. Nat. Rev. Dis. Prim. 2022, 8, 8. [Google Scholar] [CrossRef]
- Zhu, S.; Zhang, Q.; Sun, X.; Zeh, H.J., 3rd; Lotze, M.T.; Kang, R.; Tang, D. HSPA5 Regulates Ferroptotic Cell Death in Cancer Cells. Cancer Res. 2017, 77, 2064–2077. [Google Scholar] [CrossRef] [Green Version]
- Levey, A.S.; James, M.T. Acute Kidney Injury. Ann. Intern. Med. 2017, 167, Itc66–Itc80. [Google Scholar] [CrossRef] [PubMed]
- Su, L.; Zhang, J.; Gomez, H.; Kellum, J.A.; Peng, Z. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy 2023, 19, 401–414. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S. Cellular and molecular pathways of renal repair after acute kidney injury. Kidney Int. 2018, 93, 27–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapi, F.; Azoulay, L.; Yin, H.; Nessim, S.J.; Suissa, S. Concurrent use of diuretics, angiotensin converting enzyme inhibitors, and angiotensin receptor blockers with non-steroidal anti-inflammatory drugs and risk of acute kidney injury: Nested case-control study. BMJ 2013, 346, e8525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, T.; Dewitz, C.; Schmitz, J.; Schröder, A.S.; Bräsen, J.H.; Stockwell, B.R.; Murphy, J.M.; Kunzendorf, U.; Krautwald, S. Necroptosis and ferroptosis are alternative cell death pathways that operate in acute kidney failure. Cell. Mol. Life Sci. CMLS 2017, 74, 3631–3645. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D. Hallmarks of Cancer: New Dimensions. Cancer Discov. 2022, 12, 31–46. [Google Scholar] [CrossRef]
- Dai, G.; Wang, D.; Ma, S.; Hong, S.; Ding, K.; Tan, X.; Ju, W. ACSL4 promotes colorectal cancer and is a potential therapeutic target of emodin. Phytomedicine Int. J. Phytother. Phytopharm. 2022, 102, 154149. [Google Scholar] [CrossRef] [PubMed]
- Kwon, Y.S.; Lee, M.G.; Baek, J.; Kim, N.Y.; Jang, H.; Kim, S. Acyl-CoA synthetase-4 mediates radioresistance of breast cancer cells by regulating FOXM1. Biochem. Pharmacol. 2021, 192, 114718. [Google Scholar] [CrossRef] [PubMed]
- Doll, S.; Proneth, B.; Tyurina, Y.Y.; Panzilius, E.; Kobayashi, S.; Ingold, I.; Irmler, M.; Beckers, J.; Aichler, M.; Walch, A.; et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat. Chem. Biol. 2017, 13, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Liao, P.; Wang, W.; Wang, W.; Kryczek, I.; Li, X.; Bian, Y.; Sell, A.; Wei, S.; Grove, S.; Johnson, J.K.; et al. CD8+ T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell 2022, 40, 365–378.e6. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Liu, J.; Kuang, F.; Chen, X.; Zeh, H.J., 3rd; Kang, R.; Kroemer, G.; Xie, Y.; Tang, D. PDK4 dictates metabolic resistance to ferroptosis by suppressing pyruvate oxidation and fatty acid synthesis. Cell Rep. 2021, 34, 108767. [Google Scholar] [CrossRef] [PubMed]
- Maciel, F.C.; Maloberti, P.; Neuman, I.; Cano, F.; Castilla, R.; Castillo, F.; Paz, C.; Podestá, E.J. An arachidonic acid-preferring acyl-CoA synthetase is a hormone-dependent and obligatory protein in the signal transduction pathway of steroidogenic hormones. J. Mol. Endocrinol. 2005, 34, 655–666. [Google Scholar] [CrossRef] [PubMed]
- Cano, F.; Poderoso, C.; Maciel, F.C.; Castilla, R.; Maloberti, P.; Castillo, F.; Neuman, I.; Paz, C.; Podestá, E.J. Protein tyrosine phosphatases regulate arachidonic acid release, StAR induction and steroidogenesis acting on a hormone-dependent arachidonic acid-preferring acyl-CoA synthetase. J. Steroid Biochem. Mol. Biol. 2006, 99, 197–202. [Google Scholar] [CrossRef]
- Cho, Y.Y.; Kang, M.J.; Ogawa, S.; Yamashita, Y.; Fujino, T.; Yamamoto, T.T. Regulation by adrenocorticotropic hormone and arachidonate of the expression of acyl-CoA synthetase 4, an arachidonate-preferring enzyme expressed in steroidogenic tissues. Biochem. Biophys. Res. Commun. 2000, 274, 741–745. [Google Scholar] [CrossRef]
- Hu, N.; Bai, L.; Dai, E.; Han, L.; Kang, R.; Li, H.; Tang, D. Pirin is a nuclear redox-sensitive modulator of autophagy-dependent ferroptosis. Biochem. Biophys. Res. Commun. 2021, 536, 100–106. [Google Scholar] [CrossRef]
- Heimli, H.; Hollung, K.; Drevon, C.A. Eicosapentaenoic acid-induced apoptosis depends on acyl CoA-synthetase. Lipids 2003, 38, 263–268. [Google Scholar] [CrossRef]
- Covault, J.; Pettinati, H.; Moak, D.; Mueller, T.; Kranzler, H.R. Association of a long-chain fatty acid-CoA ligase 4 gene polymorphism with depression and with enhanced niacin-induced dermal erythema. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2004, 127b, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Maloberti, P.; Castilla, R.; Castillo, F.; Maciel, F.C.; Mendez, C.F.; Paz, C.; Podestá, E.J. Silencing the expression of mitochondrial acyl-CoA thioesterase I and acyl-CoA synthetase 4 inhibits hormone-induced steroidogenesis. FEBS J. 2005, 272, 1804–1814. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, A.; Ouellette, R.J.; Surette, M.E. 17β-estradiol-induced ACSL4 protein expression promotes an invasive phenotype in estrogen receptor positive mammary carcinoma cells. Carcinogenesis 2017, 38, 402–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.R.; Han, S.N. Pinolenic Acid Downregulates Lipid Anabolic Pathway in HepG2 Cells. Lipids 2016, 51, 847–855. [Google Scholar] [CrossRef] [PubMed]
- Kan, C.F.; Singh, A.B.; Stafforini, D.M.; Azhar, S.; Liu, J. Arachidonic acid downregulates acyl-CoA synthetase 4 expression by promoting its ubiquitination and proteasomal degradation. J. Lipid Res. 2014, 55, 1657–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Q.; Fu, S.; Zuo, H.; Huang, Y.; Chu, L.; Zhu, Y.; Hu, J.; Wu, Y.; Chen, S.; Wang, Y.; et al. ACSL4 is essential for radiation-induced intestinal injury by initiating ferroptosis. Cell Death Discov. 2022, 8, 332. [Google Scholar] [CrossRef]
- Cui, M.; Xiao, Z.; Sun, B.; Wang, Y.; Zheng, M.; Ye, L.; Zhang, X. Involvement of cholesterol in hepatitis B virus X protein-induced abnormal lipid metabolism of hepatoma cells via up-regulating miR-205-targeted ACSL4. Biochem. Biophys. Res. Commun. 2014, 445, 651–655. [Google Scholar] [CrossRef]
- Modi, H.R.; Basselin, M.; Rapoport, S.I. Valnoctamide, a non-teratogenic amide derivative of valproic acid, inhibits arachidonic acid activation in vitro by recombinant acyl-CoA synthetase-4. Bipolar Disord. 2014, 16, 875–880. [Google Scholar] [CrossRef] [Green Version]
- Modi, H.R.; Ma, K.; Chang, L.; Chen, M.; Rapoport, S.I. Valnoctamide, which reduces rat brain arachidonic acid turnover, is a potential non-teratogenic valproate substitute to treat bipolar disorder. Psychiatry Res. 2017, 254, 279–283. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T., Jr.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Castillo, F.; Cano, F.; Maloberti, P.; Castilla, R.; Neuman, I.; Poderoso, C.; Paz, C.; Podestá, E.J.; Maciel, F.C. Tyrosine phosphates act on steroidogenesis through the activation of arachidonic acid release. Endocr. Res. 2004, 30, 623–627. [Google Scholar] [CrossRef] [PubMed]
- Paz, C.; Maciel, F.C.; Gorostizaga, A.; Castillo, A.F.; García, M.M.M.S.; Maloberti, P.M.; Orlando, U.D.; Mele, P.G.; Poderoso, C.; Podesta, E.J. Role of Protein Phosphorylation and Tyrosine Phosphatases in the Adrenal Regulation of Steroid Synthesis and Mitochondrial Function. Front. Endocrinol. 2016, 7, 60. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, D.F.; Niu, X.L.; Hao, G.H.; Peng, N.; Wei, J.; Ning, N.; Wang, N.P. Rosiglitazone inhibits angiotensin II-induced CTGF expression in vascular smooth muscle cells—Role of PPAR-gamma in vascular fibrosis. Biochem. Pharmacol. 2007, 73, 185–197. [Google Scholar] [CrossRef]
- Orlando, U.D.; Castillo, A.F.; Medrano, M.A.R.; Solano, A.R.; Maloberti, P.M.; Podesta, E.J. Acyl-CoA synthetase-4 is implicated in drug resistance in breast cancer cell lines involving the regulation of energy-dependent transporter expression. Biochem. Pharmacol. 2019, 159, 52–63. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Chromosome Localization | Size (Number of Amino Acids) | Molecular Mass (Da) | Preferential Substrate | Tissue Expression (Most Abundant) | Subcellular Distribution (Most Abundant) | Function |
|---|---|---|---|---|---|---|---|
| ACSL1 | 4q35.1 | 698 | 77,943 | Palmitoleate, oleate and linoleate | Liver | Endoplasmic reticulum, mitochondrion | Catalyzes the conversion of long-chain fatty acids to their activated acyl-CoA forms for both cellular lipid synthesis and degradation via beta-oxidation. |
| ACSL3 | 2q36.1 | 720 | 80,420 | Myristate, laurate, arachidonate and eicosapentaenoate | Nervous system | Golgi apparatus, endoplasmic reticulum | Activates long-chain fatty acids for both cellular lipid synthesis and degradation via beta-oxidation. Promotes hepatic lipogenesis and the incorporation of fatty acids into phosphatidylcholine. |
| ACSL4 | Xq23 | 711 | 79,188 | Arachidonate and eicosapentaenoate | Eye, stomach | Endoplasmic reticulum, mitochondrion, plasma membrane | Catalyzes the conversion of long-chain fatty acids to their activated acyl-CoA forms for both cellular lipid synthesis and degradation via beta-oxidation. |
| ACSL5 | 10q25.2 | 683 | 75,991 | A wide range of saturated fatty acids and a preference for C16–C18 unsaturated fatty acids | Intestine | Endoplasmic reticulum, nucleus, mitochondrion, plasma membrane | Catalyzes the conversion of long-chain fatty acids to their activated acyl-CoA forms for both cellular lipid synthesis and degradation via beta-oxidation. Activates fatty acids from exogenous sources for the synthesis of triacylglycerol, which is destined for intracellular storage. |
| ACSL6 | 5q31 | 697 | 77,752 | Equal preference for saturated and polyunsaturated fatty acids with a backbone of C16–C20 | Nervous system | Endoplasmic reticulum, plasma membrane | Catalyzes the conversion of long-chain fatty acids to their activated acyl-CoA forms for both cellular lipid synthesis and degradation via beta-oxidation. |
| Type | Residue | Site | Ref |
|---|---|---|---|
| Phosphorylation | Ser | 140 | [64] |
| Phosphorylation | Ser | 674 | [65] |
| Phosphorylation | Ser | 95 | [64] |
| Phosphorylation | Thr | 679 | [65] |
| Acetylation | Lysine (K) | 89 | [66] |
| Type | Expression | Phenotype and Mechanism | Ref. |
|---|---|---|---|
| Obesity | Upregulation | Promotes the participation of arachidonic acid in phospholipids, leading to hepatic fat accumulation, inflammation in gonadal white adipose tissue, and insulin resistance | [112,113] |
| Cardiac remodeling and contraction | Upregulation | Short-term high-fat diet intake leads to downregulation of FUNDC1 and upregulation of ACSL4, which can result in lipid peroxidation-dependent defects in cardiac geometry and function. | [114] |
| Steroidogenesis | Normal | Promotes the formation of adrenal cholesterol esters and determines the fatty acyl composition of these esters | [115] |
| Vascular disease | Upregulation | Promotes the synthesis and metabolism of arachidonic acid and inhibits the secretion of prostaglandin E2 in vascular cells | [116] |
| Intestinal ischemia/reperfusion | Upregulation | Ischemia induces the upregulation of the SP1–ACSL4 cascade, promoting ferroptosis-dependent intestinal reperfusion injury | [33] |
| Myocardial ischemia/reperfusion | Upregulation | Promotes myocardial ischemia/reperfusion injury through lipid peroxidation-dependent ferroptosis | [117,118] |
| Pulmonary ischemia/reperfusion | Upregulation | Promotes pulmonary ischemia/reperfusion injury through lipid peroxidation-dependent ferroptosis | [119] |
| Cerebral ischemia/reperfusion | Upregulation | Promotes cerebral ischemia/reperfusion injury through the GPX4–ACSL4–ACSL3 pathway | [120] |
| Ischemic stroke | Upregulation | Thrombin-induced activation of serine protease induces ACSL4-dependent ferroptosis in neuronal cells, leading to ischemic stroke | [121] |
| Renal ischemia/reperfusion injury | Upregulation | Promotes renal damage and inflammation related to ferroptosis | [122] |
| Acute kidney injury | Upregulation | Promotes ferroptosis in renal tubular epithelial cells, leading to inflammation and acute kidney injury | [123,124,125,126] |
| Acute lung injury | Downregulation | Isoliquiritin apioside inhibits HIF1A, leading to downregulation of ACSL4 and preventing acute lung injury caused by intestinal ischemia/reperfusion | [127] |
| Non-alcoholic fatty liver disease | Upregulation | Induces the development of hepatic steatosis and fibrosis | [128,129] |
| Alzheimer’s disease | Upregulation | Induces ferroptosis-dependent brain damage and increases cytoplasmic phospholipase A2 in the mouse cortex | [130,131] |
| Parkinson’s disease | Upregulation | Induces ferroptosis in the substantia nigra brain pathway and mediates the production of cytokines | [35,132] |
| Multiple sclerosis | Upregulation | Induces ferroptosis-dependent encephalitis | [133] |
| Exertional heat stroke | Upregulation | Promotes muscle cell death induced by exertional heat stroke via ferroptosis | [134] |
| X-linked intellectual developmental disorder | Mutation | Induces X-linked intellectual developmental disorder | [24,135] |
| Type | Name | Mechanism | Ref |
|---|---|---|---|
| Inducer | Adrenocorticotropic hormone | Induces dephosphorylation of protein tyrosine phosphatase and the activity of ACOT2 | [166,167,168] |
| Inducer | RSL3; Erastin | Inhibits the SLC7A11-GPX4 pathway | [6,169] |
| Inducer | Icosapent | Increases the level of endogenous polyunsaturated fatty acids | [170,171] |
| Inducer | Acyl-CoA | Induces dephosphorylation of protein tyrosine phosphatase and the activity of ACOT2 | [168,172] |
| Inducer | 17 β-estradiol | Promotes polyunsaturated fatty-acid uptake | [173] |
| Inducer | Thrombin | Promotes the mobilization of phosphatidylethanolamine (PE) and phosphatidylcholine (PC) in the neuronal cell membrane via cPLA2α | [121] |
| Inhibitor | Pinolenic acid | Increases the level of endogenous polyunsaturated fatty acids | [174] |
| Inhibitor | Rosiglitazone | PPARγ agonist | [33,145] |
| Inhibitor | Arachidonic acid | Promotes ACSL4 ubiquitination | [175] |
| Inhibitor | Troglitazone | PPARγ agonist | [176] |
| Inhibitor | Triacsin C | Broad-spectrum ACSL inhibitor | [162,177] |
| Inhibitor | Abemaciclib | ACSL4 inhibitor | [128] |
| Inhibitor | Valnoctamide | Derivative of valproate | [178,179] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, F.; Kang, R.; Liu, J.; Tang, D. The ACSL4 Network Regulates Cell Death and Autophagy in Diseases. Biology 2023, 12, 864. https://doi.org/10.3390/biology12060864
Chen F, Kang R, Liu J, Tang D. The ACSL4 Network Regulates Cell Death and Autophagy in Diseases. Biology. 2023; 12(6):864. https://doi.org/10.3390/biology12060864
Chicago/Turabian StyleChen, Fangquan, Rui Kang, Jiao Liu, and Daolin Tang. 2023. "The ACSL4 Network Regulates Cell Death and Autophagy in Diseases" Biology 12, no. 6: 864. https://doi.org/10.3390/biology12060864
APA StyleChen, F., Kang, R., Liu, J., & Tang, D. (2023). The ACSL4 Network Regulates Cell Death and Autophagy in Diseases. Biology, 12(6), 864. https://doi.org/10.3390/biology12060864