
Autism Spectrum Disorder: A Neuro-Immunometabolic Hypothesis of the Developmental Origins

 ,
,  ,
,
Abstract
:Simple Summary
Abstract
1. Introduction
2. Methods
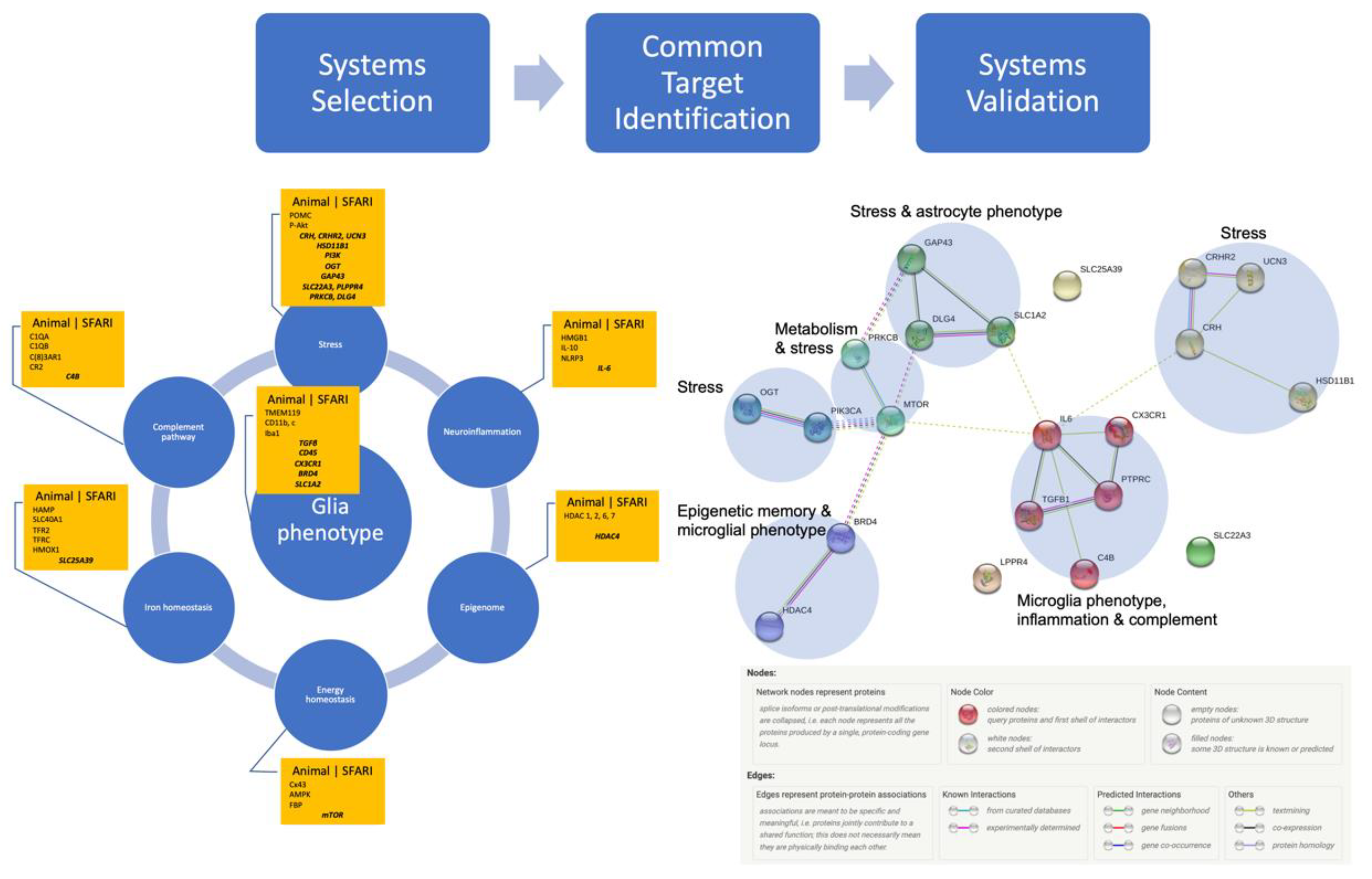
Gathering the Biomarkers across Cell Systems and Species

{kind=link}
| Groups | Genes | Function | Effect of Inflammation | Effect of Stress |
|---|---|---|---|---|
| Glial cell phenotype | TMEM119 | Transmembrane protein 119; identifies resident microglia (from blood-derived macrophages) [8,9] | Up | Up |
| TGFβ [4] | Transforming growth factor beta-1; resident microglial biomarker [10,11] | First up, then down | Down | |
| CD11b | Resident microglial biomarker [9,10,11] | Up | Up | |
| CD11c | Activated microglia [10,11] | Up | Up | |
| CD45 [4] | Protein Tyrosine Phosphatase Receptor Type C (PTPRC); Resident microglial biomarker [9,10,11] | Up | Up | |
| Iba1 | Ionized calcium binding adaptor molecule 1: Non-specific microglial/macrophage biomarker [3,9,10,11,12] | Up | Up | |
| CX3CR1 [4] | CX3C chemokine receptor 1; required for synaptic pruning during brain development [13] | Up | Up | |
| BRD4 [4] | Bromodomain containing 4; polarizes microglia toward inflammatory phenotype [14]; involved in epigenetic memory [15] | Up | Up | |
| SLC1A2 [S] | Astrocytic GLT-1 transporter required for neuron–astrocyte communication and astrocyte maturation [16] | Up | Down | |
| Inflammation | HMGB1 | Hypermobility group box protein 1, a pleiotropic signaling molecule in glia cells and neurons: a growth factor, a pro-inflammatory molecule [17]; implicated in ASD [18] | Up | Up |
| IL-10 | Key regulator of neuroimmune homeostasis via cross-talk of microglia and astrocytes [19] | Down or Up | Down | |
| IL-6 [5] | Early inflammatory cytokine | Up | Up | |
| NLRP3 | Inflammasome activated in ASD [20] | Up | Up | |
| Stress | CRH [5] | Corticotropin-releasing hormone, key hormone linking chronic stress with anxiety [21,22]; has direct effects on microglia [23] | Up | Up |
| CRHR2 [5] | Corticotropin-releasing hormone receptor 2; [23] | Up or Down, locoregional | Up | |
| HSD11B1 [4] | 11β-Hydroxysteroid dehydrogenase type 1 [24] | Up | Down | |
| POMC | Proopiomelanocortin [25] | Down | Up | |
| p-Akt | Phosphorylated-Akt [26] | Up | Up | |
| PI3K [S] | PI3K/Akt signaling pathway; e.g., PIK3CA [26,27] | Up | Up | |
| OGT [5] | O-GlcNAc transferase; a placental biomarker of maternal stress exposure related to neurodevelopmental outcomes [28,29] | Up | Up | |
| GAP43 [5] | Growth associated protein 43; in astrocytes, GAP43 mediates glial and neuronal plasticity during astrogliosis and attenuates microglial activation under LPS exposure [30] | Down | Down | |
| SLC22A3 [5] | Solute carrier family 22 member 3; modulates anxiety and social interaction [31,32] | Up | Up | |
| PLPPR4 [5] | Phospholipid Phosphatase Related 4; stress-related behaviors such as reduced resilience [33] | Up | Up | |
| PRKCB [3] | Protein Kinase C Beta; involved in stress-related behavior [34] | Up | Up | |
| UCN3 [5] | Urocortin 3; binds specifically CRHR2 [23] | Down | Down | |
| DLG4 [5] | Disks large homolog 4; modulates stress reactivity and anxiety [35,36] | Down | Down | |
| Energy homeostasis | Cx43 | Connexin 43 gap junction maintaining astrocytes’ homeostasis via metabolic cooperation [37] | Down | Down |
| AMPK | Adenosine monophosphate kinase, intracellular energy sensor [38] | Up | Up | |
| FBP | Fructo-biphosphokinase: signature of second-hit memory of inflammation in fetal microglia [3] | Up | Up | |
| mTOR [S] | Mammalian target of the rapamycin signaling pathway [39,40] | Up | Up | |
| Iron homeostasis | HAMP | Hepcidin, a regulator of iron homeostasis and inflammation; implicated, along with ferroportin and transferrin, in microglial response to endotoxin interfering with α7nAChR signaling [4] | Up | Up |
| SLC40A1 | Ferroportin | Up | Up | |
| TFR2 | Transferrin receptor 2; involved in iron sequestration | Up | Up | |
| TFRC | Transferrin receptor protein 1; needed for iron sequestration | Down | Down | |
| HMOX1 | Hemoxygenase 1, key enzyme of iron homeostasis; also serves as a signature of second-hit memory in fetal microglia and promoted by α7nAChR stimulation [3,41] | Up | Up | |
| SLC25A39 [4] | Member of the SLC25 transporter or mitochondrial carrier family of proteins; required for normal heme biosynthesis | Up | Up | |
| Complement pathway | C1QA | Aside from their traditional role in innate immunity [42], elements of the complement pathway are involved in neuronal–glial interactions; recognized as essential players in brain development, especially in synaptogenesis/synaptic pruning and predisposition for neurodegenerative diseases [7,43]; their microglial expression is also susceptible to LPS exposure in utero [4] | Up | Up |
| C1QB | Up | Up | ||
| C43AR1 | Up | Up | ||
| CR2 | Up | Up | ||
| C4B [4] | Up | Up | ||
| Epigenetic memory | HDAC [S] and HAT families | Histone acetylation/deacetylation enzymes: HDAC 1, 2, 3, 7 and 9 found in SSC database of ASD gene mutations.HDAC 1, 2, 4 and 6 involved in fetal microglial memory of LPS exposure; HDAC 1, 2, 4 and 7 increased with altered neuronal AChE signaling and increased anxiety behavior due to adult chronic stress exposure [3,4,25,27,44,45] | Up or Down | Up or Down |

3. Results
Systems Validation: Many Species, One Network
4. Discussion
4.1. Information Processing, Energy Demand and Stress
4.2. Defining a Cohesive Cell Systems Correlate of ASD as Predictive Impairment
5. Limitations and Future Directions
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Al-Haddad, B.J.S.; Jacobsson, B.; Chabra, S.; Modzelewska, D.; Olson, E.M.; Bernier, R.; Enquobahrie, D.A.; Hagberg, H.; Östling, S.; Rajagopal, L.; et al. Long-term Risk of Neuropsychiatric Disease After Exposure to Infection In Utero. JAMA Psychiatry 2019, 76, 594–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frasch, M.G.; Lobmaier, S.M.; Stampalija, T.; Desplats, P.; Pallarés, M.E.; Pastor, V.; Brocco, M.A.; Wu, H.-T.; Schulkin, J.; Herry, C.L.; et al. Non-invasive biomarkers of fetal brain development reflecting prenatal stress: An integrative multi-scale multi-species perspective on data collection and analysis. Neurosci. Biobehav. Rev. 2020, 117, 165–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, M.; Cortes, M.; Moore, C.S.; Leong, S.Y.; Durosier, L.D.; Burns, P.; Fecteau, G.; Desrochers, A.; Auer, R.N.; Barreiro, L.B.; et al. Fetal microglial phenotype in vitro carries memory of prior in vivo exposure to inflammation. Front. Cell. Neurosci. 2015, 9, 294. [Google Scholar] [CrossRef] [Green Version]
- Cortes, M.; Cao, M.; Liu, H.L.; Moore, C.S.; Durosier, L.D.; Burns, P.; Fecteau, G.; Desrochers, A.; Barreiro, L.B.; Antel, J.P.; et al. α7 nicotinic acetylcholine receptor signaling modulates the inflammatory phenotype of fetal brain microglia: First evidence of interference by iron homeostasis. Sci. Rep. 2017, 7, 10645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, Y.S.; Wollam, J.; Olefsky, J.M. An Integrated View of Immunometabolism. Cell 2018, 172, 22–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camandola, S. Astrocytes, emerging stars of energy homeostasis. Cell Stress 2018, 2, 246–252. [Google Scholar] [CrossRef] [Green Version]
- Schafer, D.P.; Lehrman, E.K.; Kautzman, A.G.; Koyama, R.; Mardinly, A.R.; Yamasaki, R.; Ransohoff, R.M.; Greenberg, M.E.; Barres, B.A.; Stevens, B. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron 2012, 74, 691–705. [Google Scholar] [CrossRef] [Green Version]
- Satoh, J.-I.; Kino, Y.; Asahina, N.; Takitani, M.; Miyoshi, J.; Ishida, T.; Saito, Y. TMEM119 marks a subset of microglia in the human brain. Neuropathology 2015, 36, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. Faculty Opinions recommendation of New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Butovsky, O.; Jedrychowski, M.P.; Moore, C.S.; Cialic, R.; Lanser, A.J.; Gabriely, G.; Koeglsperger, T.; Dake, B.; Wu, P.; Doykan, C.; et al. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat. Neurosci. 2014, 17, 131–143. [Google Scholar] [CrossRef] [Green Version]
- Abud, E.M.; Ramirez, R.N.; Martinez, E.S.; Healy, L.M.; Nguyen, C.H.; Newman, S.A.; Yeromin, A.V.; Scarfone, V.M.; Marsh, S.E.; Fimbres, C.; et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 2017, 94, 278–293.e9. [Google Scholar] [CrossRef] [Green Version]
- Frasch, M.G.; Szynkaruk, M.; Prout, A.P.; Nygard, K.; Cao, M.; Veldhuizen, R.; Hammond, R.; Richardson, B.S. Decreased neuroinflammation correlates to higher vagus nerve activity fluctuations in near-term ovine fetuses: A case for the afferent cholinergic anti-inflammatory pathway? J. Neuroinflammat. 2016, 13, 103. [Google Scholar] [CrossRef] [Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic Pruning by Microglia Is Necessary for Normal Brain Development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Chen, J.; Jin, H.; Lin, D.; Chen, Y.; Chen, X.; Wang, B.; Hu, S.; Wu, Y.; Wu, Y.; et al. BRD4 inhibition attenuates inflammatory response in microglia and facilitates recovery after spinal cord injury in rats. J. Cell. Mol. Med. 2019, 23, 3214–3223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penas, C.; Navarro, X. Epigenetic Modifications Associated to Neuroinflammation and Neuropathic Pain After Neural Trauma. Front. Cell. Neurosci. 2018, 12, 158. [Google Scholar] [CrossRef] [Green Version]
- Hasel, P.; Dando, O.; Jiwaji, Z.; Baxter, P.; Todd, A.C.; Heron, S.; Márkus, N.M.; McQueen, J.; Hampton, D.W.; Torvell, M.; et al. Neurons and neuronal activity control gene expression in astrocytes to regulate their development and metabolism. Nat. Commun. 2017, 8, 15132. [Google Scholar] [CrossRef] [Green Version]
- Frasch, M.G.; Nygard, K.L. Location, Location, Location: Appraising the Pleiotropic Function of HMGB1 in Fetal Brain. J. Neuropathol. Exp. Neurol. 2017, 76, 332–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dipasquale, V.; Cutrupi, M.C.; Colavita, L.; Manti, S.; Cuppari, C.; Salpietro, C. Neuroinflammation in Autism Spectrum Disorders: The Role of High Mobility Group Box 1 Protein. Int. J. Mol. Cell. Med. 2017, 6, 148–155. [Google Scholar] [PubMed]
- Lobo-Silva, D.; Carriche, G.M.; Gil Castro, A.; Roque, S.; Saraiva, M. Balancing the immune response in the brain: IL-10 and its regulation. J. Neuroinflammat. 2016, 13, 297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saresella, M.; Piancone, F.; Marventano, I.; Zoppis, M.; Hernis, A.; Zanette, M.; Trabattoni, D.; Chiappedi, M.; Ghezzo, A.; Canevini, M.P.; et al. Multiple inflammasome complexes are activated in autistic spectrum disorders. Brain Behav. Immun. 2016, 57, 125–133. [Google Scholar] [CrossRef]
- Tsilioni, I.; Dodman, N.; I Petra, A.; Taliou, A.; Francis, K.; Moon-Fanelli, A.; Shuster, L.; Theoharides, T.C. Elevated serum neurotensin and CRH levels in children with autistic spectrum disorders and tail-chasing Bull Terriers with a phenotype similar to autism. Transl. Psychiatry 2014, 4, e466. [Google Scholar] [CrossRef] [Green Version]
- Theoharides, T.C.; Asadi, S.; Patel, A.B. Focal brain inflammation and autism. J. Neuroinflammat. 2013, 10, 815–846. [Google Scholar] [CrossRef] [Green Version]
- Kato, T.A.; Hayakawa, K.; Monji, A.; Kanba, S. Missing and Possible Link between Neuroendocrine Factors, Neuropsychiatric Disorders, and Microglia. Front. Integr. Neurosci. 2013, 7, 53. [Google Scholar] [CrossRef] [Green Version]
- Wyrwoll, C.S.; Holmes, M.C.; Seckl, J.R. 11β-hydroxysteroid dehydrogenases and the brain: From zero to hero, a decade of progress. Front. Neuroendocrinol. 2011, 32, 265–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemche, E.; Chaban, O.S.; Lemche, A.V. Neuroendorine and Epigentic Mechanisms Subserving Autonomic Imbalance and HPA Dysfunction in the Metabolic Syndrome. Front. Neurosci. 2016, 10, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, P.-C.; Yang, C.-H.; Huang, C.-C.; Hsu, K.-S. Phosphatidylinositol 3-kinase activation is required for stress protocol-induced modification of hippocampal synaptic plasticity. J. Biol. Chem. 2008, 283, 2631–2643. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, H.-S.; Zhu, W.-L.; Liu, J.-F.; Luo, Y.-X.; Si, J.-J.; Wang, S.-J.; Xue, Y.-X.; Ding, Z.-B.; Shi, J.; Lu, L. PI3K/Akt Signaling Pathway in the Basolateral Amygdala Mediates the Rapid Antidepressant-like Effects of Trefoil Factor 3. Neuropsychopharmacology 2012, 37, 2671–2683. [Google Scholar] [CrossRef]
- Howerton, C.L.; Morgan, C.P.; Fischer, D.B.; Bale, T.L. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc. Natl. Acad. Sci. USA 2013, 110, 5169–5174. [Google Scholar] [CrossRef] [Green Version]
- Howerton, C.L.; Bale, T.L. Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2014, 111, 9639–9644. [Google Scholar] [CrossRef] [Green Version]
- Hung, C.C.; Lin, C.H.; Chang, H.; Wang, C.Y.; Lin, S.H.; Hsu, P.C.; Sun, Y.-Y.; Lin, T.-N.; Shie, F.-S.; Kao, L.-S.; et al. Astrocytic GAP43 Induced by the TLR4/NF-κB/STAT3 Axis Attenuates Astrogliosis-Mediated Microglial Activation and Neurotoxicity. J. Neurosci. 2016, 36, 2027–2043. [Google Scholar] [CrossRef] [Green Version]
- Wultsch, T.; Grimberg, G.; Schmitt, A.; Painsipp, E.; Wetzstein, H.; Breitenkamp, A.F.S.; Gründemann, D.; Schömig, E.; Lesch, K.-P.; Gerlach, M.; et al. Decreased anxiety in mice lacking the organic cation transporter 3. J. Neural Transm. 2009, 116, 689–697. [Google Scholar] [CrossRef] [PubMed]
- Garbarino, V.R.; Santos, T.A.; Nelson, A.R.; Zhang, W.Q.; Smolik, C.M.; Javors, M.A.; Daws, L.C.; Gould, G.G. Prenatal metformin exposure or organic cation transporter 3 knock-out curbs social interaction preference in male mice. Pharmacol. Res. 2018, 140, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Vogt, J.; Yang, J.; Mobascher, A.; Cheng, J.; Li, Y.; Liu, X.; Baumgart, J.; Thalman, C.; Kirischuk, S.; Unichenko, P.; et al. Molecular cause and functional impact of altered synaptic lipid signaling due to a prg-1 gene SNP. EMBO Mol. Med. 2015, 8, 25–38. [Google Scholar] [CrossRef]
- Lisowski, P.; Wieczorek, M.; Goscik, J.; Juszczak, G.R.; Stankiewicz, A.M.; Zwierzchowski, L.; Swiergiel, A.H. Effects of Chronic Stress on Prefrontal Cortex Transcriptome in Mice Displaying Different Genetic Backgrounds. J. Mol. Neurosci. 2012, 50, 33–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feyder, M.; Karlsson, R.-M.; Mathur, P.; Lyman, M.; Bock, R.; Momenan, R.; Munasinghe, J.; Scattoni, M.L.; Ihne, J.; Camp, M.; et al. Association of MouseDlg4(PSD-95) Gene Deletion and HumanDLG4Gene Variation with Phenotypes Relevant to Autism Spectrum Disorders and Williams’ Syndrome. Am. J. Psychiatry 2010, 167, 1508–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Shi, M.; Ma, Z.; Zhao, S.; Euskirchen, G.; Ziskin, J.; Urban, A.; Hallmayer, J.; Snyder, M. Integrated systems analysis reveals a molecular network underlying autism spectrum disorders. Mol. Syst. Biol. 2014, 10, 774. [Google Scholar] [CrossRef]
- Contreras, J.E.; Sánchez, H.A.; Eugenín, E.A.; Speidel, D.; Theis, M.; Willecke, K.; Bukauskas, F.F.; Bennett, M.V.L.; Sáez, J.C. Metabolic inhibition induces opening of unapposed connexin 43 gap junction hemichannels and reduces gap junctional communication in cortical astrocytes in culture. Proc. Natl. Acad. Sci. USA 2001, 99, 495–500. [Google Scholar] [CrossRef] [Green Version]
- Frasch, M.G. Putative Role of AMPK in Fetal Adaptive Brain Shut-Down: Linking Metabolism and Inflammation in the Brain. Front. Neurol. 2014, 5, 150. [Google Scholar] [CrossRef] [Green Version]
- Hui, K.; Katayama, Y.; Nakayama, K.I.; Nomura, J.; Sakurai, T. Characterizing vulnerable brain areas and circuits in mouse models of autism: Towards understanding pathogenesis and new therapeutic approaches. Neurosci. Biobehav. Rev. 2018, 110, 77–91. [Google Scholar] [CrossRef]
- Wang, H.; Doering, L.C. Reversing autism by targeting downstream mTOR signaling. Front. Cell. Neurosci. 2013, 7, 28. [Google Scholar] [CrossRef] [Green Version]
- Hua, S.; Ek, C.J.; Mallard, C.; Johansson, M.E. Perinatal hypoxia-ischemia reduces alpha 7 nicotinic receptor expression and selective alpha 7 nicotinic receptor stimulation suppresses inflammation and promotes microglial Mox phenotype. Biomed Res. Int. 2014, 2014, 718769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noris, M.; Remuzzi, G. Overview of Complement Activation and Regulation. Semin. Nephrol. 2013, 33, 479–492. [Google Scholar] [CrossRef] [Green Version]
- Hong, S.; Beja-Glasser, V.F.; Nfonoyim, B.M.; Frouin, A.; Li, S.; Ramakrishnan, S.; Merry, K.M.; Shi, Q.; Rosenthal, A.; Barres, B.A.; et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science 2016, 352, 712–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thorsell, A. Brain neuropeptide Y and corticotropin-releasing hormone in mediating stress and anxiety. Exp. Biol. Med. 2010, 235, 1163–1167. [Google Scholar] [CrossRef]
- Sailaja, B.S.; Cohen-Carmon, D.; Zimmerman, G.; Soreq, H.; Meshorer, E. Stress-induced epigenetic transcriptional memory of acetylcholinesterase by HDAC4. Proc. Natl. Acad. Sci. USA 2012, 109, E3687–E3695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.; Morris, J.; Bork, P.; et al. STRING v11: Protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [Green Version]
- Frasch, M. Martinfrasch/ASD_Origins_Hypothesis 1. 2019. Available online: https://zenodo.org/record/3402349 (accessed on 22 June 2023).
- Sinha, P.; Kjelgaard, M.M.; Gandhi, T.K.; Tsourides, K.; Cardinaux, A.L.; Pantazis, D.; Diamond, S.P.; Held, R.M. Autism as a disorder of prediction. Proc. Natl. Acad. Sci. USA 2014, 111, 15220–15225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, A.; McEwen, B.S.; Friston, K. Uncertainty and stress: Why it causes diseases and how it is mastered by the brain. Prog. Neurobiol. 2017, 156, 164–188. [Google Scholar] [CrossRef]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Boltzmann, M.L. Über die Beziehung zwischen dem zweiten Hauptsatze des mechanischen Wärmetheorie und der Wahr-scheinlichkeitsrechnung, respective den Sätzen über das Wärmegleichgewicht. On the relationship between the second main theorem of mechanical heat theory and the probability calculation with respect to the results about the heat equilibrium. Wiener Berichte 1877, 76, 373–435. [Google Scholar]
- McEwen, B.S.; Stellar, E. Stress and the individual. Mechanisms leading to disease. Arch. Intern. Med. 1993, 153, 2093–2101. [Google Scholar] [CrossRef]
- D’urso, A.; Brickner, J.H. Mechanisms of epigenetic memory. Trends Genet. 2014, 30, 230–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donohoe, D.R.; Bultman, S.J. Metaboloepigenetics: Interrelationships between energy metabolism and epigenetic control of gene expression. J. Cell. Physiol. 2012, 227, 3169–3177. [Google Scholar] [CrossRef] [Green Version]
- Etchegaray, J.-P.; Mostoslavsky, R. Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 2016, 62, 695–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Long, H.; Chang, C.; Zhao, M.; Lu, Q. Crosstalk between metabolism and epigenetic modifications in autoimmune diseases: A comprehensive overview. Cell. Mol. Life Sci. 2018, 75, 3353–3369. [Google Scholar] [CrossRef]
- Thakkar, K.N.; Polli, F.E.; Joseph, R.M.; Tuch, D.S.; Hadjikhani, N.; Barton, J.J.; Manoach, D.S. Response monitoring, repetitive behaviour and anterior cingulate abnormalities in autism spectrum dis-orders (ASD). Brain 2008, 131, 2464–2478. [Google Scholar] [CrossRef] [Green Version]
- Ide, J.S.; Shenoy, P.; Yu, A.J.; Li, C.-S.R. Bayesian Prediction and Evaluation in the Anterior Cingulate Cortex. J. Neurosci. 2013, 33, 2039–2047. [Google Scholar] [CrossRef] [Green Version]
- Einarsson, E.; Nader, K. Involvement of the anterior cingulate cortex in formation, consolidation, and reconsolidation of recent and remote contextual fear memory. Learn. Mem. 2012, 19, 449–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Velzen, L.S.; Schmaal, L.; Milaneschi, Y.; van Tol, M.J.; van der Wee, N.J.; Veltman, D.J.; Penninx, B.W. Immunometabolic dysregulation is associated with reduced cortical thickness of the anterior cingulate cortex. Brain Behav. Immun. 2017, 60, 361–368. [Google Scholar] [CrossRef]
- Halder, R.; Hennion, M.; O Vidal, R.; Shomroni, O.; Rahman, R.-U.; Rajput, A.; Centeno, T.P.; Van Bebber, F.; Capece, V.; Vizcaino, J.C.G.; et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat. Neurosci. 2015, 19, 102–110. [Google Scholar] [CrossRef]
- Sharma, R.; Frasch, M.G.; Zelgert, C.; Zimmermann, P.; Fabre, B.; Wilson, R.; Waldenberger, M.; MacDonald, J.W.; Bammler, T.K.; Lobmaier, S.M.; et al. Maternal–fetal stress and DNA methylation signatures in neonatal saliva: An epigenome-wide association study. Clin. Epigenetics 2022, 14, 87. [Google Scholar] [CrossRef]
- Huang, Z.; Wang, Y.; Hu, G.; Zhou, J.; Mei, L.; Xiong, W.-C. YAP Is a Critical Inducer of SOCS3, Preventing Reactive Astrogliosis. Cereb. Cortex 2015, 26, 2299–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Gayed, E.M.A.; Rizk, M.S.; Ramadan, A.N.; Bayomy, N.R. mRNA Expression of the CUB and Sushi Multiple Domains 1 (CSMD1) and Its Serum Protein Level as Predictors for Psychosis in the Familial High-Risk Children and Young Adults. ACS Omega 2021, 6, 24128–24138. [Google Scholar] [CrossRef]
- Liu, Y.; Fu, X.; Tang, Z.; Li, C.; Xu, Y.; Zhang, F.; Zhou, D.; Zhu, C. Altered expression of the CSMD1 gene in the peripheral blood of schizophrenia patients. BMC Psychiatry 2019, 19, 113. [Google Scholar] [CrossRef] [Green Version]
- Cukier, H.N.; Dueker, N.D.; Slifer, S.H.; Lee, J.M.; Whitehead, P.L.; Lalanne, E.; Leyva, N.; Konidari, I.; Gentry, R.C.; Hulme, W.F.; et al. Exome sequencing of extended families with autism reveals genes shared across neurodevelopmental and neuropsychiatric disorders. Mol. Autism 2014, 5, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Peng, Y.; Hu, Z.; Li, Y.; Xun, G.; Ou, J.; Sun, L.; Xiong, Z.; Liu, Y.; Wang, T.; et al. Genome-wide copy number variation analysis in a Chinese autism spectrum disorder cohort. Sci. Rep. 2017, 7, 44155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deciphering Developmental Disorders Study. Large-scale discovery of novel genetic causes of developmental disorders. Nature 2015, 519, 223–228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoelper, D.; Huang, H.; Jain, A.Y.; Patel, D.J.; Lewis, P.W. Structural and mechanistic insights into ATRX-dependent and -independent functions of the histone chaperone DAXX. Nat. Commun. 2017, 8, 1193. [Google Scholar] [CrossRef] [Green Version]
- Rubin, A.N.; Malik, R.; Cho, K.K.A.; Lim, K.J.; Lindtner, S.; Schwartz, S.E.R.; Vogt, D.; Sohal, V.S.; Rubenstein, J.L.R. Regulatory Elements Inserted into AAVs Confer Preferential Activity in Cortical Interneurons. Eneuro 2020, 7. [Google Scholar] [CrossRef]
- Han, V.X.; Patel, S.; Jones, H.F.; Dale, R.C. Maternal immune activation and neuroinflammation in human neurodevelopmental disorders. Nat. Rev. Neurol. 2021, 17, 564–579. [Google Scholar] [CrossRef]
- Jin, Y.; Kong, J. Transcutaneous Vagus Nerve Stimulation: A Promising Method for Treatment of Autism Spectrum Disorders. Front. Neurosci. 2017, 10, 609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lobmaier, S.M.; Müller, A.; Zelgert, C.; Shen, C.; Su, P.C.; Schmidt, G.; Haller, B.; Berg, G.; Fabre, B.; Weyrich, J.; et al. Fetal heart rate variability responsiveness to maternal stress, non-invasively detected from maternal transabdominal ECG. Arch. Gynecol. Obstet. 2019, 301, 405–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonelli, M.C.; Frasch, M.G.; Rumi, M.; Sharma, R.; Zimmermann, P.; Molinet, M.S.; Lobmaier, S.M. Early Biomarkers and Intervention Programs for the Infant Exposed to Prenatal Stress. Curr. Neuropharmacol. 2022, 20, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Tracey, K.J. Reflex Principles of Immunological Homeostasis. Annu. Rev. Immunol. 2012, 30, 313–335. [Google Scholar] [CrossRef] [Green Version]
- Frasch, M.; Prout, A.; Szynkaruk, M.; Gagnon, R.; Richardson, B. Cholinergic anti-inflammatory pathway mechanisms may be active in the pre-term ovine fetus. Reprod. Sci. 2009, 16, 137A. [Google Scholar]
- Frasch, M.G.; Nygard, K.; Vittal, P.; Zhao, L.; Regnault, T.R.; Richardson, B.S. Translocation of neuronal high-mobility group box1 protein in relation to microglial activation in fetal sheep following repetitive umbilical cord occlusions with severe hypoxic-acidemia. Reprod. Sci. 2010, 17, 51A. [Google Scholar]
- Nygard, K.; Vittal, P.; Richardson, B.S.; Frasch, M.G. Fetal cholinergic anti-inflammatory pathway and the neuronal and astrocytic high-mobility group box 1 (HMGB1) protein release during cerebral inflammatory response. Reprod. Sci. 2011, 18, 161A. [Google Scholar]
- Furukawa, S.; Sameshima, H.; Yang, L.; Ikenoue, T. Activation of acetylcholine receptors and microglia in hypoxic-ischemic brain damage in newborn rats. Brain Dev. 2012, 35, 607–613. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, S.; Sameshima, H.; Yang, L.; Ikenoue, T. Acetylcholine receptor agonist reduces brain damage induced by hypoxia-ischemia in newborn rats. Reprod. Sci. 2011, 18, 172–179. [Google Scholar] [CrossRef]
- Cheyuo, C.; Jacob, A.; Wu, R.; Zhou, M.; Coppa, G.F.; Wang, P. The Parasympathetic Nervous System in the Quest for Stroke Therapeutics. J. Cereb. Blood Flow Metab. 2011, 31, 1187–1195. [Google Scholar] [CrossRef]
- Cao, J.; Lu, K.-H.; Powley, T.L.; Liu, Z. Vagal nerve stimulation triggers widespread responses and alters large-scale func-tional connectivity in the rat brain. PLoS ONE 2017, 12, e0189518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, B.; Wang, J.; Shahed, M.; Jelfs, B.; Chan, R.H.M.; Li, Y. Vagus Nerve Stimulation Alters Phase Synchrony of the Anterior Cingulate Cortex and Facilitates Decision Making in Rats. Sci. Rep. 2016, 6, 35135. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Yoon, B.-J.; Dougherty, E.R. Accurate and Reliable Cancer Classification Based on Probabilistic Inference of Pathway Activity. PLoS ONE 2009, 4, e8161. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jeong, H.; Yoon, B.-J.; Qian, X. ClusterM: A scalable algorithm for computational prediction of conserved protein complexes across multiple protein interaction networks. BMC Genom. 2020, 21, 1–14. [Google Scholar] [CrossRef]
- Maddouri, O.; Qian, X.; Yoon, B.-J. Deep graph representations embed network information for robust disease marker identification. Bioinformatics 2021, 38, 1075–1086. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frasch, M.G.; Yoon, B.-J.; Helbing, D.L.; Snir, G.; Antonelli, M.C.; Bauer, R. Autism Spectrum Disorder: A Neuro-Immunometabolic Hypothesis of the Developmental Origins. Biology 2023, 12, 914. https://doi.org/10.3390/biology12070914
Frasch MG, Yoon B-J, Helbing DL, Snir G, Antonelli MC, Bauer R. Autism Spectrum Disorder: A Neuro-Immunometabolic Hypothesis of the Developmental Origins. Biology. 2023; 12(7):914. https://doi.org/10.3390/biology12070914
Chicago/Turabian StyleFrasch, Martin G., Byung-Jun Yoon, Dario Lucas Helbing, Gal Snir, Marta C. Antonelli, and Reinhard Bauer. 2023. "Autism Spectrum Disorder: A Neuro-Immunometabolic Hypothesis of the Developmental Origins" Biology 12, no. 7: 914. https://doi.org/10.3390/biology12070914
APA StyleFrasch, M. G., Yoon, B. -J., Helbing, D. L., Snir, G., Antonelli, M. C., & Bauer, R. (2023). Autism Spectrum Disorder: A Neuro-Immunometabolic Hypothesis of the Developmental Origins. Biology, 12(7), 914. https://doi.org/10.3390/biology12070914