
Whole Genome Screening Procures a Holistic Hold of the Russian Chicken Gene Pool Heritage and Demographic History

 , , ,
, , ,  ,
,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Breeds, Sampling and Ethics Statement
2.2. DNA Isolation
2.3. SNP Markers and Genotyping Quality Control
2.4. Genetic Diversity, Population Structure and Split and Admixture
2.5. Runs of Homozygosity Analysis
2.6. Demographic History Inference
3. Results
3.1. Genetic Diversity
3.2. ROH Distribution in Chicken Breed Genomes
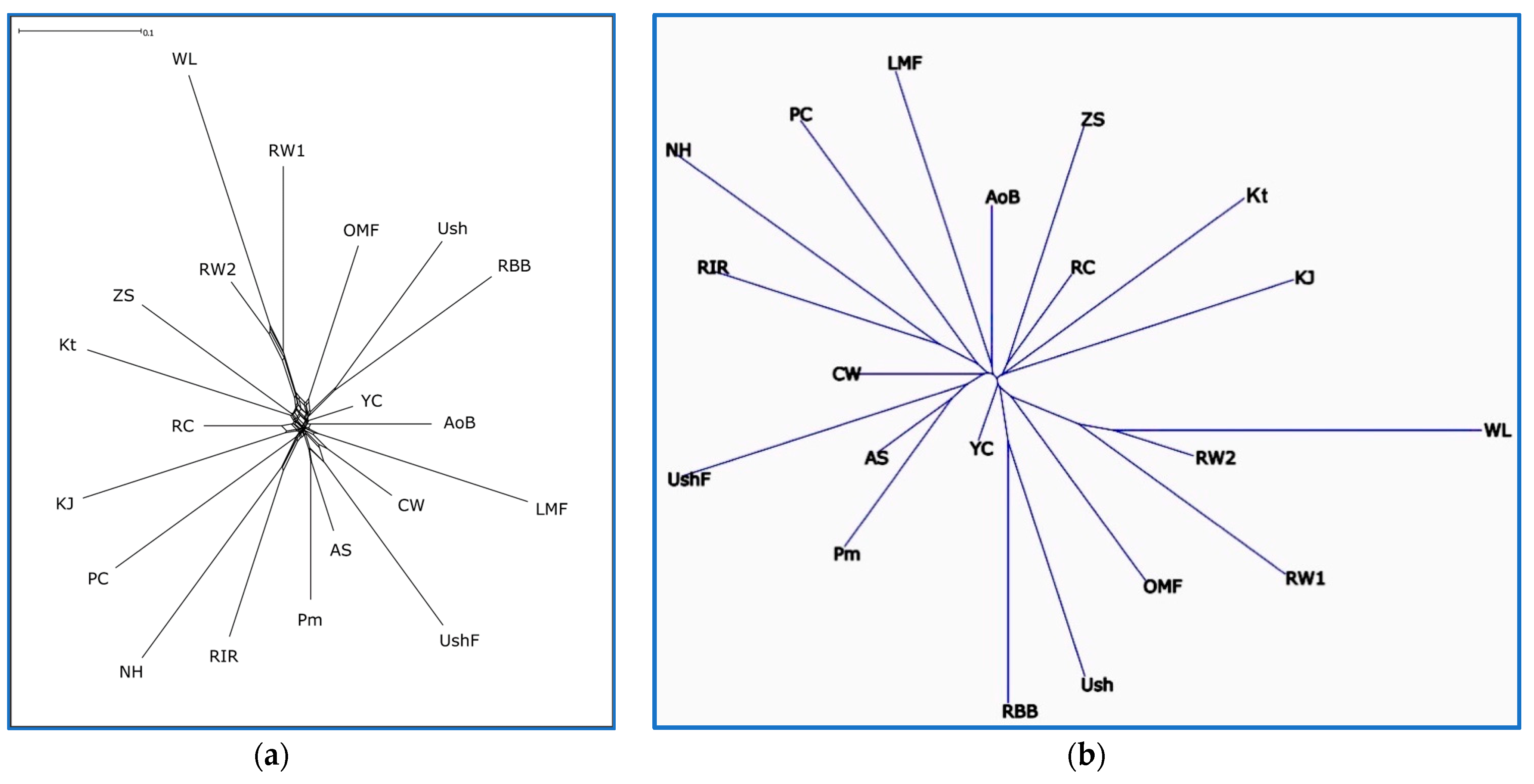
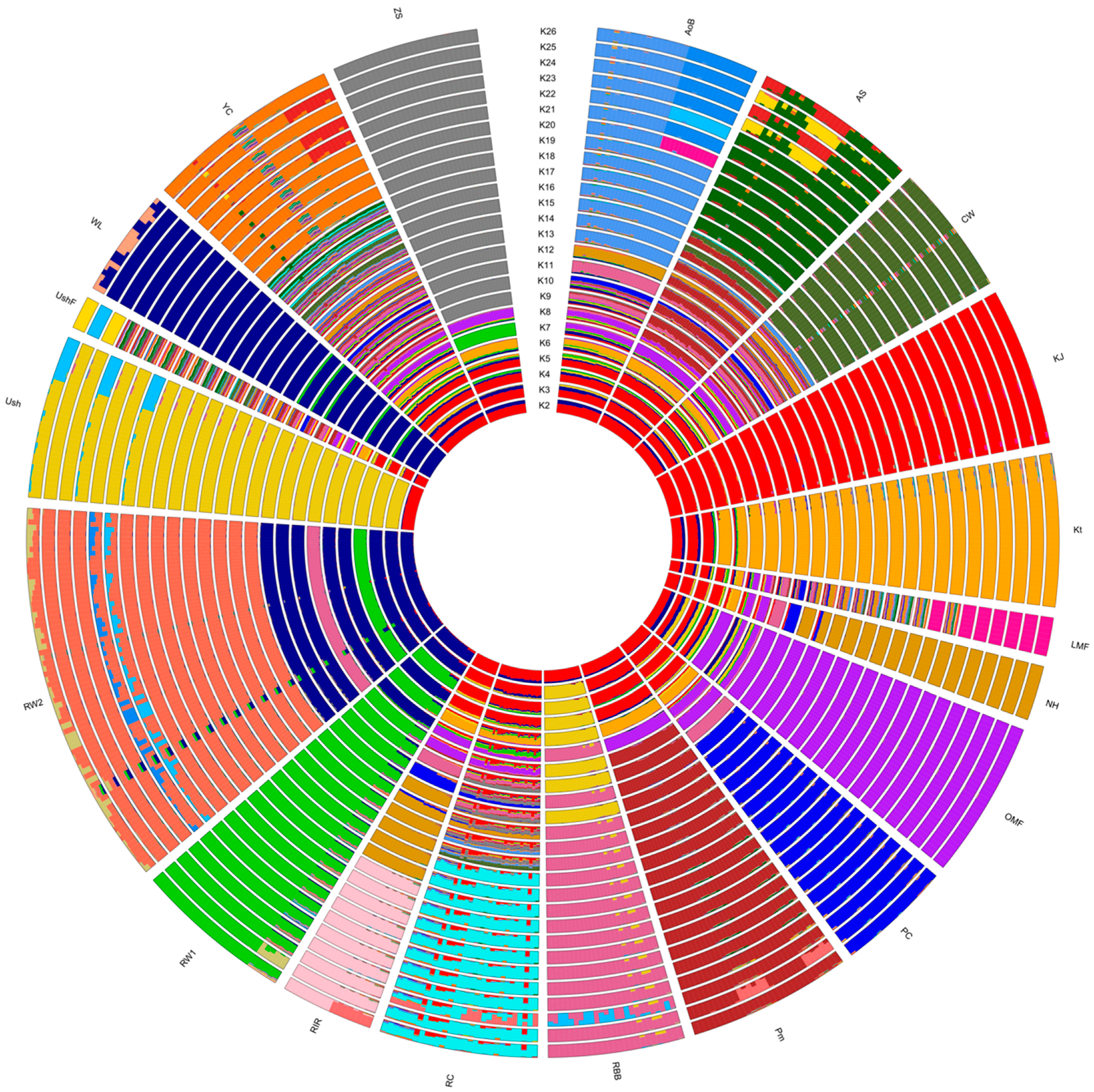
3.3. Breed Relationship and Admixture
3.4. Demographic History Inference
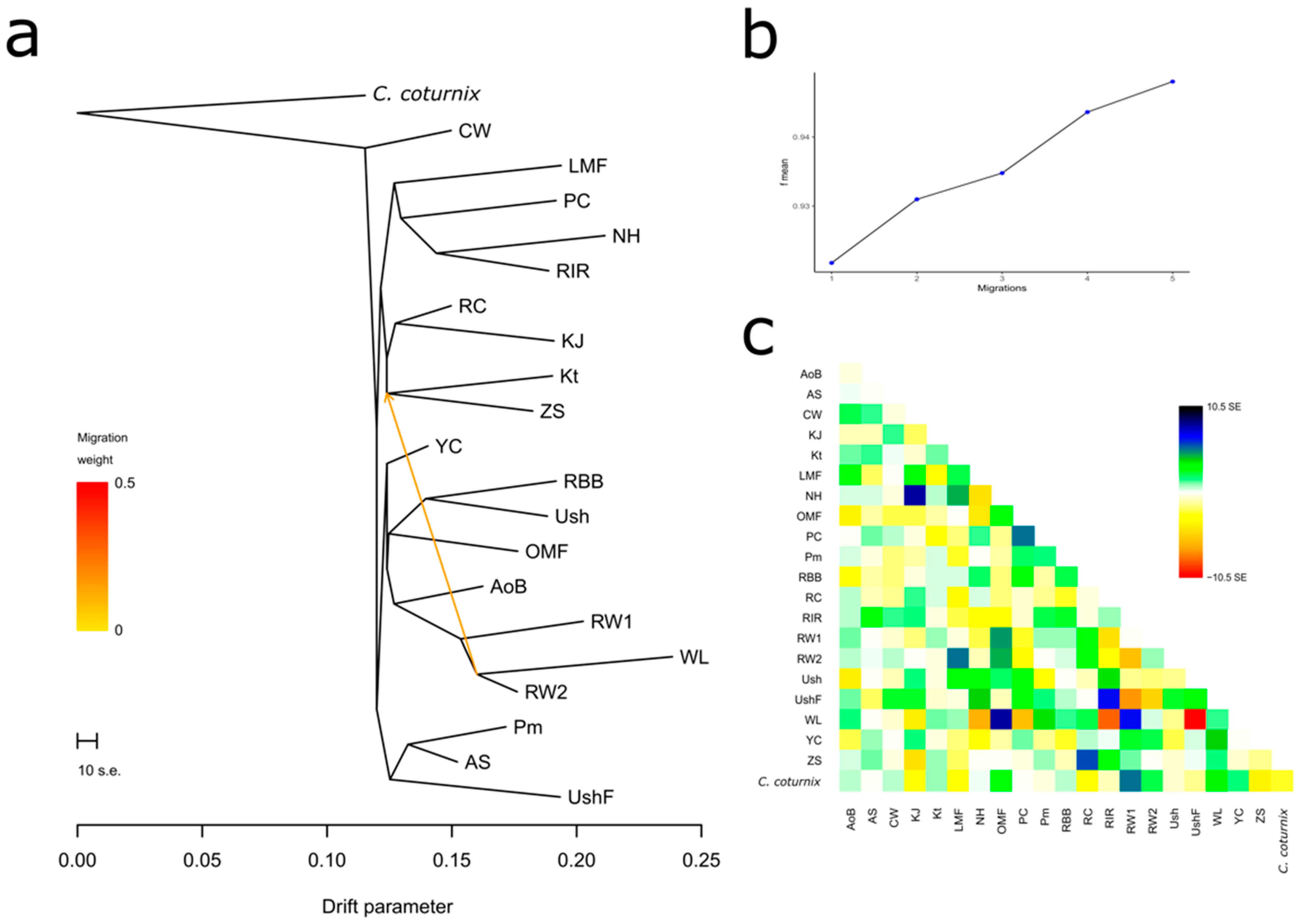
3.4.1. Population Divergence and Gene Flow
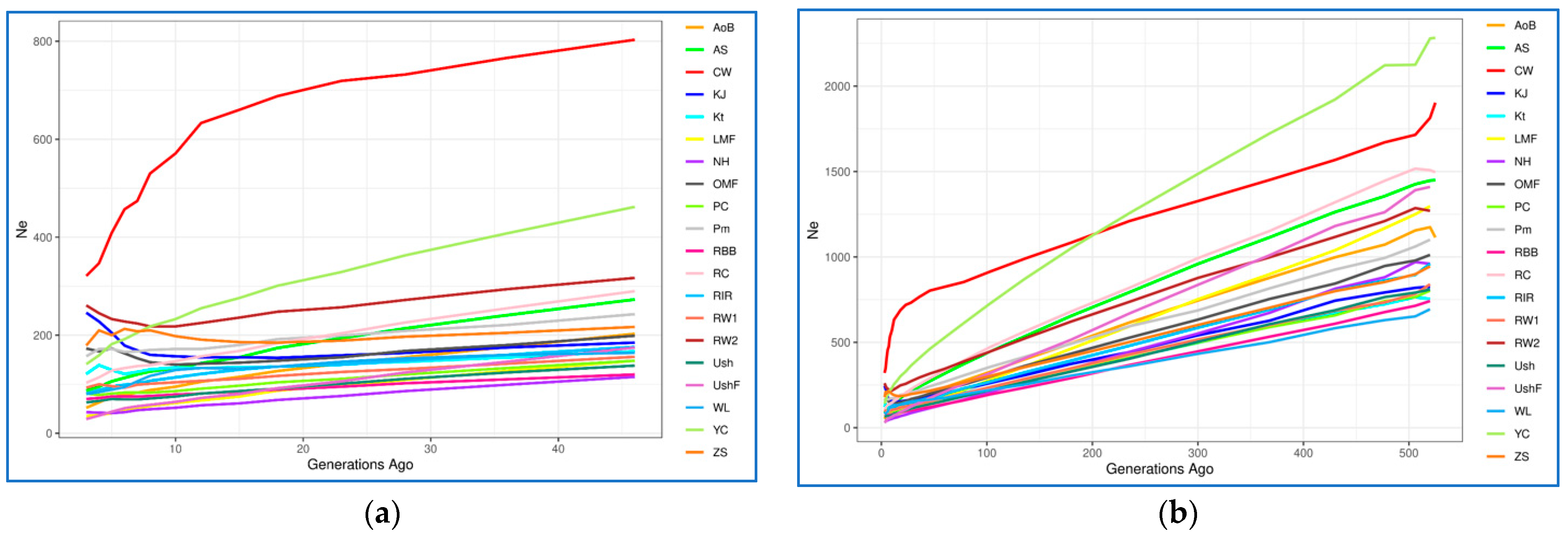
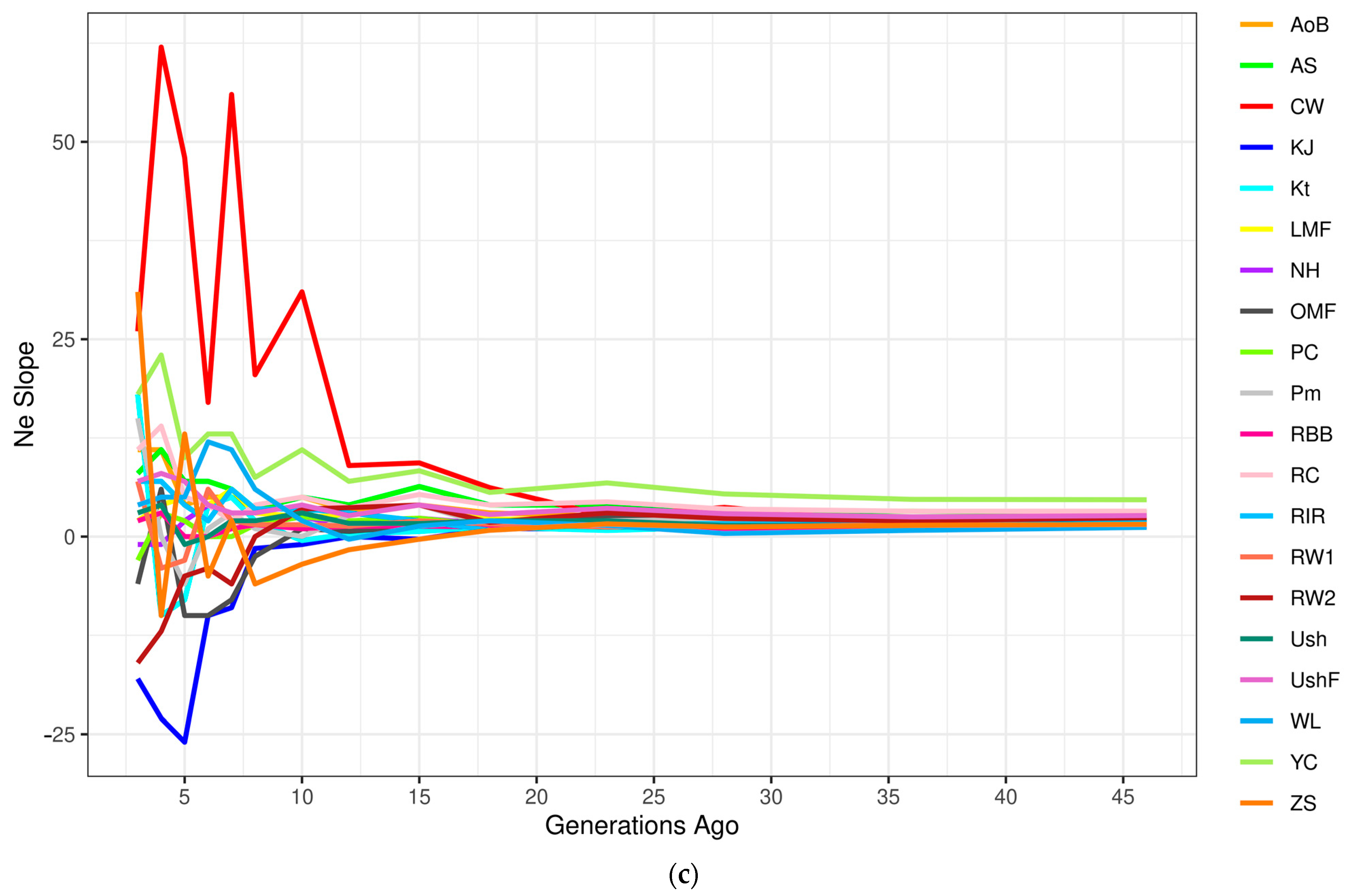
3.4.2. Effective Population Size
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Schmidt, C.J.; Kim, D.K.; Pendarvis, G.K.; Abasht, B.; McCarthy, F.M. Proteomic insight into human directed evolution of the domesticated chicken Gallus gallus. bioRxiv 2023. preprint. [Google Scholar] [CrossRef]
- Puetz, L.C.; Delmont, T.O.; Aizpurua, O.; Guo, C.; Zhang, G.; Katajamaa, R.; Jensen, P.; Gilbert, M.T.P. Gut microbiota linked with reduced fear of humans in Red Junglefowl has implications for early domestication. Adv. Genet. 2021, 2, 2100018. [Google Scholar] [CrossRef] [PubMed]
- Moiseyeva, I.G.; Nikiforov, A.A.; Romanov, M.N.; Balanovsky, O.P. Dynamics of domestic animal diversity as influenced by anthropogenic factor. In Integration of Archaeological and Ethnographic Studies: Collection of Scientific Papers; Korusenko, M.A., Tikhonov, S.S., Tomilov, N.A., Eds.; Nauka-Omsk: Omsk, Russia, 2003; pp. 284–287. [Google Scholar]
- Deng, J.; Xie, X.L.; Wang, D.F.; Zhao, C.; Lv, F.H.; Li, X.; Yang, J.; Yu, J.L.; Shen, M.; Gao, L.; et al. Paternal origins and migratory episodes of domestic sheep. Curr. Biol. 2020, 30, 4085–4095. [Google Scholar] [CrossRef] [PubMed]
- Feuerborn, T.R.; Carmagnini, A.; Losey, R.J.; Nomokonova, T.; Askeyev, A.; Askeyev, I.; Askeyev, O.; Antipina, E.E.; Appelt, M.; Bachura, O.P.; et al. Modern Siberian dog ancestry was shaped by several thousand years of Eurasian-wide trade and human dispersal. Proc. Natl. Acad. Sci. USA 2021, 118, e2100338118. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, C.; Peng, Y.; Cai, X.; Hu, X.; Bosse, M.; Zhao, Y. Whole-genome analysis reveals the hybrid formation of Chinese indigenous DHB pig following human migration. Evol. Appl. 2022, 15, 501–514. [Google Scholar] [CrossRef]
- Manunza, A.; Ramirez Diaz, J.; Bobbo, T.; Cozzi, P.; Lazzari, B.; Biscarini, F.; Biffani, S.; Johansson, A.M.; Grøva, L.; Stella, A. Following the Hidden Footsteps of Past Human Migrations Via the Goat Genome. In Proceedings of 12th World Congress on Genetics Applied to Livestock Production (WCGALP): Technical and Species Orientated Innovations in Animal Breeding, and Contribution of Genetics to Solving Societal Challenges, Part 14: Genetic Diversity and Inbreeding; Veerkamp, R.F., de Haas, Y., Eds.; Wageningen Academic Publishers: Wageningen, The Netherlands, 2022; pp. 906–909. [Google Scholar] [CrossRef]
- Carson, A.; Elliott, M.; Groom, J.; Winter, A.; Bowles, D. Geographical isolation of native sheep breeds in the UK—Evidence of endemism as a risk factor to genetic resources. Livest. Sci. 2009, 123, 288–299. [Google Scholar] [CrossRef]
- Meseret, S.; Mekonnen, Y.A.; Brenig, B.; Schütz, E.; Hanotte, O.; Gültas, M.; Schmitt, A.O. Genetic diversity and population structure of six Ethiopian cattle breeds from different geographical regions using high density single nucleotide polymorphisms. Livest. Sci. 2020, 234, 103979. [Google Scholar] [CrossRef]
- Campos, E.; Cuéllar, J.; Salvador, O.; García-Trejo, E.A.; Pereira, F. The genetic diversity and phylogeography of Mexican domestic sheep. Small Rumin. Res. 2020, 187, 106109. [Google Scholar] [CrossRef]
- Hata, A.; Takenouchi, A.; Kinoshita, K.; Hirokawa, M.; Igawa, T.; Nunome, M.; Suzuki, T.; Tsudzuki, M. Geographic origin and genetic characteristics of Japanese indigenous chickens inferred from mitochondrial D-loop region and microsatellite DNA markers. Animals 2020, 10, 2074. [Google Scholar] [CrossRef]
- Al-Jumaili, A.S.; Hanotte, O. The usefulness of maternally inherited genetic markers for phylogeographic studies in village chicken. Anim. Biotechnol. 2022, 1–19. [Google Scholar] [CrossRef]
- VarGoats Consortium; Nijman, I.J.; Rosen, B.D.; Bardou, P.; Faraut, T.; Cumer, T.; Daly, K.G.; Zheng, Z.; Cai, Y.; Asadollahpour, H.; et al. Geographical contrasts of Y-chromosomal haplogroups from wild and domestic goats reveal ancient migrations and recent introgressions. Mol. Ecol. 2022, 31, 4364–4380. [Google Scholar] [CrossRef] [PubMed]
- Ahbara, A.M.; Musa, H.H.; Robert, C.; Abebe, A.; Al-Jumaili, A.S.; Kebede, A.; Latairish, S.; Agoub, M.O.; Clark, E.; Hanotte, O.; et al. Natural adaptation and human selection of northeast African sheep genomes. Genomics 2022, 114, 110448. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Bosse, M.; Rochus, C.M.; Groenen, M.A.M.; Crooijmans, R.P.M.A. Genomic insight into the influence of selection, crossbreeding, and geography on population structure in poultry. Genet. Sel. Evol. 2023, 55, 5. [Google Scholar] [CrossRef]
- Tixier-Boichard, M.; Coquerelle, G.; Vilela-Lamego, C.; Weigend, S.; Barre-Dirrie, A.; Groenen, M.; Crooijmans, R.; Vignal, A.; Hillel, J.; Freidlin, P.; et al. Contribution of Data on History, Management and Phenotype to the Description of the Diversity between Chicken Populations Sampled within the AVIANDIV Project. In Proceedings of the Poultry Genetics Symposium, Mariensee, Germany, 6–8 October 1999; Preisinger, R., Ed.; Working Group 3 of WPSA, Lohmann Tierzucht: Cuxhaven, Germany, 1999; pp. 15–21. [Google Scholar]
- Guo, Y.; Ou, J.H.; Zan, Y.; Wang, Y.; Li, H.; Zhu, C.; Chen, K.; Zhou, X.; Hu, X.; Carlborg, Ö. Researching on the fine structure and admixture of the worldwide chicken population reveal connections between populations and important events in breeding history. Evol. Appl. 2022, 15, 553–564. [Google Scholar] [CrossRef]
- Crooijmans, R.P.M.A.; Groen, A.F.; Van Kampen, A.J.A.; Van Der Beek, S.; Van Der Poel, J.J.; Groenen, M.A.M. Microsatellite polymorphism in commercial broiler and layer lines estimated using pooled blood samples. Poult. Sci. 1996, 75, 904–909. [Google Scholar] [CrossRef] [PubMed]
- Semyenova, S.K.; Moiseyeva, I.G.; Vasil’ev, V.A.; Filenko, A.L.; Nikiforov, A.A.; Sevast’yanova, A.A.; Ryskov, A.P. Genetic Polymorphisms by DNA and Biochemical Markers in Old Russian Chicken Breeds. In Proceedings of the Biodiversity and Dynamics of Ecosystems in North Eurasia: First International Conference, Novosibirsk, Russia, 21–26 August 2000; Russian Academy of Sciences, Siberian Branch: Novosibirsk, Russia, 2000; Volume 1, Part 3. pp. 101–102. [Google Scholar]
- Semyenova, S.K.; Moiseev, I.G.; Vasil’ev, V.A.; Filenko, A.L.; Nikiforov, A.A.; Sevast’yanova, A.A.; Ryskov, A.P. Genetic polymorphism of Russian, European, and Asian chicken breeds as revealed with DNA and protein markers. Russ. J. Genet. 2002, 38, 1109–1112. [Google Scholar] [CrossRef]
- Weigend, S.; Romanov, M.N. Current strategies for the assessment and evaluation of genetic diversity in chicken resources. Worlds Poult. Sci. J. 2001, 57, 275–288. [Google Scholar] [CrossRef]
- Romanov, M.N. Centres of Domestication, Breed Formation and Dispersion of Geese and Ducks. In Abstracts of the 1st Ukrainian Poultry Conference, Borky, Simferopol, Ukraine, 1993; World’s Poultry Science Association, Ukrainian Branch, Poultry Research Institute, UAAS: Borky, Ukraine, 1993; pp. 11–14, 122–123. [Google Scholar]
- Romanov, M.N. Evolution of domestic geese. Centres of domestication and dispersion. In New Investigations on Palearctic Geese; Gudina, A.N., Ed.; Zaporizhya Branch of the Ukrainian Ornithological Society, Zaporizhya State University: Zaporizhya, Ukraine, 1995; pp. 120–126. [Google Scholar]
- Katerinich, O.A.; Zakharchenko, O.P.; Bondarenko, Y.V. Alternative poultry—Chickens of the meat and egg direction of productivity. Ptitsa i Ptitseprodukty 2014, 3, 66–67. [Google Scholar]
- Tereshchenko, O.V.; Pankova, S.M.; Katerynych, O.O. Directions of development of poultry industry. Vìsnik Agrar. Nauk 2015, 93, 27–30. [Google Scholar]
- Bondarenko, Y.V.; Khvostik, V.P. Improving the productivity of meat and egg chickens of domestic selection. Visnyk Sumsʹkoho Natsionalʹnoho Ahrarnoho Universytetu Seriya Tvarynnytstvo 2020, 2, 29–32. [Google Scholar] [CrossRef]
- Tereshchenko, O.V.; Katerinich, O.O.; Pankova, S.M.; Borodai, V.P. Formation of genetic resources of domestic breeds of poultry in the context of food security of the state. Sučasne Ptahìvnictvo 2015, 7–8, 19–21. [Google Scholar]
- Kutnyuk, P.I.; Bondarenko, Y.V.; Ivanova, T.V. Karyometric method of determining the genetic potential of laying hens. Ptakhivnytstvo 2001, 50, 75–81. [Google Scholar]
- Tagirov, M.T.; Tereshchenko, L.V.; Tereshchenko, A.V. Substantiation of the possibility of using primary germ cells as material for the preservation of poultry genetic resources. Ptakhivnytstvo 2006, 58, 464–473. [Google Scholar]
- Linnik, T.P.; Dyubko, T.S.; Martynyuk, I.N.; Tereschenko, A.V. Interactions of cryoprotectants with liposomes from total lipids of the fowl spermatozoa. Probl. Cryobiol. Cryomedicine 2010, 20, 34–46. [Google Scholar]
- Sakhatsky, N.I.; Tereshchenko, A.V.; Artemenko, A.B. An express-method of estimation of fertilizing ability of freezing-thawing of poultry spermatozoa. Sel’skokhozyaistvennaya Biol. 1987, 22, 7780. [Google Scholar]
- Genofond. Catalogue of Breeds: Chickens. Official Site of the Company Genofond. Sergiev Posad, Russia. 2015. Available online: http://www.genofond-sp.ru/hen.html (accessed on 27 May 2023).
- Larkina, T.A.; Barkova, O.Y.; Peglivanyan, G.K.; Mitrofanova, O.V.; Dementieva, N.V.; Stanishevskaya, O.I.; Vakhrameev, A.B.; Makarova, A.V.; Shcherbakov, Y.S.; Pozovnikova, M.V.; et al. Evolutionary subdivision of domestic chickens: Implications for local breeds as assessed by phenotype and genotype in comparison to commercial and fancy breeds. Agriculture 2021, 11, 914. [Google Scholar] [CrossRef]
- Moiseyeva, I.G.; Nikiforov, A.A.; Romanov, M.N.; Aleksandrov, A.V.; Moysyak, Y.V.; Semyenova, S.K. Origin, history, genetics and economic traits of the Yurlov Crower chicken breed. In Elektronnyi Zhurnal (Electronic Journal); Laboratory of Animal Comparative Genetics, N.I. Vavilov Institute of General Genetics: Moscow, Russia, 2007; Available online: https://web.archive.org/web/20120210170800/http://www.lab-cga.ru/articles/Yurlovskaya/Yurlovskaya.htm (accessed on 10 February 2012).
- Moiseyeva, I.G.; Romanov, M.N.; Aleksandrov, A.V.; Nikiforov, A.A.; Sevastyanova, A.A. Evolution and genetic diversity of old domestic hen’s breed—Yurlovskaya Golosistaya: System analysis of variability forms. Izv. Timiryazev. S-Kh. Akad. 2009, 3, 132–147. [Google Scholar]
- Moiseyeva, I.G.; Sevastyanova, A.A.; Aleksandrov, A.V.; Romanov, M.N. Singing breeds of hens. Priroda 2011, 4, 10–18. [Google Scholar]
- Romanov, M.N.; Abdelmanova, A.S.; Fisinin, V.I.; Gladyr, E.A.; Volkova, N.A.; Koshkina, O.A.; Rodionov, A.N.; Vetokh, A.N.; Gusev, I.V.; Anshakov, D.V.; et al. Selective footprints and genes relevant to cold adaptation and other phenotypic traits are unscrambled in the genomes of divergently selected chicken breeds. J. Anim. Sci. Biotechnol. 2023, 14, 35. [Google Scholar] [CrossRef]
- Romanov, M.N.; Bondarenko, Y.V. Introducing the Ukrainian indigenous poultry—The Poltava chickens. Fancy Fowl 1994, 14, 8–9. [Google Scholar]
- Moiseyeva, I.G.; Kovalenko, A.T.; Mosyakina, T.V.; Romanov, M.N.; Bondarenko, Y.V.; Kutnyuk, P.I.; Podstreshny, A.P.; Nikiforov, A.A.; Tkachik, T.E. Origin, history, genetics and economic traits of the Poltava chicken breed. In Elektronnyi zhurnal (Electronic Journal); Laboratory of Animal Comparative Genetics, N.I. Vavilov Institute of General Genetics: Moscow, Russia, 2006; Available online: https://web.archive.org/web/20120205195904/http://www.lab-cga.ru/articles/Jornal04/Statia2.htm (accessed on 5 February 2012).
- Moiseyeva, I.G.; Romanov, M.N.; Kovalenko, A.T.; Mosyakina, T.V.; Bondarenko, Y.V.; Kutnyuk, P.I.; Podstreshny, A.P.; Nikiforov, A.A. The Poltava chicken breed of Ukraine: Its history, characterization and conservation. Anim. Genet. Resour. Inf. 2007, 40, 71–78. [Google Scholar] [CrossRef]
- Kulibaba, R.A.; Tereshchenko, A.V. Transforming growth factor β1, pituitary-specific transcriptional factor 1 and insulin-like growth factor I gene polymorphisms in the population of the Poltava clay chicken breed: Association with productive traits. Agric. Sci. Pract. 2015, 2, 67–72. [Google Scholar] [CrossRef]
- Abdelmanova, A.S.; Dotsev, A.V.; Romanov, M.N.; Stanishevskaya, O.I.; Gladyr, E.A.; Rodionov, A.N.; Vetokh, A.N.; Volkova, N.A.; Fedorova, E.S.; Gusev, I.V.; et al. Unveiling comparative genomic trajectories of selection and key candidate genes in egg-type Russian White and meat-type White Cornish chickens. Biology 2021, 10, 876. [Google Scholar] [CrossRef]
- Moiseyeva, I.G.; Sevastyanova, A.A.; Aleksandrov, A.V.; Vakhrameev, A.B.; Romanov, M.N.; Dmitriev, Y.I.; Semenova, S.K.; Sulimova, G.E. Orloff chicken breed: History, current status and studies. Izv. Timiryazev. S-Kh. Akad. 2016, 1, 78–96. [Google Scholar]
- Dementeva, N.V.; Romanov, M.N.; Kudinov, A.A.; Mitrofanova, O.V.; Stanishevskaya, O.I.; Terletsky, V.P.; Fedorova, E.S.; Nikitkina, E.V.; Plemyashov, K.V. Studying the structure of a gene pool population of the Russian White chicken breed by genome-wide SNP scan. Sel’skokhozyaistvennaya Biol. 2017, 52, 1166–1174. [Google Scholar] [CrossRef]
- Dementieva, N.V.; Fedorova, E.S.; Krutikova, A.A.; Mitrofanova, O.V.; Stanishevskaya, O.I.; Pleshanov, N.V.; Smaragdov, M.G.; Kudinov, A.A.; Terletsky, V.P.; Romanov, M.N. Genetic variability of indels in the prolactin and dopamine receptor D2 genes and their association with the yield of allanto-amniotic fluid in Russian White laying hens. Tarım Bilim. Derg.—J. Agric. Sci. 2020, 26, 373–379. [Google Scholar] [CrossRef]
- Kudinov, A.A.; Dementieva, N.V.; Mitrofanova, O.V.; Stanishevskaya, O.I.; Fedorova, E.S.; Larkina, T.A.; Mishina, A.I.; Plemyashov, K.V.; Griffin, D.K.; Romanov, M.N. Genome-wide association studies targeting the yield of extraembryonic fluid and production traits in Russian White chickens. BMC Genomics 2019, 20, 270. [Google Scholar] [CrossRef]
- Romanov, M.N. Analysis of Down and Plumage Colour and Pattern in Rhode Island Red-like Breeder Hens. In Proceedings of the Abstracts of the 2nd Baltic Poultry Conference, Vilnius, Lithuania, 15–16 September 1994; World’s Poultry Science Association, Lithuanian Branch: Vilnius, Lithuania, 1994; pp. 81–82. [Google Scholar]
- Bondarenko, Y.V. The Genetic Structure of White Leghorn Hen Plumage Colouring. In Abstracts of the 5th Congress of Geneticists and Breeders of Ukraine, Part 5: Genetic Basis of Animal Breeding, Kiev, Ukraine, 1986; Naukova Dumka: Kiev, Ukraine, 1986; p. 16. [Google Scholar]
- Eusemann, B.K.; Patt, A.; Schrader, L.; Weigend, S.; Thöne-Reineke, C.; Petow, S. The role of egg production in the etiology of keel bone damage in laying hens. Front. Vet. Sci. 2020, 7, 81. [Google Scholar] [CrossRef]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.R.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.W.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023; Available online: https://www.r-project.org/ (accessed on 27 May 2023).
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar] [CrossRef]
- Kalinowski, S.T. Counting alleles with rarefaction: Private alleles and hierarchical sampling designs. Conserv. Genet. 2004, 5, 539–543. [Google Scholar] [CrossRef]
- Keenan, K.; McGinnity, P.; Cross, T.F.; Crozier, W.W.; Prodohl, P.A. diveRsity: An R package for the estimation and exploration of population genetics parameters and their associated errors. Methods Ecol. Evol. 2013, 4, 782–788. [Google Scholar] [CrossRef]
- Kamvar, Z.N.; Brooks, J.C.; Grünwald, N.J. Novel R tools for analysis of genome-wide population genetic data with emphasis on clonality. Front. Genet. 2015, 6, 208. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009. [Google Scholar] [CrossRef]
- Zenkova, D.; Kamenev, V.; Sablina, R.; Artyomov, M.; Sergushichev, A. Phantasus: Visual and Interactive Gene Expression Analysis; Roswell Park Comprehensive Cancer Center: Buffalo, NY, USA, 2022. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Boc, A.; Diallo Alpha, B.; Makarenkov, V. T-REX: A web server for inferring, validating and visualizing phylogenetic trees and networks. Nucleic Acids Res. 2012, 40, W573–W579. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Reynolds, J.; Weir, B.S.; Cockerham, C.C. Estimation of the coancestry coefficient: Basis for a short-term genetic distance. Genetics 1983, 105, 767–779. [Google Scholar] [CrossRef]
- Sattath, S.; Tversky, A. Additive similarity trees. Psychometrika 1977, 42, 319–345. [Google Scholar] [CrossRef]
- Gascuel, O. Concerning the NJ algorithm and its unweighted version UNJ. In Mathematical Hierarchies and Biology; Mirkin, B., McMorris, F.R., Roberts, F., Rzhetsky, A., Eds.; DIMACS Series in Discrete Mathematics and Theoretical Computer Science; American Mathematical Society: Providence, RI, USA, 1997; Volume 37, pp. 149–171. [Google Scholar] [CrossRef]
- Metsalu, T.; Vilo, J. ClustVis: A web tool for visualizing clustering of multivariate data using Principal Component Analysis and heatmap. Nucleic Acids Res. 2015, 43, W566–W570. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef] [PubMed]
- Milanesi, M.; Capomaccio, S.; Vajana, E.; Bomba, L.; Garcia, J.F.; Ajmone-Marsan, P.; Colli, L. BITE: An R package for biodiversity analyses. bioRxiv 2017, 181610. [Google Scholar] [CrossRef]
- Marras, G.; Gaspa, G.; Sorbolini, S.; Dimauro, C.; Ajmone-Marsam, P.; Valentini, A.; Williams, J.L.; Macciotta, N.P.P. Analysis of runs of homozygosity and their relationship with inbreeding in five cattle breeds farmed in Italy. Anim. Genet. 2014, 46, 110–121. [Google Scholar] [CrossRef] [PubMed]
- Biscarini, F.; Paolo Cozzi, P.; Gaspa, G.; Marras, G. detectRUNS: Detect Runs of Homozygosity and Runs of Heterozygosity in Diploid Genomes. R Package Version 0.9.6. Available online: https://cran.r-project.org/web/packages/detectRUNS/index.html (accessed on 27 May 2023).
- Ferenčaković, M.; Sölkner, J.; Curik, I. Estimating autozygosity from high-throughput information: Effects of SNP density and genotyping errors. Genet. Sel. Evol. 2013, 45, 42. [Google Scholar] [CrossRef]
- Wall, J.D.; Pritchard, J.K. Haplotype blocks and linkage disequilibrium in the human genome. Nat. Rev. Genet. 2003, 4, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Lencz, T.; Lambert, C.; DeRosse, P.; Burdick, K.E.; Morgan, T.V.; Kane, J.M.; Kucherlapati, R.; Malhotra, A.K. Runs of homozygosity reveal highly penetrant recessive loci in schizophrenia. Proc. Natl. Acad. Sci. USA 2007, 104, 19942–19947. [Google Scholar] [CrossRef] [PubMed]
- Purfield, D.C.; Berry, D.P.; McParland, S.; Bradley, D.G. Runs of homozygosity and population history in cattle. BMC Genet. 2012, 13, 70. [Google Scholar] [CrossRef]
- Peripolli, E.; Stafuzza, N.B.; Munari, D.P.; Lima, A.L.F.; Irgang, R.; Machado, M.A.; Panetto, J.C.D.C.; Ventura, R.V.; Baldi, F.; da Silva, M.V.G.B. Assessment of runs of homozygosity islands and estimates of genomic inbreeding in Gyr (Bos indicus) dairy cattle. BMC Genom. 2018, 19, 34. [Google Scholar] [CrossRef]
- Grilz-Seger, G.; Neuditschko, M.; Ricard, A.; Velie, B.; Lindgren, G.; Mesarič, M.; Cotman, M.; Horna, M.; Dobretsberger, M.; Brem, G.; et al. Genome-wide homozygosity patterns and evidence for selection in a set of European and Near Eastern horse breeds. Genes 2019, 10, 491. [Google Scholar] [CrossRef]
- Talebi, R.; Szmatoła, T.; Mészáros, G.; Qanbari, S. Runs of homozygosity in modern chicken revealed by sequence data. G3 2020, 10, 4615–4623. [Google Scholar] [CrossRef]
- Pickrell, J.K.; Pritchard, J.K. Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 2012, 8, e1002967. [Google Scholar] [CrossRef]
- Fitak, R.R. OptM: Estimating the optimal number of migration edges on population trees using Treemix. Biol. Methods Protocol. 2021, 6, bpab017. [Google Scholar] [CrossRef]
- Barbato, M.; Orozco-terWengel, P.; Tapio, M.; Bruford, M.W. SNeP: A tool to estimate trends in recent effective population size trajectories using genome-wide SNP data. Front Genet. 2015, 6, 109. [Google Scholar] [CrossRef] [PubMed]
- Sved, J.A. Linkage disequilibrium and homozygosity of chromosome segments in finite populations. Theor. Popul. Biol. 1971, 2, 125–141. [Google Scholar] [CrossRef]
- Hill, W. Estimation of effective population size from data on linkage disequilibrium. Genet. Res. 1981, 38, 209–216. [Google Scholar] [CrossRef]
- Waples, R.S.; Do, C. Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: A largely untapped resource for applied conservation and evolution. Evol. Appl. 2010, 3, 244–262. [Google Scholar] [CrossRef]
- Sved, J.A.; Feldman, M.W. Correlation and probability methods for one and two loci. Theor. Popul. Biol. 1973, 4, 129–132. [Google Scholar] [CrossRef] [PubMed]
- Pitt, D.; Bruford, M.W.; Barbato, M.; Orozco-terWengel, W.; Martínez, R.; Sevane, N. Demography and rapid local adaptation shape Creole cattle genome diversity in the tropics. Evol. Appl. 2019, 12, 105–122. [Google Scholar] [CrossRef]
- Cervantes, I.; Goyache, F.; Molina, A.; Valera, M.; Gutiérrez, J.P. Estimation of effective population size from the rate of coancestry in pedigreed populations. J. Anim. Breed. Genet. 2011, 128, 56–63. [Google Scholar] [CrossRef]
- Miniwebtool.com. Online Tools and Calculators. Available online: https://miniwebtool.com/ (accessed on 27 May 2023).
- GraphPad Software. Dotmatics. Available online: https://www.graphpad.com/ (accessed on 27 May 2023).
- Liu, W.-Y.; Zhao, C.-J. Comprehensive genetic analysis with mitochondrial DNA data reveals the population evolution relationship between Chinese gamecocks and their neighboring native chicken breeds. Asian J. Anim. Vet. Adv. 2010, 5, 388–401. [Google Scholar] [CrossRef]
- Moiseyeva, I.G.; Romanov, M.N.; Nikiforov, A.A.; Sevastyanova, A.A.; Semyenova, S.K. Evolutionary relationships of Red Jungle Fowl and chicken breeds. Genet. Sel. Evol. 2003, 35, 403–423. [Google Scholar] [CrossRef] [PubMed]
- Dementieva, N.V.; Mitrofanova, O.V.; Dysin, A.P.; Kudinov, A.A.; Stanishevskaya, O.I.; Larkina, T.A.; Plemyashov, K.V.; Griffin, D.K.; Romanov, M.N.; Smaragdov, M.G. Assessing the effects of rare alleles and linkage disequilibrium on estimates of genetic diversity in the chicken populations. Animal 2021, 15, 100171. [Google Scholar] [CrossRef] [PubMed]
- Sokolova, A.N. Genetic and Selection Methods of Creation of a Chicken Population with an Increased Resistance to Neoplasms: Author’s Abstract. Ph.D. Thesis, RRIFAGB, Pushkin, Russia, 1999. [Google Scholar]
- Crawford, R.D. (Ed.) Poultry Breeding and Genetics; Elsevier: Amsterdam, The Netherlands; New York, NY, USA, 1990. [Google Scholar]
- Romanov, M.N.; Weigend, S. Genetic Diversity in Chicken Populations Based on Microsatellite Markers. In Proceedings of the Conference “From Jay Lush to Genomics: Visions for Animal Breeding and Genetics”, Ames, IA, USA, 16–18 May 1999; Dekkers, J.C.M., Lamont, S.J., Rothschild, M.F., Eds.; Iowa State University, Department of Animal Science: Ames, IA, USA, 1999; p. 174. [Google Scholar]
- Romanov, M.N.; Weigend, S. Using RAPD markers for assessment of genetic diversity in chickens. Arch. Geflugelkd. 2001, 65, 145–148. [Google Scholar]
- Romanov, M.N.; Weigend, S.; Bondarenko, Y.V.; Podstreshny, A.P.; Kutnyuk, P.I.; Sakhatsky, N.I. Studies on Poultry Germplasm Diversity and Conservation in Ukraine. In Proceedings of the Poultry Genetics Symposium, Mariensee, Germany, 6–8 October 1999; Working Group 3 of WPSA: Mariensee, Germany, 1999; p. 140. [Google Scholar]
- Weigend, S.; Romanov, M.N.; Rath, D. Methodologies to Identify, Evaluate and Conserve Poultry Genetic Resources. In Proceedings of the XXII World’s Poultry Congress & Exhibition, Participant List & Full Text CD + Book of Abstracts, Istanbul, Turkey, 8–13 June 2004; WPSA—Turkish Branch: Istanbul, Turkey, 2004; p. 84. [Google Scholar]
- Dementieva, N.V.; Kudinov, A.A.; Larkina, T.A.; Mitrofanova, O.V.; Dysin, A.P.; Terletsky, V.P.; Tyshchenko, V.I.; Griffin, D.K.; Romanov, M.N. Genetic variability in local and imported germplasm chicken populations as revealed by analyzing runs of homozygosity. Animals 2020, 10, 1887. [Google Scholar] [CrossRef] [PubMed]
- Dementieva, N.V.; Shcherbakov, Y.S.; Tyshchenko, V.I.; Terletsky, V.P.; Vakhrameev, A.B.; Nikolaeva, O.A.; Ryabova, A.E.; Azovtseva, A.I.; Mitrofanova, O.V.; Peglivanyan, G.K.; et al. Comparative analysis of molecular RFLP and SNP markers in assessing and understanding the genetic diversity of various chicken breeds. Genes 2022, 13, 1876. [Google Scholar] [CrossRef]
- Buggiotti, L.; Yurchenko, A.A.; Yudin, N.S.; Vander Jagt, C.J.; Vorobieva, N.V.; Kusliy, M.A.; Vasiliev, S.K.; Rodionov, A.N.; Boronetskaya, O.I.; Zinovieva, N.A.; et al. Demographic history, adaptation, and NRAP convergent evolution at amino acid residue 100 in the world northernmost cattle from Siberia. Mol. Biol. Evol. 2021, 38, 3093–3110. [Google Scholar] [CrossRef]
- Abdelmanova, A.S.; Kharzinova, V.R.; Volkova, V.V.; Mishina, A.I.; Dotsev, A.V.; Sermyagin, A.A.; Boronetskaya, O.I.; Petrikeeva, L.V.; Chinarov, R.Y.; Brem, G.; et al. Genetic diversity of the historical and modern populations of Russian cattle breeds revealed by microsatellite analysis. Genes 2020, 11, 940. [Google Scholar] [CrossRef]
- Abdelmanova, A.; Mishina, A.I.; Volkova, V.; Dotsev, A.V.; Sermyagin, A.A.; Boronetskaya, O.; Petrikeeva, L.; Chinarov, R.; Brem, G.; Zinovieva, N.A. PSXII-27 Microsatellite analysis of the historical and modern populations of the Russian local cattle breeds. J. Anim. Sci. 2020, 98 (Suppl. S4), 240–241. [Google Scholar] [CrossRef]
- Abdelmanova, A.S.; Kharzinova, V.R.; Volkova, V.; Dotsev, A.V.; Sermyagin, A.A.; Boronetskaya, O.; Chinarov, R.; Brem, G.; Zinovieva, N.A. PSXII-1 Tracing the historical genetic components in turano-mongolian cattle breeds based on the microsatellite analysis of modern and museum samples. J. Anim. Sci. 2021, 99 (Suppl. S3), 253–254. [Google Scholar] [CrossRef]
- Abdelmanova, A.S.; Kharzinova, V.R.; Dotsev, A.V.; Sermyagin, A.A.; Boronetskaya, O.; Chinarov, R.; Brem, G.; Zinovieva, N.A. PSXV-10 Study of allelic pattern of PLAG1 gene in the historical and modern populations of two oldest Russian dairy cattle breeds. J. Anim. Sci. 2021, 99 (Suppl. S3), 259–260. [Google Scholar] [CrossRef]
- Markov, N.; Economov, A.; Hjeljord, O.; Rolandsen, C.M.; Bergqvist, G.; Danilov, P.; Dolinin, V.; Kambalin, V.; Kondratov, A.; Krasnoshapka, N.; et al. The wild boar Sus scrofa in northern Eurasia: A review of range expansion history, current distribution, factors affecting the northern distributional limit, and management strategies. Mamm. Rev. 2022, 52, 519–537. [Google Scholar] [CrossRef]
- Weigend, S.; Romanov, M.N. Current Strategies for the Assessment and Evaluation of Genetic Diversity in Chicken Resources. In Proceedings of the Abstracts and Proceedings of the XXI World’s Poultry Congress, Montreal, QC, Canada, 20–24 August 2000; CD-ROM. World’s Poultry Science Association: Montreal, QC, Canada, 2000. [Google Scholar]
- Vakhrameev, A.B.; Narushin, V.G.; Larkina, T.A.; Barkova, O.Y.; Peglivanyan, G.K.; Dysin, A.P.; Dementieva, N.V.; Makarova, A.V.; Shcherbakov, Y.S.; Pozovnikova, M.V.; et al. Selection-driven chicken phenome and phenomenon of pectoral angle variation across different chicken phenotypes. Livest. Sci. 2022, 264, 105067. [Google Scholar] [CrossRef]
- Vakhrameev, A.B.; Narushin, V.G.; Larkina, T.A.; Barkova, O.Y.; Peglivanyan, G.K.; Dysin, A.P.; Dementieva, N.V.; Makarova, A.V.; Shcherbakov, Y.S.; Pozovnikova, M.V.; et al. Disentangling clustering configuration intricacies for divergently selected chicken breeds. Sci. Rep. 2023, 13, 3319. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Breed (Population) | Code | n | Breed Type | Origin | Refs | Image Source | |
|---|---|---|---|---|---|---|---|
| Old Russian indigenous breeds | |||||||
 | Orloff Mille Fleur | OMF | 29 | DPB/MEB, fancy and semi-game breed (GB), cold and heat tolerant, chicks are late feathering and cannot withstand cold and humidity, incubation instinct | Central Russia, late 18th century, from local chickens, Gilian and Old English Game | [32,33,37,43] | a |
 | Poltava Clay | PC | 21 | DPB/MEB | Poltava Governorate, Russia; mid 19th century, from local chickens, Buff Orpington and possibly RIR and NH, Wyandotte, etc., bred at the Ukrainian Poultry Research Institute, USSR since 1951 | [32,33,38,39,40,41] | a |
 | Russian Black Bearded (Galan) | RBB | 25 | DPB/MEB and fancy, cold tolerant, incubation instinct | Kursk, Oryol and neighboring governorates, Russia, 19th century (2nd half), from (1) Wyandotte and Crevecoeur or (2) Orloff Black and Wyandotte | [32] | b |
 | Russian Crested | RC | 29 | DPB/MEB and fancy, cold tolerant, incubation instinct | Russia, 19th century, from local chickens and possibly Asian breeds | [32] | a |
 | Ushanka (or Ukrainian Muffed) | Ush | 30 | DPB/MEB and fancy, sex-linked early feathering in chicks, cold tolerant, incubation instinct | South Russia and territory of Ukraine, 17th century to 1880s, from local chickens, probable random mating to other breeds | [32,33,37] | a |
 | Yurlov Crower | YC | 36 | DPB/MEB, long crowing, of two varieties (silver and golden), sex-linked late feathering in chicks | Russia, 19th century (2nd half), from local and GB chickens, Brahma, Cochin and Langshan, almost extinct in 1941–1945, brought to ARPRTI, Zagorsk, USSR in 1948 | [32,33,34,35,36] | a |
| Improved Russian breeds | |||||||
 | Adler Silver | AS | 30 | DPB/MEB | Adler Poultry Farm, Krasnodar Krai, USSR, 1951–1965, from Pm, RW, NH, White Plymouth Rock and YC | [32] | a |
 | Kotlyarevsky | Kt | 28 | DPB/MEB | Kotlyarevsky Breeding Farm, Kabardino-Balkarian ASSR, USSR, 20th century (2nd half), from NH, RW, ZS, Naked Neck and Barred Plymouth Rock | [32] | b |
 | Kuchino Jubilee | KJ | 29 | DPB/MEB, sex-linked early feathering in chicks | Kuchinsky State Breeding Farm, Moscow Oblast, USSR, 1947–1990, from RW, NH, RIR, AoB, Plymouth Rock White, YC, Brown Leghorn (Italian Partridge) and Livny | [32] | a |
 | Leningrad Mille Fleur | LMF | 7 | DPB/EMB | RRIFAGB, Pushkin, Leningrad Oblast, USSR, 1985, from Australorp Black Speckled, NH and PC | [33] | a |
 | Pervomai | Pm | 30 | DPB/MEB | Pervoe Maya State Farm, Kharkov Oblast, Ukrainian SSR, USSR, 1935–1941; Pachelma State Farm, Penza Oblast, RSFSR, USSR, 1942–1963; from White Wyandotte, RIR and YC | [32,33] | a |
 | Russian White (ARPRTI) | RW1 | 29 | ETB | USSR, 1929–1953, from local white chickens and WL, bred at ARPRTI, Zagorsk/Sergiev Posad, USSR/Russia | [32,37,42] | b |
 | Russian White (RRIFAGB) | RW2 | 71 | ETB, cold tolerant | Bred at RRIFAGB, Pushkin, USSR/Russia since 1952, a single telic mating of an RRIFAGB inbred line of RW, selected for cold tolerance, to WL | [32,33,37,42,44,45,46] | a |
 | Ushanka Foot-feathered | UshF | 6 | DPB/MEB and fancy, cold tolerant | ARPRTI, Sergiev Posad, Russia, 20th century (more recently), from Pavlov, Orloff and possibly Ush | [32] | b |
 | Zagorsk Salmon | ZS | 27 | DPB/MEB, sex-linked late feathering and down color in chicks | Zagorsk, USSR, 1950–1959, from RW, NH, RIR and YC | [32,33] | a |
| Specialized foreign breeds | |||||||
 | Australorp Black | AoB | 30 | DPB/MEB | Australia, 1890s to 1929, from Black Orpington, RIR, Black Minorca, WL, Langshan and Barred Plymouth Rock, bred in Russia since 1946 | [32,33] | a |
 | Cornish White | CW | 24 | MTB | England, 1886, from local GB chickens, Asil, White Malay, Indian Game and Cochin, used as paternal stock in commercial broiler crosses, bred in Russia | [33,37,42] | a |
 | New Hampshire | NH | 10 | DPB/MEB | USA, early 20th century to 1935, from RIR | [32,33] | a |
 | Rhode Island Red | RIR | 17 | DPB/EMB | States of Rhode Island and Massachusetts, USA, 1880s to 1904, from Cochin, Java, Malay, Shanghai and Brown Leghorn | [32,47] | a |
 | White Leghorn | WL | 20 | ETB, the only Mediterranean breed of economic importance today | Tuscany, Italy, 19th century, from light rural breeds, brought to USA in 1828, a white variety imported to USA in 1853, line G11 bred in Germany since 1965 | [48,49] | a |
| Breed | HO (M ± SE) | HE (M ± SE) | UHE (M ± SE) | AR (M ± SE) | FIS [CI 95%] | UFIS [CI 95%] |
|---|---|---|---|---|---|---|
| Old Russian indigenous breeds | ||||||
| OMF | 0.291 ± 0.001 | 0.286 ± 0.001 | 0.291 ± 0.001 | 1.709 ± 0.002 | −0.019 [−0.021; −0.017] | −0.001 [−0.003; 0.001] |
| PC | 0.257 ± 0.001 | 0.249 ± 0.001 | 0.255 ± 0.001 | 1.655 ± 0.002 | −0.029 [−0.031; −0.027] | −0.003 [ −0.005; −0.001] |
| RBB | 0.250 ± 0.001 | 0.247 ± 0.001 | 0.252 ± 0.001 | 1.624 ± 0.002 | −0.008 [−0.010; −0.006] | 0.012 [0.010; 0.014] |
| RC | 0.325 ± 0.001 | 0.327 ± 0.001 | 0.333 ± 0.001 | 1.819 ± 0.002 | 0.005 [0.003; 0.007] | 0.022 [0.020; 0.024] |
| Ush | 0.249 ± 0.001 | 0.255 ± 0.001 | 0.259 ± 0.001 | 1.647 ± 0.002 | 0.016 [0.014; 0.018] | 0.033 [0.031; 0.035] |
| YC | 0.347 ± 0.001 | 0.358 ± 0.001 | 0.363 ± 0.001 | 1.867 ± 0.001 | 0.029 [0.027; 0.031] | 0.043 [0.041; 0.045] |
| Av. | 0.287 ± 0.017 | 0.287 ± 0.019 | 0.292 ± 0.019 | 1.720 ± 0.041 | −0.001 ± 0.009 [−0.031; 0.031] | 0.018 ± 0.008 [−0.005; 0.045] |
| Improved Russian breeds | ||||||
| AS | 0.321 ± 0.001 | 0.329 ± 0.001 | 0.335 ± 0.001 | 1.811 ± 0.002 | 0.020 [0.018; 0.022] | 0.037 [0.035; 0.039] |
| KJ | 0.256 ± 0.001 | 0.252 ± 0.001 | 0.256 ± 0.001 | 1.665 ± 0.002 | −0.018 [−0.020; −0.016] | 0.000 [−0.002; 0.002] |
| Kt | 0.257 ± 0.001 | 0.256 ± 0.001 | 0.261 ± 0.001 | 1.650 ± 0.002 | −0.006 [−0.008; −0.004] | 0.012 [0.010; 0.014] |
| LMF | 0.249 ± 0.001 | 0.228 ± 0.001 | 0.246 ± 0.001 | 1.606 ± 0.002 | −0.090 [−0.094; −0.086] | −0.011 [−0.015; −0.007] |
| Pm | 0.291 ± 0.001 | 0.290 ± 0.001 | 0.295 ± 0.001 | 1.729 ± 0.002 | −0.006 [−0.008; −0.004] | 0.011 [0.009; 0.013] |
| RW1 | 0.255 ± 0.001 | 0.248 ± 0.001 | 0.252 ± 0.001 | 1.632 ± 0.002 | −0.016 [−0.018; −0.014] | 0.001 [−0.001; 0.003] |
| RW2 | 0.306 ± 0.001 | 0.305 ± 0.001 | 0.307 ± 0.001 | 1.763 ± 0.002 | −0.001 [−0.002; 0.000] | 0.006 [0.005; 0.007] |
| UshF | 0.252 ± 0.001 | 0.232 ± 0.001 | 0.253 ± 0.001 | 1.645 ± 0.003 | −0.088 [−0.092; −0.084] | 0.003 [−0.001; 0.007] |
| ZS | 0.275 ± 0.001 | 0.271 ± 0.001 | 0.276 ± 0.001 | 1.688 ± 0.002 | −0.015 [−0.017; −0.013] | 0.004 [0.002; 0.006] |
| Av. | 0.274 ± 0.009 | 0.268 ± 0.011 | 0.276 ± 0.010 | 1.688 ± 0.022 | −0.024 ± 0.013 [−0.094; 0.022] | 0.007 ± 0.004 [−0.015; 0.039] |
| Specialized foreign breeds | ||||||
| AoB | 0.296 ± 0.001 | 0.314 ± 0.001 | 0.319 ± 0.001 | 1.777 ± 0.002 | 0.045 [0.043; 0.047] | 0.061 [0.059; 0.063] |
| CW | 0.365 ± 0.001 | 0.368 ± 0.001 | 0.371 ± 0.001 | 1.876 ± 0.001 | 0.001 [−0.001; 0.003] | 0.022 [0.020; 0.024] |
| NH | 0.233 ± 0.001 | 0.205 ± 0.001 | 0.216 ± 0.001 | 1.542 ± 0.002 | −0.122 [−0.125; −0.119] | −0.067 [−0.070; −0.064] |
| RIR | 0.256 ± 0.001 | 0.256 ± 0.001 | 0.264 ± 0.001 | 1.661 ± 0.002 | −0.003 [−0.006; 0.000] | 0.027 [0.024; 0.030] |
| WL | 0.164 ± 0.001 | 0.185 ± 0.001 | 0.185 ± 0.001 | 1.452 ± 0.002 | 0.080 [0.077; 0.083] | 0.103 [0.100; 0.106] |
| Av. | 0.263 ± 0.033 | 0.266 ± 0.033 | 0.271 ± 0.034 | 1.662 ± 0.077 | 0.000 ± 0.034 [−0.125; 0.083] | 0.029 ± 0.028 [−0.070; 0.106] |
| Source of Variation | df 1 | Sum of Squares | Variance Components | Percentage of Variation | p-Value of Fixation |
|---|---|---|---|---|---|
| Among groups | 2 | 450,114,870.5 | 0.014 | 1.36 | 0.011 |
| Among populations within group | 17 | 4,945,631,101 | 0.151 | 14.93 | 0.001 |
| Within populations | 508 | 27,729,967,336 | 0.163 | 83.71 | 0.001 |
| Total | 527 | 33,125,713,307 | 0.328 | 100 |
| Breed | ROH Length, Mb (M ± SE) | ROH No. (M ± SE) | FROH (M ± SE) |
|---|---|---|---|
| Old Russian indigenous breeds | |||
| OMF | 266.42 ± 12.42 | 125.17 ± 4.03 | 0.279 ± 0.013 |
| PC | 337.93 ± 15.74 | 145.00 ± 6.50 | 0.354 ± 0.017 |
| RBB | 369.41 ± 16.07 | 141.32 ± 5.01 | 0.387 ± 0.017 |
| RC | 197.84 ± 19.08 | 89.28 ± 6.88 | 0.207 ± 0.020 |
| Ush | 375.63 ± 14.23 | 127.13 ± 2.70 | 0.394 ± 0.015 |
| YC | 146.91 ± 11.94 | 70.11 ± 2.62 | 0.154 ± 0.013 |
| Average (M ± SE) | 282.36 ± 38.78 | 121.75 ± 12.26 | 0.296 ± 0.041 |
| Improved Russian breeds | |||
| AS | 205.05 ± 9.33 | 94.87 ± 2.86 | 0.215 ± 0.010 |
| KJ | 368.96 ± 7.20 | 142.00 ± 2.71 | 0.387 ± 0.008 |
| Kt | 360.79 ± 9.22 | 143.86 ± 3.83 | 0.378 ± 0.010 |
| LMF | 375.11 ± 18.37 | 112.14 ± 4.13 | 0.393 ± 0.019 |
| Pm | 274.02 ± 9.01 | 139.87 ± 3.77 | 0.287 ± 0.009 |
| RW1 | 323.45 ± 15.34 | 151.38 ± 2.41 | 0.339 ± 0.016 |
| RW2 | 195.24 ± 5.89 | 130.83 ± 2.74 | 0.205 ± 0.006 |
| UshF | 357.71 ± 35.25 | 116.00 ± 4.56 | 0.375 ± 0.037 |
| ZS | 301.06 ± 9.48 | 154.04 ± 4.9 | 0.316 ± 0.010 |
| Average (M ± SE) | 306.82 ± 23.04 | 131.67 ± 6.66 | 0.322 ± 0.024 |
| Specialized foreign breeds | |||
| AoB | 260.03 ± 10.18 | 107.03 ± 2.89 | 0.273 ± 0.011 |
| CW | 154.61 ± 5.98 | 126.29 ± 3.28 | 0.162 ± 0.006 |
| NH | 403.39 ± 11.15 | 120.80 ± 1.43 | 0.423 ± 0.012 |
| RIR | 352.15 ± 14.42 | 151.94 ± 8.01 | 0.369 ± 0.015 |
| WL | 525.64 ± 6.12 | 249.75 ± 2.48 | 0.551 ± 0.006 |
| Average (M ± SE) | 339.16 ± 63.00 | 151.16 ± 25.70 | 0.356 ± 0.066 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanov, M.N.; Abdelmanova, A.S.; Fisinin, V.I.; Gladyr, E.A.; Volkova, N.A.; Anshakov, D.V.; Stanishevskaya, O.I.; Vakhrameev, A.B.; Dotsev, A.V.; Griffin, D.K.; et al. Whole Genome Screening Procures a Holistic Hold of the Russian Chicken Gene Pool Heritage and Demographic History. Biology 2023, 12, 979. https://doi.org/10.3390/biology12070979
Romanov MN, Abdelmanova AS, Fisinin VI, Gladyr EA, Volkova NA, Anshakov DV, Stanishevskaya OI, Vakhrameev AB, Dotsev AV, Griffin DK, et al. Whole Genome Screening Procures a Holistic Hold of the Russian Chicken Gene Pool Heritage and Demographic History. Biology. 2023; 12(7):979. https://doi.org/10.3390/biology12070979
Chicago/Turabian StyleRomanov, Michael N., Alexandra S. Abdelmanova, Vladimir I. Fisinin, Elena A. Gladyr, Natalia A. Volkova, Dmitry V. Anshakov, Olga I. Stanishevskaya, Anatoly B. Vakhrameev, Arsen V. Dotsev, Darren K. Griffin, and et al. 2023. "Whole Genome Screening Procures a Holistic Hold of the Russian Chicken Gene Pool Heritage and Demographic History" Biology 12, no. 7: 979. https://doi.org/10.3390/biology12070979
APA StyleRomanov, M. N., Abdelmanova, A. S., Fisinin, V. I., Gladyr, E. A., Volkova, N. A., Anshakov, D. V., Stanishevskaya, O. I., Vakhrameev, A. B., Dotsev, A. V., Griffin, D. K., & Zinovieva, N. A. (2023). Whole Genome Screening Procures a Holistic Hold of the Russian Chicken Gene Pool Heritage and Demographic History. Biology, 12(7), 979. https://doi.org/10.3390/biology12070979