

Molecular Modeling Insights into Metal-Organic Frameworks (MOFs) as a Potential Matrix for Immobilization of Lipase: An In Silico Study

 ,
, 
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Method
2.1. Structure Retrieval and Preparation
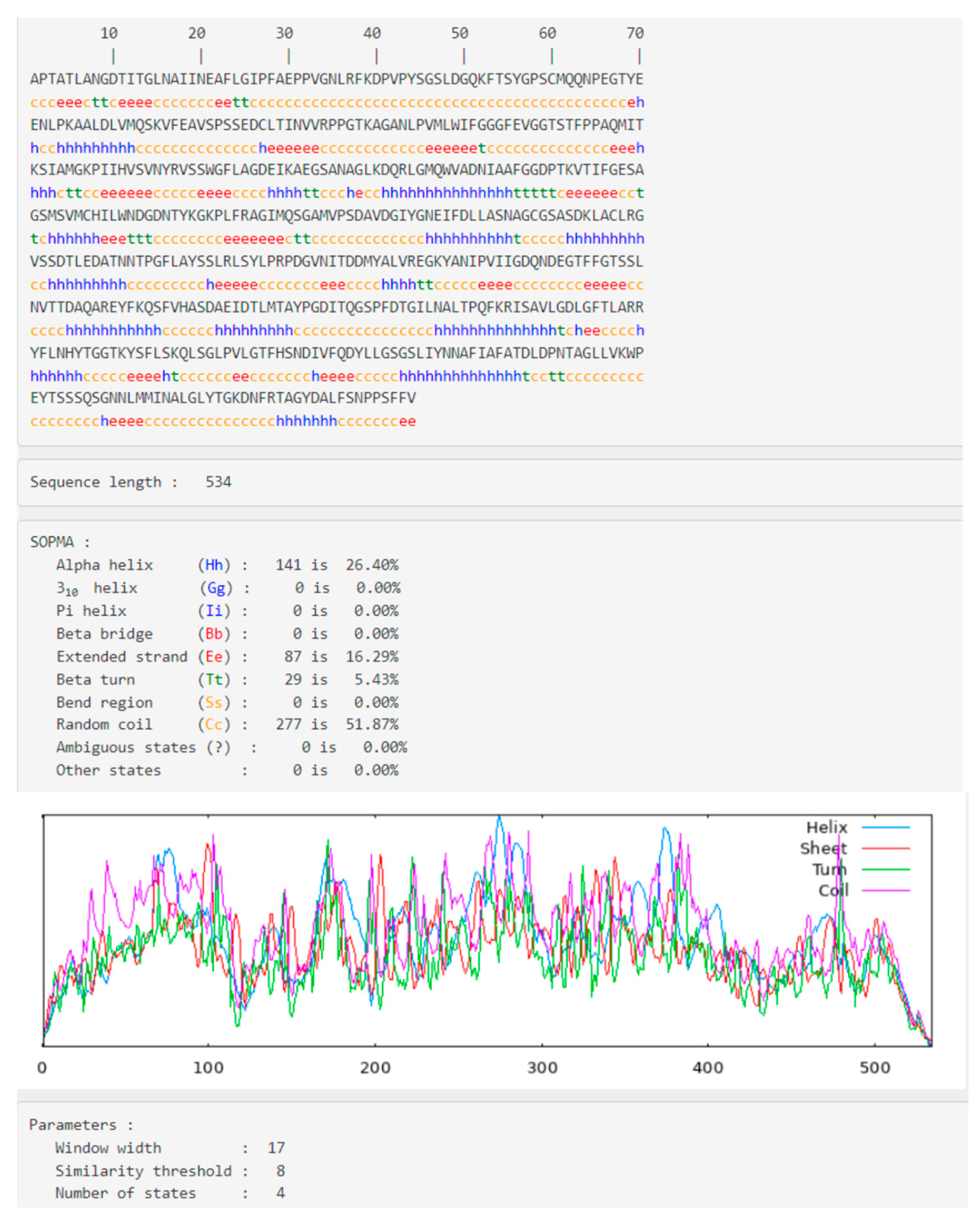
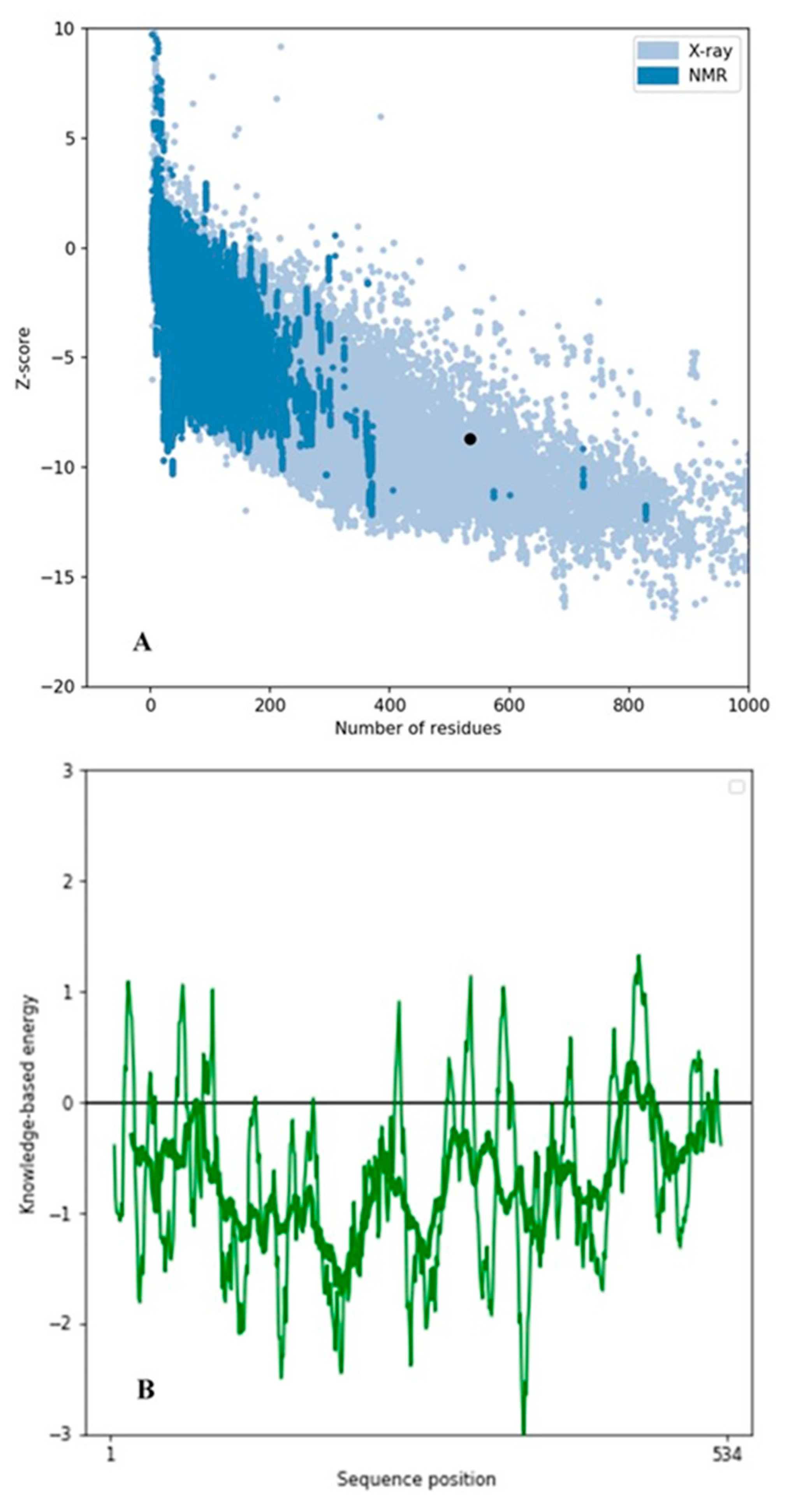
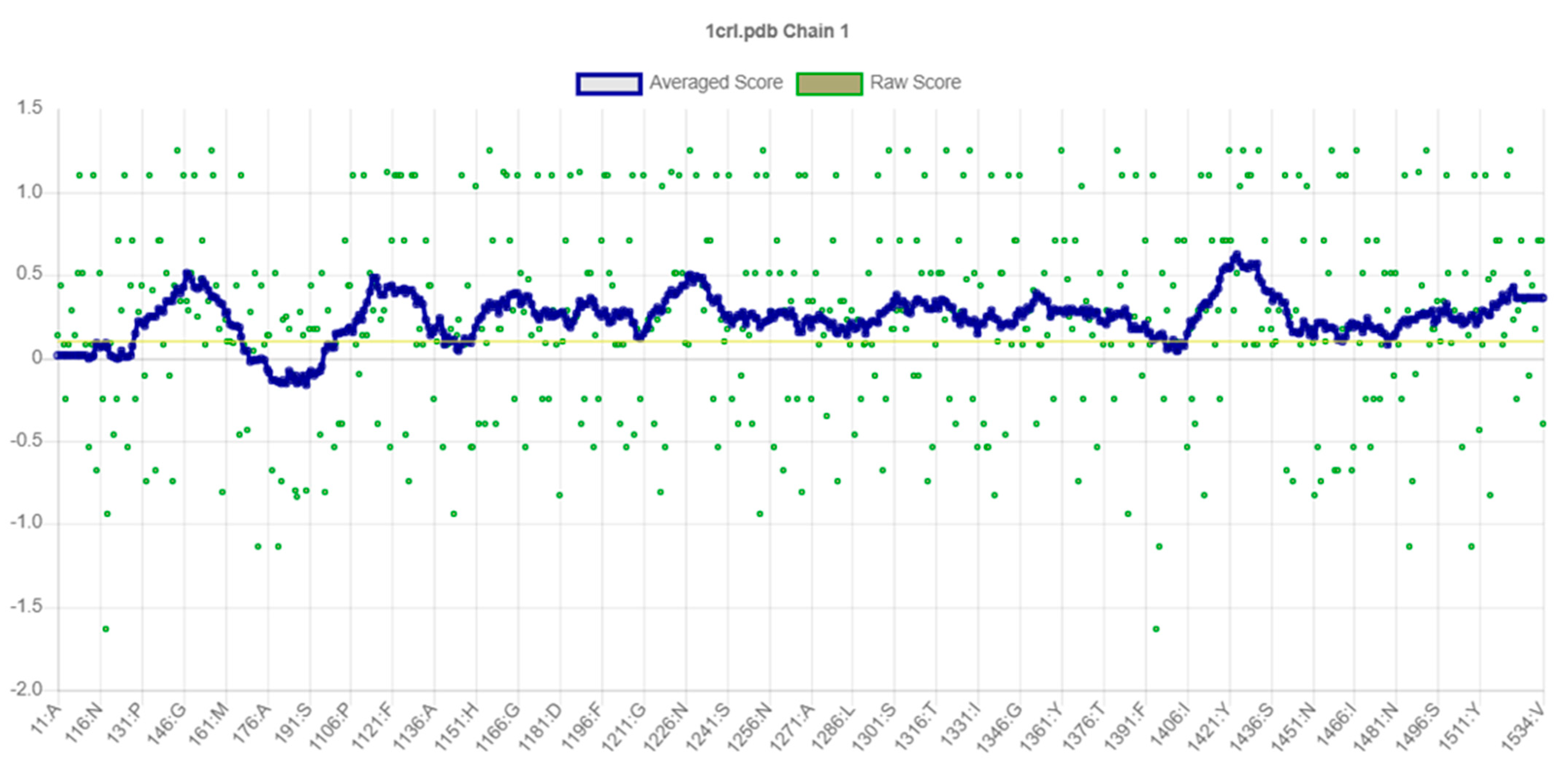
2.2. Secondary Structural Analysis and Homology Modeling Studies of CRL
2.3. Molecular Docking Analysis of CRL with ZIF-8
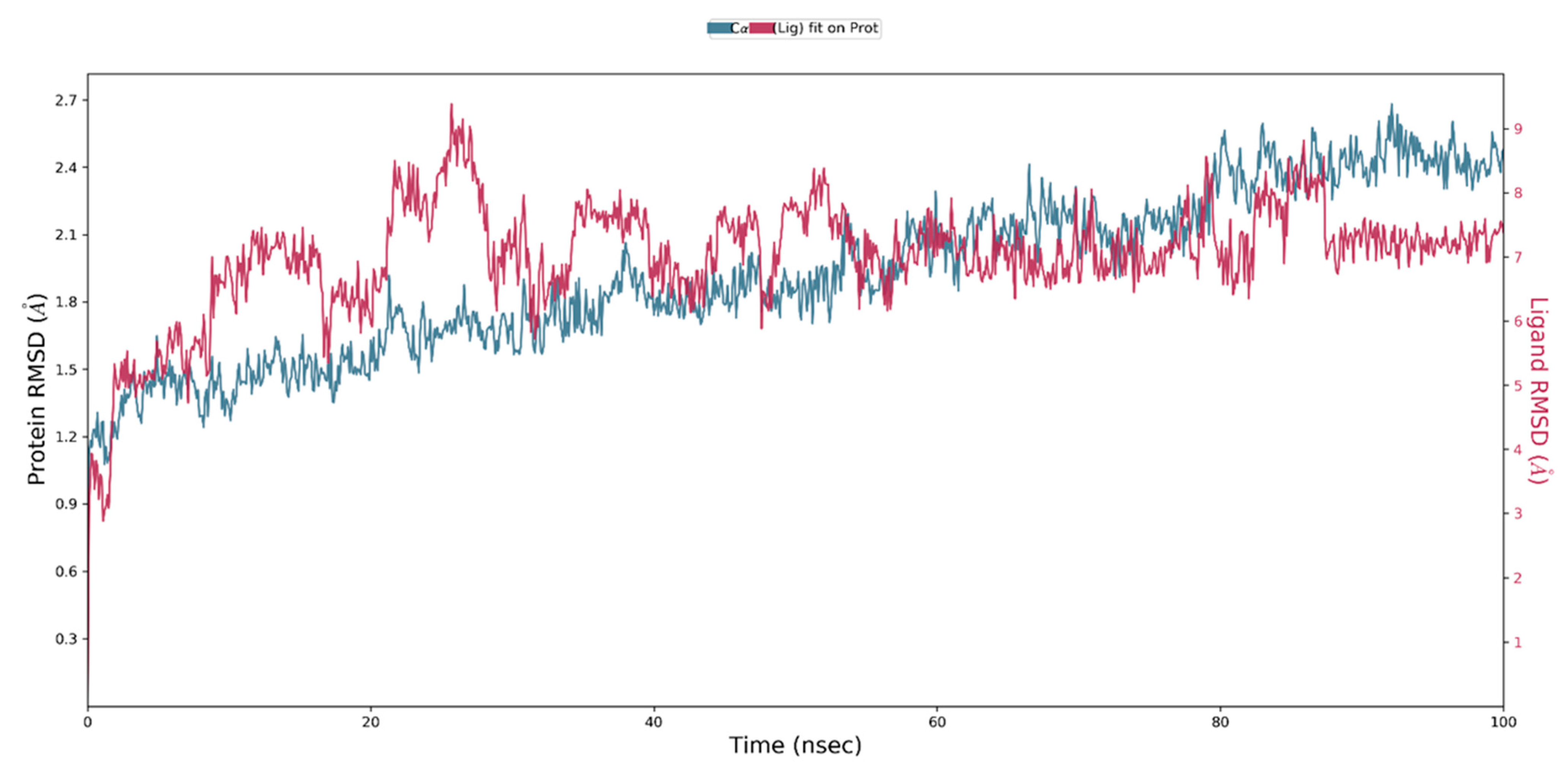
2.4. MD Simulation of a Docked Complex of CRL and ZIF-8
3. Results
3.1. Secondary Structural Analysis and Homology Modeling Studies of CRL
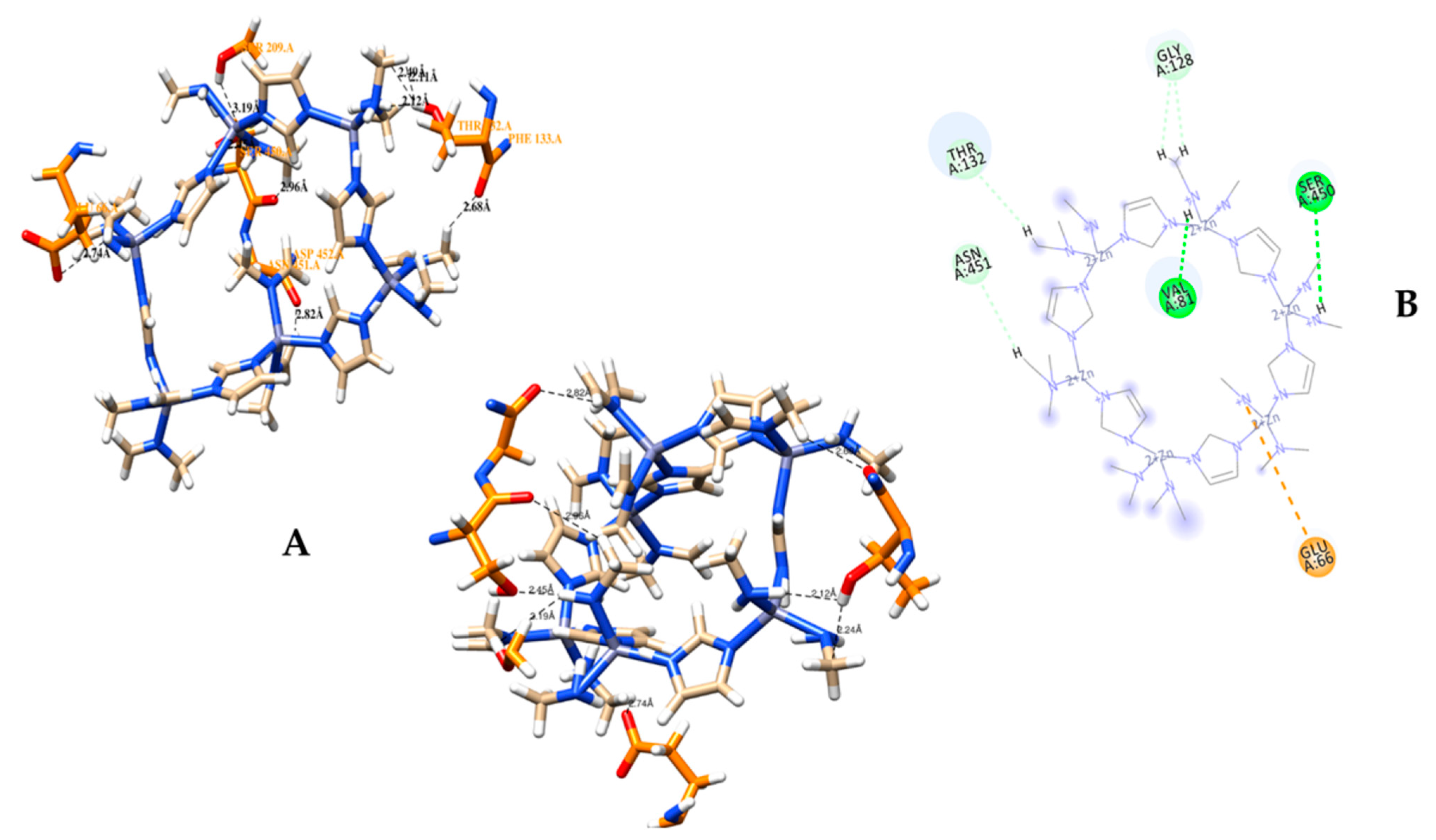
3.2. Molecular Docking Analysis of CRL with ZIF-8
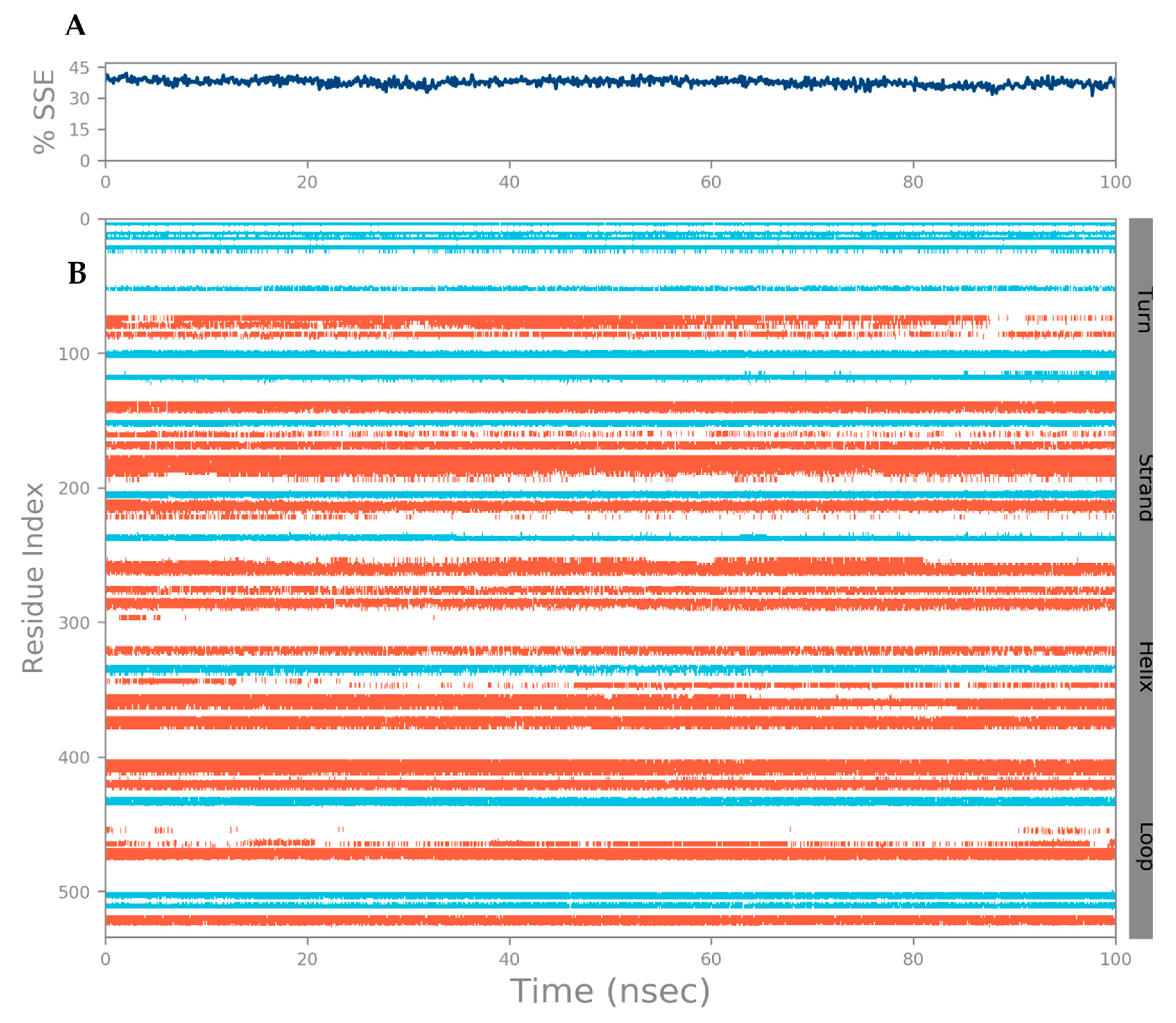
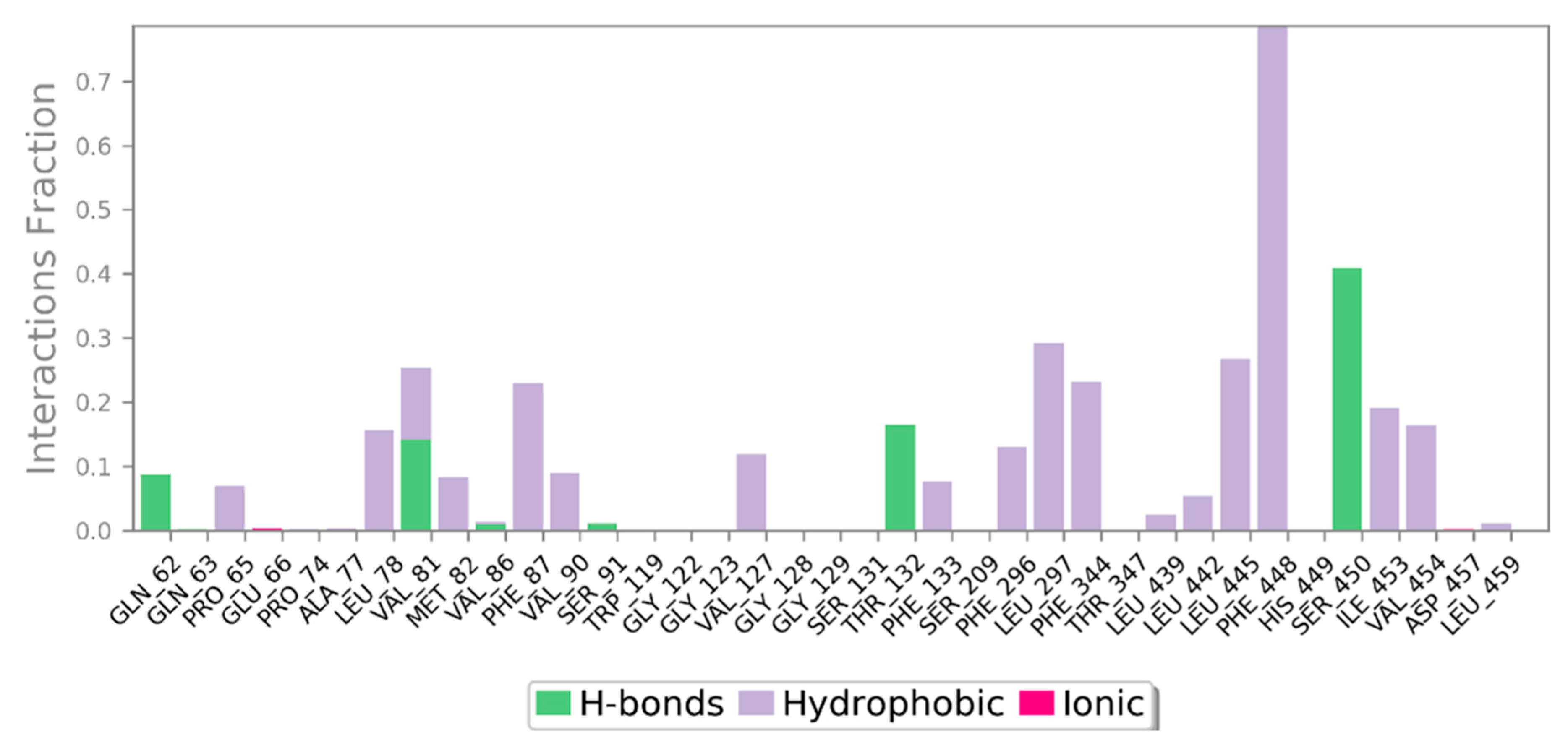
3.3. MD Simulation of a Docked Complex of CRL and ZIF-8
4. Discussion
- (1)
- One strategy for dealing with this problem in simulation is to employ a coarse-grained enzyme model. By doing so, the enzyme’s size will decrease, and it will be simpler to fit through the pores of the ZIF-8 framework [46,47]. Adding constraints or positioning constraints to the enzyme during the simulation will also replicate the experimental restraint imposed by the framework. In order to limit the mobility of the enzyme within the MOF, this can entail constraining specific regions or atoms of the enzyme. The investigation of the enzyme-MOF system can be facilitated through the development of a comprehensive model that enables the examination of the temporal dynamics and interactions of the enzyme. The enzyme’s confinement within the MOF structure can be effectively replicated by employing external restraints or constraints, such as harmonic potentials or position restraints, which are defined by suitable force field parameters. Measurements such as cavity occupancy, residence times, and others can be used to determine how much confinement the enzyme faces while housed in the MOF. This will shed light on how much restriction and confinement the framework imposes on the enzyme.
- (2)
- The following strategies should be taken into account in experiments:
- (a)
- Enzymatic assays, kinetic studies, or other appropriate techniques can be used to examine the stability and activity of the enzyme-MOF system. This will make it easier to evaluate how the MOF framework affects how the enzyme works and how active it is.
- (b)
- Modifying MOF characteristics:
- (bi)
- Pore size modification: MOF’s pore size is too small for the enzyme; we can use different MOFs with larger pores that still have a high surface area and stability.
- (bii)
- Functionalization: Changing the surface functional groups of the MOF to enhance interactions with the enzyme. Enzyme-MOF interactions may be improved, and dramatic constraints may be lessened.
- (c)
- Examining alternative MOF synthesis techniques: To create a MOF structure with bigger pores or more accessible areas for the enzyme in the experimental in-situ encapsulation, different crystallization techniques might be investigated.
- (d)
- Time-resolved methods: Using time-resolved methods, such as in-situ spectroscopy or time-resolved X-ray diffraction, to track the enzyme immobilization procedure in real time. In doing so, we can see how the MOF confines and controls the enzyme as it crystallizes.
- (e)
- Techniques for characterizing: Using several characterization methods to examine the shape and distribution of the enzyme within the MOF, such as scanning electron microscopy (SEM) or transmission electron microscopy (TEM). This can assist in determining how efficiently the enzyme is restricted and immobilized.
- (f)
- Catalytic activity tests: Calculating the immobilized enzyme’s catalytic activity and contrasting it with the free enzyme’s activity. This reveals the MOF’s efficiency in inhibiting the enzyme’s mobility and functionality.
- Size and complexity: Enzymes are characterized by their substantial molecular size, composed of numerous atoms. In contrast, MOFs exhibit a wide range of structural complexity, encompassing varying quantities of metal ions and organic ligands, typically ranging from hundreds to thousands. Simulating large systems with atomistic precision necessitates substantial computational resources and time. The computational costs experience exponential growth as the size of the system increases, thereby imposing constraints on the feasible timescales and the number of replicates that can be simulated.
- Force field parametrization: The precise determination of force fields for enzymes and MOFs is of utmost importance to ensure simulation reliability. Nevertheless, parameterizing force fields for biomolecules and MOF components presents significant challenges. Enzymes frequently necessitate specialized force fields due to their distinctive functional groups and catalytic mechanisms. In a similar vein, MOFs exhibit a wide range of metal-ligand coordination environments, thereby requiring precise parameterization in order to characterize their properties effectively.
- Differences in binding free energy: Due to the small difference in free energy between substrate enantiomers, predicting an enzyme’s enantioselectivity towards these compounds is difficult. One probable explanation is that enzymes and MOF have different binding free energies, which are commonly used to quantify the affinity of biomolecular interactions [50].
- Metal-enzyme interactions: Metal ions are frequently necessary as cofactors for the catalytic activity of enzymes. Accurately representing metal-enzyme interactions in MD simulations poses a challenge, primarily stemming from the requirement for advanced force fields capable of accurately describing the intricate nature of metal coordination chemistry. The process of parameterizing metal centers can present significant challenges, particularly when dealing with transition metal ions that possess intricate electronic configurations.
- Enzyme flexibility: Enzymes are highly dynamic biomolecules that undergo conformational modifications to execute their essential biological functions. Effectively simulating these conformational changes poses significant computational challenges. The comprehensive exploration of an enzyme’s conformational space within a MOF is frequently difficult due to the restricted timescales that can be accessed through MD simulations.
- Treatment of solvents and ions: MOF systems are commonly subjected to simulation in the presence of solvents and ions in order to replicate the conditions observed in experimental settings. Precisely representing the solvent environment, encompassing water molecules or organic solvents, is imperative; however, it introduces complexities to the simulation. Furthermore, the selection of ion parameters and concentrations can impact the stability and dynamics of the enzyme-MOF system.
- Enzyme-MOF interface: The interaction between the enzyme and MOF can exhibit complexity, encompassing various types of interactions, including hydrogen bonding, hydrophobic contacts, and electrostatic interactions. The task of capturing the complex intricacies of these interactions and comprehending the impact of the MOF on enzyme dynamics can present considerable difficulties.
- System stability and timescales: Enzyme-MOF systems possess inherent instability owing to the possibility of structural rearrangements and destruction of the MOF framework upon binding with enzymes. The challenge lies in simulating the long-term stability of the enzyme-MOF system while ensuring an accurate representation of the system. Numerous enzymatic reactions take place within time frames that surpass the capabilities of conventional MD simulations. The investigation of rare occurrences, such as substrate binding or enzymatic reactions, necessitates the utilization of sophisticated sampling methodologies, such as enhanced sampling or hybrid techniques that integrate MD with other complementary strategies.
- Limited experimental data: The experimental characterization of enzyme-MOF systems presents significant challenges, resulting in the limited availability of benchmarking data to validate simulation outcomes. The absence of empirical data poses a significant obstacle to the advancement and verification of precise simulation models.
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Lian, X.; Fang, Y.; Joseph, E.; Wang, Q.; Li, J.; Banerjee, S.; Lollar, C.; Wang, X.; Zhou, H.C. Enzyme-MOF (Metal-Organic Framework) Composites. Chem. Soc. Rev. 2017, 46, 3386–3401. [Google Scholar] [CrossRef]
- Rezaei, A.; Akhavan, O.; Hashemi, E.; Shamsara, M. Ugi Four-Component Assembly Process: An Efficient Approach for One-Pot Multifunctionalization of Nanographene Oxide in Water and Its Application in Lipase Immobilization. Chem. Mater. 2016, 28, 3004–3016. [Google Scholar] [CrossRef]
- Ismail, A.R.; Baek, K.H. Lipase Immobilization with Support Materials, Preparation Techniques, and Applications: Present and Future Aspects. Int. J. Biol. Macromol. 2020, 163, 1624–1639. [Google Scholar] [CrossRef] [PubMed]
- Melani, N.B.; Tambourgi, E.B.; Silveira, E. Lipases: From Production to Applications. Sep. Purif. Rev. 2020, 49, 143–158. [Google Scholar] [CrossRef]
- Chapman, J.; Zoica Dinu, C. Assessment of Enzyme Functionality at Metal-Organic Framework Interfaces Developed through Molecular Simulations. Langmuir 2023, 39, 1750–1763. [Google Scholar] [CrossRef]
- Shomal, R.; Du, W.; Al-Zuhair, S. Immobilization of Lipase on Metal-Organic Frameworks for Biodiesel Production. J. Environ. Chem. Eng. 2022, 10, 107265. [Google Scholar] [CrossRef]
- Liang, W.; Wied, P.; Carraro, F.; Sumby, C.J.; Nidetzky, B.; Tsung, C.K.; Falcaro, P.; Doonan, C.J. Metal-Organic Framework-Based Enzyme Biocomposites. Chem. Rev. 2021, 121, 1077–1129. [Google Scholar] [CrossRef] [PubMed]
- Liang, S.; Wu, X.L.; Xiong, J.; Zong, M.H.; Lou, W.Y. Metal-Organic Frameworks as Novel Matrices for Efficient Enzyme Immobilization: An Update Review. Coord. Chem. Rev. 2020, 406, 213149. [Google Scholar] [CrossRef]
- Du, Y.; Jia, X.; Zhong, L.; Jiao, Y.; Zhang, Z.; Wang, Z.; Feng, Y.; Bilal, M.; Cui, J.; Jia, S. Metal-Organic Frameworks with Different Dimensionalities: An Ideal Host Platform for Enzyme@MOF Composites. Coord. Chem. Rev. 2022, 454, 214327. [Google Scholar] [CrossRef]
- Zhang, P.; Chen, J.; Sun, B.; Sun, C.; Xu, W.; Tang, K. Enhancement of the Catalytic Efficiency of Candida Antarctica Lipase A in Enantioselective Hydrolysis through Immobilization onto a Hydrophobic MOF Support. Biochem. Eng. J. 2021, 173, 108066. [Google Scholar] [CrossRef]
- Moayed Mohseni, M.; Jouyandeh, M.; Mohammad Sajadi, S.; Hejna, A.; Habibzadeh, S.; Mohaddespour, A.; Rabiee, N.; Daneshgar, H.; Akhavan, O.; Asadnia, M.; et al. Metal-Organic Frameworks (MOF) Based Heat Transfer: A Comprehensive Review. Chem. Eng. J. 2022, 449, 137700. [Google Scholar] [CrossRef]
- Rabiee, N.; Atarod, M.; Tavakolizadeh, M.; Asgari, S.; Rezaei, M.; Akhavan, O.; Pourjavadi, A.; Jouyandeh, M.; Lima, E.C.; Hamed Mashhadzadeh, A.; et al. Green Metal-Organic Frameworks (MOFs) for Biomedical Applications. Microporous Mesoporous Mater. 2022, 335, 111670. [Google Scholar] [CrossRef]
- Wu, M.X.; Yang, Y.W. Metal–Organic Framework (MOF)-Based Drug/Cargo Delivery and Cancer Therapy. Adv. Mater. 2017, 29, 1606134. [Google Scholar] [CrossRef]
- Zhao, J.; Kan, Y.; Chen, Z.; Li, H.; Zhang, W. MOFs-Modified Electrochemical Sensors and the Application in the Detection of Opioids. Biosensors 2023, 13, 284. [Google Scholar] [CrossRef] [PubMed]
- Souza, J.E.D.S.; Oliveira, G.P.D.; Alexandre, J.Y.; Neto, J.G.; Sales, M.B.; Junior, P.G.D.S.; Oliveira, A.L.B.d.; Souza, M.C.M.d; Santos, J.C.S.d. A Comprehensive Review on the Use of Metal-Organic Frameworks (MOFs) Coupled with Enzymes as Biosensors. Electrochem 2022, 3, 89–113. [Google Scholar] [CrossRef]
- Qin, X.; Zhong, J.; Wang, Y. A Mutant T1 Lipase Homology Modeling, and Its Molecular Docking and Molecular Dynamics Simulation with Fatty Acids. J. Biotechnol. 2021, 337, 24–34. [Google Scholar] [CrossRef]
- Cavalcante, F.T.T.; da Fonseca, A.M.; Holanda Alexandre, J.Y.N.; dos Santos, J.C.S. A Stepwise Docking and Molecular Dynamics Approach for Enzymatic Biolubricant Production Using Lipase Eversa® Transform as a Biocatalyst. Ind. Crops Prod. 2022, 187, 115450. [Google Scholar] [CrossRef]
- Rad, M.; Ebrahimipour, G.; Bandehpour, M.; Akhavan, O.; Yarian, F. SOEing PCR/Docking Optimization of Protein A-G/ScFv-Fc-Bioconjugated Au Nanoparticles for Interaction with Meningitidis Bacterial Antigen. Catalysts 2023, 13, 790. [Google Scholar] [CrossRef]
- Wang, S.; Liu, S.; Liu, C.; Tang, S.; Gu, D.; Tian, J.; Yang, Y. Affinity Screening of Potential Anti-Obesity and Anti-Diabetic Component from Pomegranate Peel by Co-Immobilization of Lipase and α-Amylase Using Carbon Nanotube and Hydrogel. Process Biochem. 2023, 126, 51–60. [Google Scholar] [CrossRef]
- Kamble, S.; Barale, S.; Dhanavade, M.; Sonawane, K. Structural Significance of Neprylysin from Streptococcus Suis GZ1 in the Degradation of Aβ Peptides, a Causative Agent in Alzheimer’s Disease. Comput. Biol. Med. 2021, 136, 104691. [Google Scholar] [CrossRef]
- Taghizadeh, T.; Ameri, A.; Talebian-Kiakalaieh, A.; Mojtabavi, S.; Ameri, A.; Forootanfar, H.; Tarighi, S.; Faramarzi, M.A. Lipase@zeolitic Imidazolate Framework ZIF-90: A Highly Stable and Recyclable Biocatalyst for the Synthesis of Fruity Banana Flavour. Int. J. Biol. Macromol. 2021, 166, 1301–1311. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lan, P.C.; Ma, S. Metal-Organic Frameworks for Enzyme Immobilization: Beyond Host Matrix Materials. ACS Cent. Sci. 2020, 6, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Vanleeuw, E.; Winderickx, S.; Thevissen, K.; Lagrain, B.; Dusselier, M.; Cammue, B.P.A.; Sels, B.F. Substrate-Specificity of Candida Rugosa Lipase and Its Industrial Application. ACS Sustain. Chem. Eng. 2019, 7, 15828–15844. [Google Scholar] [CrossRef]
- Vulichi, S.R.; Runthala, A.; Rachamreddy, S.K.; Yaramanedi, R.S.P.; Sahoo, P.S.; Burra, P.V.L.S.; Kaur, N.; Akkiraju, S.; Kanala, S.R.; Chippada, A.R.; et al. Appraisal of Pancreatic Lipase Inhibitory Potential of Ziziphus Oenoplia (L.)Mill. Leaves by In Vitro and In Silico Approaches. ACS Omega 2023, 8, 16630–16646. [Google Scholar] [CrossRef]
- Kumar, R. Structural Dynamics and Mechanistic Action Guided Engineering of Lipolytic Enzymes. J. Cell. Biochem. 2023, 124, 877–888. [Google Scholar] [CrossRef] [PubMed]
- Mittal, A.; Gandhi, S.; Roy, I. Mechanistic Interaction Studies of Synthesized ZIF-8 Nanoparticles with Bovine Serum Albumin Using Spectroscopic and Molecular Docking Approaches. Sci. Rep. 2022, 12, 10331. [Google Scholar] [CrossRef]
- Combet, C.; Blanchet, C.; Geourjon, C.; Deleage, G. NPS@: Network Protein Sequence Analysis. Trends Biochem. Sci. 2000, 25, 147–150. [Google Scholar] [CrossRef]
- Zhang, M.; Huang, C.; Wang, Z.; Lv, H.; Li, X. In Silico Analysis of Non-Synonymous Single Nucleotide Polymorphisms (NsSNPs) in the Human GJA3 Gene Associated with Congenital Cataract. BMC Mol. Cell Biol. 2020, 21, 12. [Google Scholar] [CrossRef] [Green Version]
- Ayinla, Z.A.; Ademakinwa, A.N.; Agboola, F.K. Comparative Modelling, Molecular Docking and Immobilization Studies on Rhizopus Oryzae Lipase: Evaluation of Potentials for Fatty Acid Methyl Esters Synthesis. J. Biomol. Struct. Dyn. 2022, 2119279. [Google Scholar] [CrossRef]
- Mohammed, A.A.; Barale, S.S.; Kamble, S.A.; Paymal, S.B.; Sonawane, K.D. Molecular Insights into the Inhibition of Early Stages of Aβ Peptide Aggregation and Destabilization of Alzheimer’s Aβ Protofibril by Dipeptide D-Trp-Aib: A Molecular Modelling Approach. Int. J. Biol. Macromol. 2023, 242, 124880. [Google Scholar] [CrossRef]
- Barale, S.S.; Parulekar, R.S.; Fandilolu, P.M.; Dhanavade, M.J.; Sonawane, K.D. Molecular Insights into Destabilization of Alzheimer’s Aβ Protofibril by Arginine Containing Short Peptides: A Molecular Modeling Approach. ACS Omega 2019, 4, 892–903. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Patel, S.; Tulsian, K.; Kumar, P.; Vyas, V.K.; Ghate, M. Design of 2-Amino-6-Methyl-Pyrimidine Benzoic Acids as ATP Competitive Casein Kinase-2 (CK2) Inhibitors Using Structure- and Fragment-Based Design, Docking and Molecular Dynamic Simulation Studies. SAR QSAR Environ. Res. 2023, 34, 211–230. [Google Scholar] [CrossRef] [PubMed]
- Harikrishna, A.S.; Venkitasamy, K. Identification of Novel Human Nicotinamide N-Methyltransferase Inhibitors: A Structure-Based Pharmacophore Modeling and Molecular Dynamics Approach. J. Biomol. Struct. Dyn. 2023, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Qasim, A.; Jaan, S.; Wara, T.U.; Shehroz, M.; Nishan, U.; Shams, S.; Shah, M.; Ojha, S.C. Computer-Aided Genomic Data Analysis of Drug-Resistant Neisseria Gonorrhoeae for the Identification of Alternative Therapeutic Targets. Front. Cell. Infect. Microbiol. 2023, 13, 1017315. [Google Scholar] [CrossRef] [PubMed]
- Man, V.H.; He, X.; Gao, J.; Wang, J. Phosphorylation of Tau R2 Repeat Destabilizes Its Binding to Microtubules: A Molecular Dynamics Simulation Study. ACS Chem. Neurosci. 2023, 14, 458–467. [Google Scholar] [CrossRef]
- Nóbrega, C.S.; Carvalho, A.L.; Romão, M.J.; Pauleta, S.R. Structural Characterization of Neisseria Gonorrhoeae Bacterial Peroxidase—Insights into the Catalytic Cycle of Bacterial Peroxidases. Int. J. Mol. Sci. 2023, 24, 6246. [Google Scholar] [CrossRef]
- Sahoo, R.K.; Sanket, A.S.; Gaur, M.; Das, A.; Subudhi, E. Insight into the Structural Configuration of Metagenomically Derived Lipase from Diverse Extreme Environment. Biocatal. Agric. Biotechnol. 2019, 22, 101404. [Google Scholar] [CrossRef]
- Skariyachan, S.; Praveen, P.K.U.; Uttarkar, A.; Niranjan, V. Computational Design of Prospective Molecular Targets for Burkholderia Cepacia Complex by Molecular Docking and Dynamic Simulation Studies. Proteins Struct. Funct. 2023, 91, 724–738. [Google Scholar] [CrossRef]
- Arasu, M.V.; Vijayaragavan, P.; Purushothaman, S.; Rathi, M.A.; Al-Dhabi, N.A.; Gopalakrishnan, V.K.; Choi, K.C.; Ilavenil, S. Molecular Docking of Monkeypox (Mpox) Virus Proteinase with FDA Approved Lead Molecules. J. Infect. Public. Health 2023, 16, 784–791. [Google Scholar] [CrossRef]
- Yves Nunes Holanda Alexandre, J.; Thálysson Tavares Cavalcante, F.; Matias Freitas, L.; Prudêncio Castro, A.; Tavares Borges, P.; Gonçalves de Sousa Junior, P.; Nazareno Ribeiro Filho, M.; Amelia Sanders Lopes, A.; Marques da Fonseca, A.; Lomonaco, D.; et al. A Theoretical and Experimental Study for Enzymatic Biodiesel Production from Babassu Oil (Orbignya Sp.) Using Eversa Lipase. Catalysts 2022, 12, 1322. [Google Scholar] [CrossRef]
- Chapman, R.; Stenzel, M.H. All Wrapped up: Stabilization of Enzymes within Single Enzyme Nanoparticles. J. Am. Chem. Soc. 2019, 141, 2754–2769. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, N.; Alonso-Cotchico, L.; Lucas, M.F. Enzyme Immobilization Studied through Molecular Dynamic Simulations. Front. Bioeng. Biotechnol. 2023, 11, 1200293. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Yue, H.; Zhang, Y.; Gao, X.; Li, X.; Wang, L.; Cao, Y.; Hou, M.; An, H.; Zhang, L.; et al. Packaging and Delivering Enzymes by Amorphous Metal-Organic Frameworks. Nat. Commun. 2019, 10, 5165. [Google Scholar] [CrossRef] [Green Version]
- Wu, X.; Ge, J.; Yang, C.; Hou, M.; Liu, Z. Facile Synthesis of Multiple Enzyme-Containing Metal-Organic Frameworks in a Biomolecule-Friendly Environment. Chem. Commun. 2015, 51, 13408–13411. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.T.; Yi, J.T.; Zhao, Y.Y.; Chu, X. Biomineralized Metal-Organic Framework Nanoparticles Enable Intracellular Delivery and Endo-Lysosomal Release of Native Active Proteins. J. Am. Chem. Soc. 2018, 140, 9912–9920. [Google Scholar] [CrossRef]
- Li, Y.; Ogorzalek, T.L.; Wei, S.; Zhang, X.; Yang, P.; Jasensky, J.; Brooks, C.L.; Marsh, E.N.G.; Chen, Z. Effect of Immobilization Site on the Orientation and Activity of Surface-Tethered Enzymes. Phys. Chem. Chem. Phys. 2018, 20, 1021–1029. [Google Scholar] [CrossRef]
- Zhang, S.; Yu, S.; Wang, M.; Cui, Z.; Chen, B.; Tan, T. Enzyme Self-Aggregation in Supramolecular Self-Assembly of Glucose Oxidase and Catalase: Insight from Molecular Dynamics Simulation Based on Coarse-Grained Method. Chem. Phys. 2022, 552, 111366. [Google Scholar] [CrossRef]
- Wang, C.; Liao, K. Recent Advances in Emerging Metal- And Covalent-Organic Frameworks for Enzyme Encapsulation. ACS Appl. Mater. Interfaces 2021, 13, 56752–56776. [Google Scholar] [CrossRef]
- Tuan Kob, T.N.A.; Ismail, M.F.; Abdul Rahman, M.B.; Cordova, K.E.; Mohammad Latif, M.A. Unraveling the Structural Dynamics of an Enzyme Encapsulated within a Metal-Organic Framework. J. Phys. Chem. B 2020, 124, 3678–3685. [Google Scholar] [CrossRef]
- Raza, S. Enantioselectivity in Candida Antarctica Lipase B: A Molecular Dynamics Study. Protein Sci. 2001, 10, 329–338. [Google Scholar] [CrossRef]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sr. No. | Possible Hydrogen Bonding Interactions between Active Site Residues of CRL and ZIF-8 before MD | Distance in Å |
|---|---|---|
| 1 | THR 347 HG1 ------------ ZIF 1 C: | 2.87 |
| 2 | PHE 296 CE1 ------------ ZIF 1 N: | 3.13 |
| 3 | ZIF 1 C ------------ PHE 87 CE1: | 3.20 |
| 4 | PHE 344 CE1 ------------ ZIF 1 C: | 3.30 |
| 5 | LEU 297 CD1 ------------ ZIF 1 N: | 3.30 |
| 6 | ZIF 1 C ------------ VAL 81 CG2: | 3.30 |
| 7 | ZIF 1 C ------------ THR 347 HG1: | 3.32 |
| 8 | ZIF 1 C ------------ SER 91 HG: | 3.33 |
| 9 | ZIF 1 C ------------ VAL 81 CG1: | 3.92 |
| Sr. No. | Hydrogen Bond Interactions between Amino Acid Residues of CRL with ZIF-8 after MD | Distance in Å |
|---|---|---|
| 1 | ZIF.het H__2------------THR 132.A HG1 | 2.12 |
| 2 | ZIF.het H__1------------SER 209.A HG | 3.19 |
| 3 | ZIF.het H__1------------SER 450.A HG | 2.45 |
| 4 | ZIF.het 1HXT------------GLU 66.A OE2 | 2.74 |
| 5 | ZIF.het 3HXT------------THR 132.A O | 2.68 |
| 6 | ZIF.het 2HXT------------SER 450.A O | 2.96 |
| 7 | ZIF.het 1HXT------------ASN 451.A O | 2.82 |
| 8 | ZIF.het 2HXT------------THR 132.A HG1 | 2.11 |
| 9 | ZIF.het 3HXT------------THR 132.An HG1 | 2.49 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Patil, P.J.; Kamble, S.A.; Dhanavade, M.J.; Liang, X.; Zhang, C.; Li, X. Molecular Modeling Insights into Metal-Organic Frameworks (MOFs) as a Potential Matrix for Immobilization of Lipase: An In Silico Study. Biology 2023, 12, 1051. https://doi.org/10.3390/biology12081051
Patil PJ, Kamble SA, Dhanavade MJ, Liang X, Zhang C, Li X. Molecular Modeling Insights into Metal-Organic Frameworks (MOFs) as a Potential Matrix for Immobilization of Lipase: An In Silico Study. Biology. 2023; 12(8):1051. https://doi.org/10.3390/biology12081051
Chicago/Turabian StylePatil, Prasanna J., Subodh A. Kamble, Maruti J. Dhanavade, Xin Liang, Chengnan Zhang, and Xiuting Li. 2023. "Molecular Modeling Insights into Metal-Organic Frameworks (MOFs) as a Potential Matrix for Immobilization of Lipase: An In Silico Study" Biology 12, no. 8: 1051. https://doi.org/10.3390/biology12081051
APA StylePatil, P. J., Kamble, S. A., Dhanavade, M. J., Liang, X., Zhang, C., & Li, X. (2023). Molecular Modeling Insights into Metal-Organic Frameworks (MOFs) as a Potential Matrix for Immobilization of Lipase: An In Silico Study. Biology, 12(8), 1051. https://doi.org/10.3390/biology12081051