
Metabolic Heterogeneity, Plasticity, and Adaptation to “Glutamine Addiction” in Cancer Cells: The Role of Glutaminase and the GTωA [Glutamine Transaminase—ω-Amidase (Glutaminase II)] Pathway



Abstract
:Simple Summary
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Discovery of Enzymes That Convert l-Glutamine Amide to Ammonium and Notes on Nomenclature
3. Enzymes That Catalyze l-Glutamine Transamination and Subcellular Localization
4. l-Glutamine Transaminases Catalyze l-Cysteine S-Conjugate β-Lyase (CCBL) and l-Selenocysteine Se-Conjugate β-Lyase Side Reactions
5. Relationship of l-Glutamine Transaminases to Kynurenine Aminotransferases (KATs)
6. Evidence That Enzymes of GTωA Pathway Operate Extensively In Vivo
6.1. ω-Amidase Is Widespread in Mammalian Tissues
6.2. High Inherent l-Glutamine Transaminase Activities in Mammalian/Human Tissues
6.3. Occurrence of KGM in Rat Tissues and Clinical Samples
6.4. Possible Involvement of KGM in Urea Nitrogen Formation and in Acid–Base Balance
7. Additional Proposed Biological Roles of the GTωA Pathway
7.1. Closure of the Methionine Salvage Pathway by Transamination of KMB
7.2. Salvage/Detoxification of α-Keto Acids
7.3. Possible Anti-Oxidant Role of the GTωA Pathway and Its Response to Hypoxia
8. Possible Role of the GTωA Pathway in Transferring α-Keto Acid/l-Amino Acid Carbon between Cellular and Subcellular Compartments
9. “Glutamine Addiction” and the Canonical Pathway for α-Ketoglutarate Formation in Cancer Cells
9.1. The Canonical Pathway for Satisfying “Glutamine Addiction” in Cancer Cells
9.2. What Are the Transaminases (Aminotransferases) That Can Potentially Supply α-Ketoglutarate to the TCA Cycle in the Canonical Pathway [Equation (8)]?
9.2.1. Alanine Aminotransferase (AlaAT; Glutamate Pyruvate Transaminase (GPT))
9.2.2. Aspartate Aminotransferase (AspAT; Glutamate Oxaloacetate Transaminase (GOT))
9.2.3. Branched-Chain Amino Acid (BCAA) Aminotransferases (BCATs)
9.2.4. Conclusions Regarding Contribution of l-Glutamate-Utilizing Transaminases to the Canonical Pathway
9.3. Possible Role of Glutamate Dehydrogenase (GDH) in the Production of α-Ketoglutarate in the Canonical Pathway
10. “Glutamine Addiction” and the GTωA Pathway in Cancer Cells
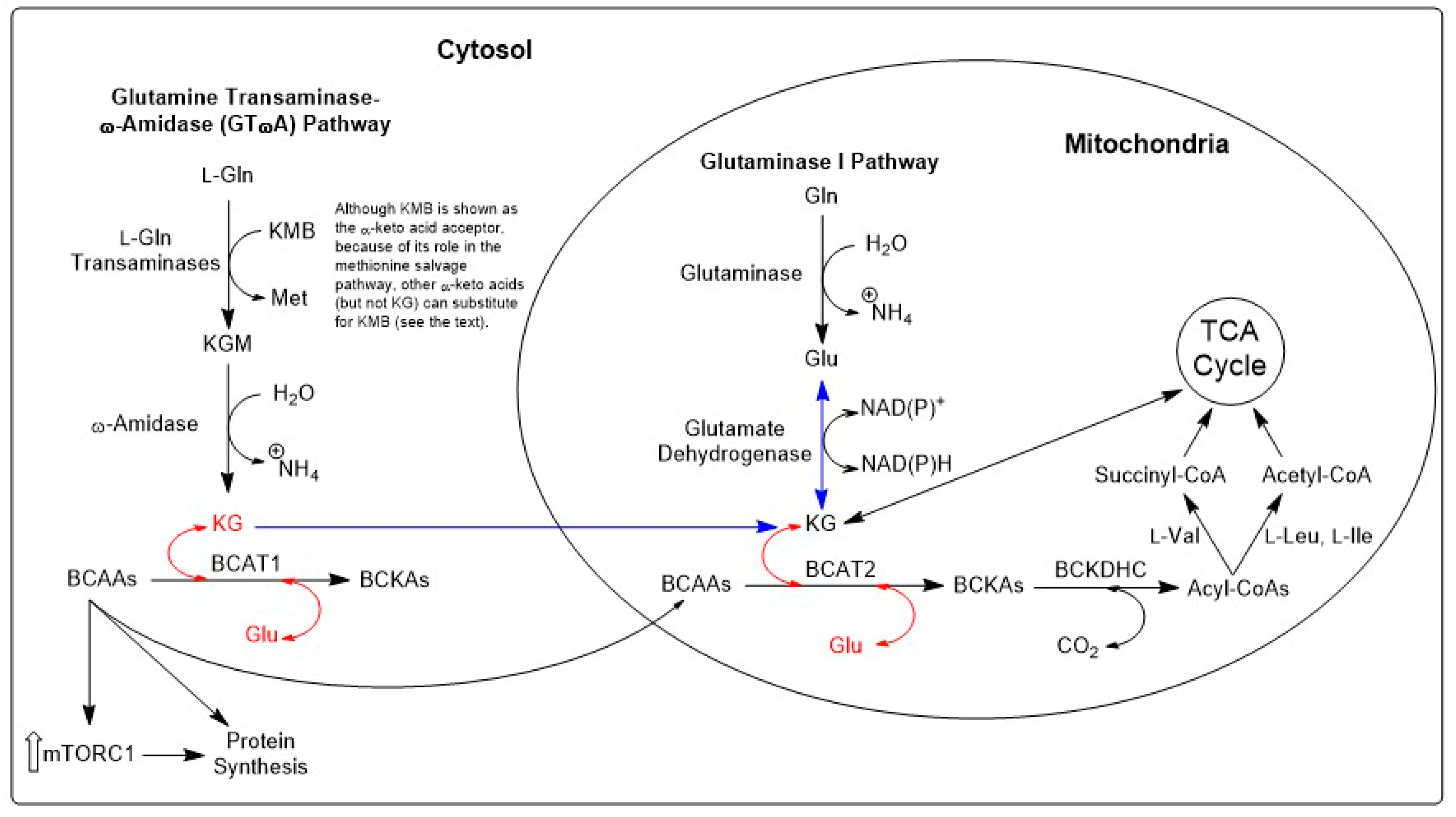
10.1. Sequence of Enzymatic Reactions in the GTωA Pathway
10.2. Why Has the GTwA Pathway Been Largely Overlooked by Biochemists/Oncologists?
11. Glutaminase II (GTωA) Pathway Enzymes in Cancer
11.1. Background
11.2. Is Nit2/ω-Amidase a Tumor Suppressor or Promoter?
11.3. The GTωA Pathway in Pancreatic Cancer and in Medulloblastoma Tumors
12. Studies from the Authors’ Laboratories Showing the Importance of the GTωA Pathway in Glutamine-Addicted Cancers, with Special Reference to Prostate Cancer
12.1. Background
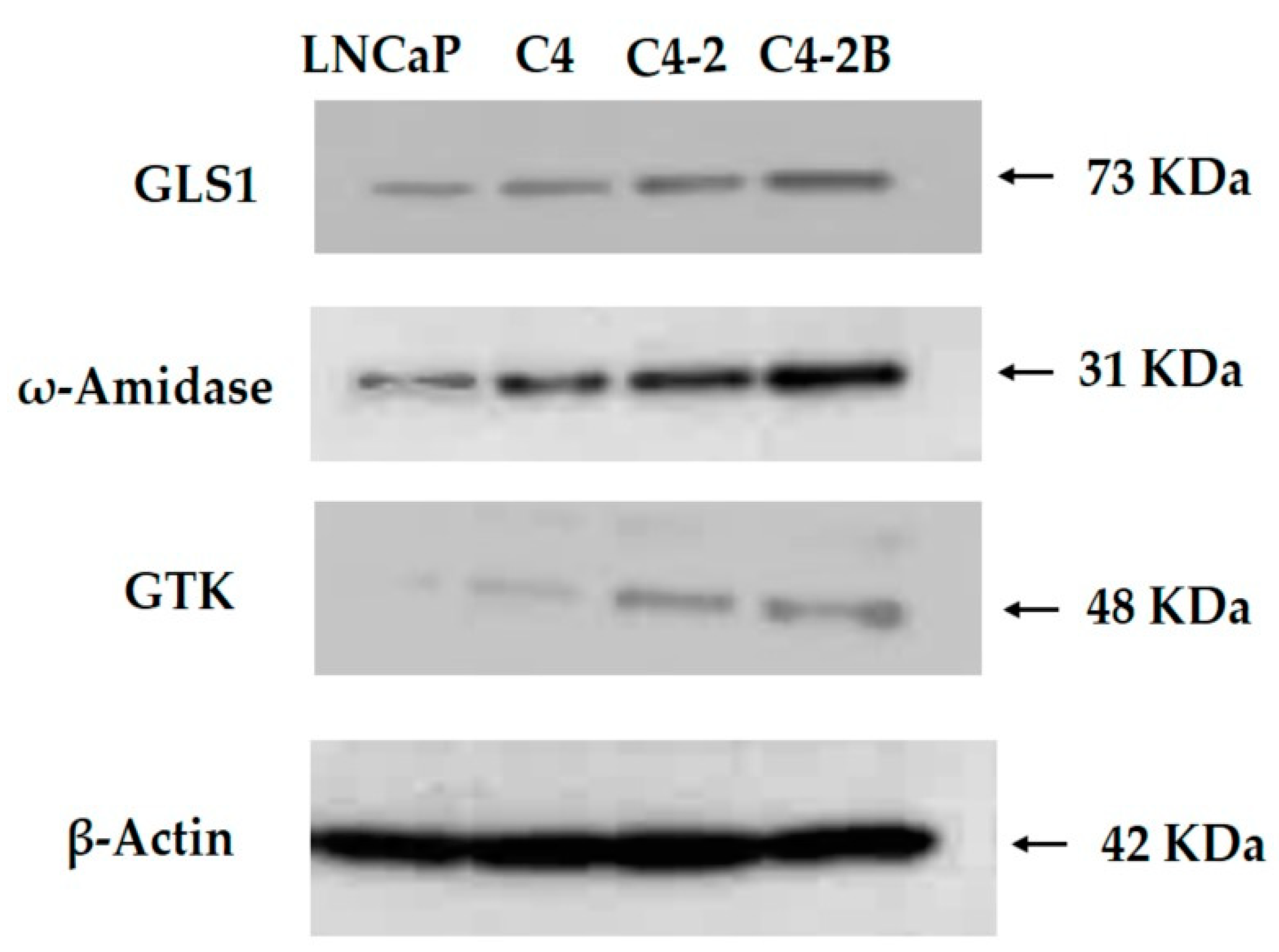
12.2. The GLS1 and GTωA Pathway Enzymes in Human Prostate Cancer Cells in Culture
12.3. Expression of GLS1 and GTωA Pathway Enzymes in Relation to Metabolic Cross-Talk between Supporting Stromal Cells and Prostate Cancer Cells
13. l-Glutamine Addiction and Metabolic Plasticity in Cancer Cells
13.1. Facets of Metabolic Plasticity—I. Integration of GLS1 and GTωA Pathways
13.2. Facets of Metabolic Plasticity—II. Role of Branched-Chain Amino Acids in Cancer and Their Relationship to the GLS1 and GTωA Pathways
13.3. Facets of Metabolic Plasticity—III. Potential Role of ω-Amidase and l-Asparagine in Metastatic Colonization
13.4. Facets of Metabolic Plasticity—IV. Role of GLS1/GTωA Pathways in Tumor–Stroma Collaboration
14. Will Inhibitors of GTωA Enzymes, Perhaps in Combination with Inhibitors of GLS1, Glutamine Transporters, or Other Proteins/Enzymes, Be Clinically Useful in Cancer Patients?
15. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dorai, T.; Dorai, B.; Pinto, J.T.; Grasso, M.; Cooper, A.J.L. High levels of glutaminase II pathway enzymes in normal and cancerous prostate suggest a role in ‘Glutamine Addiction’. Biomolecules 2019, 10, 2. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.J.L.; Shurubor, Y.I.; Dorai, T.; Pinto, J.T.; Isakova, E.P.; Deryabina, Y.I.; Denton, T.T.; Krasnikov, B.F. ω-Amidase: An underappreciated, but important enzyme in l-glutamine and l-asparagine metabolism; relevance to sulfur and nitrogen metabolism, tumor biology and hyperammonemic diseases. Amino Acids 2015, 48, 1–20. [Google Scholar] [CrossRef]
- Dorai, T.; Pinto, J.T.; Denton, T.T.; Krasnikov, B.F.; Cooper, A.J.L. The metabolic importance of the glutaminase II pathway in normal and cancerous cells. Anal. Biochem. 2020, 644, 114083. [Google Scholar] [CrossRef]
- Cooper, A.J.L.; Dorai, T.; Pinto, J.T.; Denton, T.T. α-Ketoglutaramate—A key metabolite contributing to glutamine addiction in cancer cells. Front. Med. 2022, 13, 1035335. [Google Scholar] [CrossRef]
- Carter, C.E.; Greenstein, J.P. Acceleration of enzymatic desamidation of glutamine by several inorganic anions. J. Natl. Cancer Inst. 1947, 7, 433–436. [Google Scholar] [PubMed]
- Errera, M.; Greenstein, J.P. Phosphate-activated glutaminase in kidney and other tissues. J. Biol. Chem. 1949, 178, 495–502. [Google Scholar] [CrossRef]
- Greenstein, J.P.; Carter, C.E. Influence of α-keto acids on the desamidation of amino acid amides. J. Natl. Cancer Inst. 1946, 7, 57–60. [Google Scholar] [CrossRef]
- Perera, S.Y.; Chen, T.C.; Curthoys, N.P. Biosynthesis and processing of renal mitochondrial glutaminase in cultured proximal tubular epithelial-cells and in isolated-mitochondria. J. Biol. Chem. 1990, 265, 17764–17770. [Google Scholar] [CrossRef] [PubMed]
- Mates, J.M.; Segura, J.A.; Martin-Rufian, M.; Campos-Sandoval, J.A.; Alonso, F.J.; Marquez, J. Glutaminase isoenzymes as key regulators in metabolic and oxidative stress against cancer. Curr. Mol. Med. 2013, 13, 514–534. [Google Scholar] [CrossRef]
- Ferreira, I.M.; Quesñay, J.E.N.; Bastos, A.C.; Rodrigues, C.T.; Vollmar, M.; Krojer, T.; Strain-Damerell, C.; Burgess-Brown, N.A.; von Delft, F.; Yue, W.W.; et al. Structure and activation mechanism of the human liver-type glutaminase GLS2. Biochimie 2021, 185, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Cederkvist, H.; Kolan, S.S.; Wik, J.A.; Sener, Z.; Skålhegg, B.S. Identification and characterization of a novel glutaminase inhibitor. FEBS Open Bio. 2022, 12, 163–174. [Google Scholar] [CrossRef]
- Aledo, J.C.; Gómez-Fabre, P.M.; Olalla, L.; Márquez, J. Identification of two human glutaminase loci and tissue-specific expression of the two related genes. Mamm. Genome 2000, 11, 1107–1110. [Google Scholar] [CrossRef]
- Szweda, L.I.; Atkinson, D.E. Response of rat liver glutaminase to pH, ammonium, and citrate. Possible regulatory role of glutaminase in ureagenesis. J. Biol. Chem. 1990, 265, 20869–20873. [Google Scholar] [CrossRef]
- Häussinger, D.; Schliess, F. Glutamine metabolism and signaling in the liver. Front. Biosci. 2007, 12, 371–391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, X.; Chen, H.; Allen, Z.; Lin, A.Z.; Kim, S.; Burczynski, M.E.; Na, E.; Halasz, G.; Sleeman, M.W.; Murphy, A.J.; et al. Glutaminase 2 knockdown reduces hyperammonemia and associated lethality of urea cycle disorder mouse model. J. Inherit. Metab. Dis. 2022, 45, 470–480. [Google Scholar] [CrossRef] [PubMed]
- Greenstein, J.P.; Price, V.E. α-Keto acid-activated glutaminase and asparaginase. J. Biol. Chem. 1949, 178, 695–705. [Google Scholar] [CrossRef] [PubMed]
- Meister, A.; Tice, S.V. Transamination from glutamine to α-keto acids. J. Biol. Chem. 1950, 187, 173–187. [Google Scholar] [CrossRef]
- Meister, A.; Sober, H.A.; Tice, S.V.; Fraser, P.E. Transamination and associated deamidation of asparagine and glutamine. J. Biol. Chem. 1952, 197, 319–330. [Google Scholar] [CrossRef]
- Meister, A. Preparation of enzymatic reactions of the keto analogues of asparagine and glutamine. J. Biol. Chem. 1953, 200, 571–589. [Google Scholar] [CrossRef]
- Meister, A.; Otani, T.T. ω-Amide and ω-amino acid derivatives of α-ketoglutaric and oxalacetic acids. J. Biol. Chem. 1957, 224, 137–148. [Google Scholar]
- Hersh, L.B. Rat liver ω-amidase. Purification and properties. Biochemistry 1971, 10, 2884–2891. [Google Scholar] [CrossRef]
- Cooper, A.J.L. Glutamine aminotransferases and ω-amidases. In Glutamine and Glutamate in Mammals; Kvamme, E., Ed.; CRC Press Inc.: Boca Raton, FL, USA, 1988; Volume 1, pp. 33–52. [Google Scholar]
- Cooper, A.J.L.; Meister, A. Comparative studies of glutamine transaminases from rat tissues. Comp. Biochem. Physiol. 1981, 69, 137–145. [Google Scholar] [CrossRef]
- Duffy, T.E.; Cooper, A.J.L.; Meister, A. Identification of α-ketoglutaramate in rat liver, kidney, and brain. Relationship to glutamine transaminase and ω-amidase activities. J. Biol. Chem. 1974, 249, 7603–7606. [Google Scholar] [CrossRef] [PubMed]
- Shurubor, Y.I.; Cooper, A.J.L.; Isakova, E.P.; Deryabina, Y.I.; Beal, M.F.; Krasnikov, B.F. HPLC determination of α-ketoglutaramate [5-amino-2,5-dioxopentanoate] in biological samples. Anal. Biochem. 2016, 494, 52–54. [Google Scholar] [CrossRef] [PubMed]
- Vergara, F.; Plum, F.; Duffy, T.E. α-Ketoglutaramate: Increased concentrations in the cerebrospinal fluid of patients in hepatic coma. Science 1974, 183, 81–83. [Google Scholar] [CrossRef]
- Cooper, A.J.L.; Meister, A. Isolation and properties of highly purified glutamine transaminase. Biochemistry 1972, 11, 661–671. [Google Scholar] [CrossRef]
- Cooper, A.J.L.; Meister, A. Isolation and properties of a new glutamine transaminase from rat kidney. J. Biol. Chem. 1974, 249, 2554–2561. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, L.; Han, Q.; Liao, C.; Lan, J.; Ding, H.; Zhou, H.; Diao, X.; Li, J. Kynurenine aminotransferase 3/glutamine transaminase L/cysteine conjugate β-lyase 2 is a major glutamine transaminase in the mouse kidney. Biochem. Biophys. Rep. 2016, 8, 234–241. [Google Scholar] [CrossRef] [Green Version]
- Malherbe, P.; Alberati-Giani, D.; Köhler, C.; Cesura, A.M. Identification of a mitochondrial form of kynurenine aminotransferase/glutamine transaminase K from rat brain. FEBS Lett. 1995, 367, 141–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, J.T.; Krasnikov, B.F.; Alcutt, S.; Jones, M.E.; Dorai, T.; Villar, M.T.; Artigues, A.; Li, J.; Cooper, A.J.L. Kynurenine aminotransferase III and glutamine transaminase L are identical enzymes that have cysteine S-conjugate β-lyase activity and can transaminate l-selenomethionine. J. Biol. Chem. 2014, 289, 30950–30961. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.J.L.; Dorai, T.; Dorai, B.; Krasnikov, B.F.; Li, J.; Hallen, A.; Pinto, J.T. Role of glutamine transaminases in nitrogen, sulfur, selenium and 1-carbon metabolism: Glutamine transaminases in normal and cancer cells. In Glutamine in Clinical Nutrition; Nutrition and Health Series; Rajendram, R., Preedy, V.R., Patel, V.B., Bendich, A., Eds.; Humana Press: New York, NY, USA, 2015; pp. 37–54. [Google Scholar]
- Cooper, A.J.L.; Krasnikov, B.F.; Niatsetskaya, Z.V.; Pinto, J.T.; Callery, P.S.; Villar, M.T.; Artigues, A.; Bruschi, S.A. Cysteine S-conjugate β-lyases: Important roles in the metabolism of naturally occurring sulfur and selenium-containing compounds, xenobiotics and anticancer agents. Amino Acids 2011, 41, 7–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowsher, R.R.; Henry, D.P. Purification, characterization and identification of rat brain cytosolic tyrosine transaminase as glutamine transaminase-K. Neurochem. Int. 2020, 133, 104653. [Google Scholar] [CrossRef] [PubMed]
- Han, Q.; Cai, T.; Tagle, D.A.; Li, J. Structure, expression, and function of kynurenine aminotransferases in human and rodent brains. Cell Mol Life Sci. 2010, 67, 353–368. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Li, J.; Li, J. pH dependence, substrate specificity and inhibition of human kynurenine aminotransferase I. Eur. J. Biochem. 2004, 271, 4804–4814. [Google Scholar] [CrossRef]
- Han, Q.; Robinson, H.; Cai, T.; Tagle, D.A.; Li, J. Biochemical and structural properties of mouse KAT III. Mol. Cell Biol. 2009, 29, 784–793. [Google Scholar] [CrossRef] [Green Version]
- Rossi, F.; Miggiano, R.; Ferraris, D.M.; Rizzi, M. The synthesis of kynurenic acid in mammals: An updated kynurenine aminotransferase structural KATalogue. Front. Mol. Biosci. 2019, 6, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, Q.; Cai, T.; Tagle, D.A.; Robinson, H.; Li, J. Substrate specificity and structure of human aminoadipate aminotransferase/kynurenine aminotransferase II. Biosci. Rep. 2008, 28, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Yang, C.; Lu, J.; Zhang, Y.; Li, J. Metabolism of oxalate in humans: A potential role kynurenine aminotransferase/glutamine transaminase/cysteine conjugate β-lyase plays in hyperoxaluria. Curr. Med. Chem. 2019, 26, 4944–4963. [Google Scholar] [CrossRef]
- Hanna, P.E.; Anders, M.W. The mercapturic acid pathway. Crit. Rev. Toxicol. 2019, 49, 819–929. [Google Scholar] [CrossRef] [Green Version]
- Commandeur, J.N.M.; Andreadou, I.; Rooseboom, M.; Out, M.; de Leur, L.J.; Groot, E.; Vermeulen, N.P.E. Bioactivation of selenocysteine Se-conjugates by a highly purified rat renal cysteine conjugate β-lyase/glutamine transaminase K. J. Pharmacol. Exp. Ther. 2000, 294, 753–761. [Google Scholar]
- Lee, J.I.; Nian, H.; Cooper, A.J.L.; Sinha, R.; Dai, J.; Bisson, W.H.; Dashwood, R.H.; Pinto, J.T. α-Keto acid metabolites of naturally occurring organoselenium compounds as inhibitors of histone deacetylase in human prostate cancer cells. Cancer Prev. Res. 2009, 2, 683–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, J.T.; Lee, J.I.; Sinha, R.; MacEwan, M.E.; Cooper, A.J.L. Chemopreventive mechanisms of α-keto acid metabolites of naturally occurring organoselenium compounds. Amino Acids 2011, 41, 29–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selvam, A.K.; Jawad, R.; Gramignoli, R.; Achour, A.; Salter, H.; Björnstedt, M.A. Novel mRNA-mediated and microRNA-guided approach to specifically eradicate drug-resistant hepatocellular carcinoma cell lines by Se-methylselenocysteine. Antioxidants 2021, 10, 1094. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L.; Pinto, J.T.; Krasnikov, B.F.; Niatsetskaya, Z.V.; Han, Q.; Li, J.; Vauzour, D.; Spencer, J.P. Substrate specificity of human glutamine transaminase K as an aminotransferase and as a cysteine S-conjugate β-lyase. Arch. Biochem. Biophys. 2008, 474, 72–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruzat, V.; Macedo Rogero, M.; Keane, K.N.; Curi, R.; Newsholme, P. Glutamine: Metabolism and immune function, supplementation and clinical translation. Nutrients 2018, 10, 1564. [Google Scholar] [CrossRef] [Green Version]
- Ohta, Y.; Kubo, H.; Yashiro, K.; Ohashi, K.; Tsuzuki, Y.; Wada, N.; Yamamoto, Y.; Saito, K. Effect of water-immersion restraint stress on tryptophan catabolism through the kynurenine pathway in rat tissues. J. Physiol. Sci. 2017, 67, 361–372. [Google Scholar] [CrossRef]
- Ostapiuk, A.; Urbanska, E.M. Kynurenic acid in neurodegenerative disorders-unique neuroprotection or double-edged sword? CNS Neurosci. Ther. 2022, 28, 19–35. [Google Scholar] [CrossRef]
- Jacobs, K.R.; Castellano-Gonzalez, G.; Guillemin, G.J.; Lovejoy, D.B. Major developments in the design of inhibitors along the kynurenine pathway. Curr. Med. Chem. 2017, 24, 2471–2495. [Google Scholar] [CrossRef] [Green Version]
- Nematollahi, A.; Sun, G.; Jayawickrama, G.S.; Church, W.B. Kynurenine aminotransferase isozyme inhibitors: A review. Int. J. Mol. Sci. 2016, 17, 946. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.H.; Chung, M.Y.; Chen, W.B.; Chien, C.H. Growth inhibitory effect of the human NIT2 gene and its allelic imbalance in cancer. FEBS J. 2007, 274, 2946–2956. [Google Scholar] [CrossRef]
- Krasnikov, B.F.; Nostramo, R.; Pinto, J.T.; Cooper, A.J.L. Assay and purification of ω-amidase/Nit2, a ubiquitously expressed putative tumor suppressor, that catalyzes the deamidation of the α-keto acid analogues of glutamine and asparagine. Anal. Biochem. 2009, 391, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Jaisson, S.; Veiga-da-Cunha, M.; Van Schaftingen, E. Molecular identification of ω-amidase, the enzyme that is functionally coupled with glutamine transaminases, as the putative tumor suppressor Nit2. Biochimie 2009, 91, 1066–1071. [Google Scholar] [CrossRef]
- Krasnikov, B.F.; Chien, C.-H.; Nostramo, R.; Pinto, J.T.; Nieves, E.; Callaway, M.; Sun, J.; Huebner, K.; Cooper, A.J.L. Identification of the putative tumor suppressor Nit2 as ω-amidase, an enzyme metabolically linked to glutamine and asparagine transamination. Biochimie 2009, 91, 1072–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chien, C.H.; Gao, Q.Z.; Cooper, A.J.L.; Lyu, J.H.; Sheu, S.Y. Structural insights into the catalytic active site and activity of human Nit2/ω-amidase: Kinetic assay and molecular dynamics simulation. J. Biol. Chem. 2012, 287, 25715–25726. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epova, E.Y.; Shevelev, A.B.; Shurubor, Y.I.; Cooper, A.J.L.; Biryukova, Y.K.; Bogdanova, E.S.; Tyno, Y.Y.; Lebedeva, A.A.; Krasnikov, B.F. A novel efficient producer of human ω-amidase (Nit2) in Escherichia coli. Anal. Biochem. 2021, 632, 114332. [Google Scholar] [CrossRef]
- Häussinger, D.; Stehle, T.; Gerok, W. Glutamine metabolism in isolated perfused rat liver. The transamination pathway. Biol. Chem. Hoppe Seyler 1985, 366, 527–536. [Google Scholar] [CrossRef]
- Botman, D.; Tigchelaar, W.; Van Noorden, C.J. Determination of phosphate-activated glutaminase activity and its kinetics in mouse tissues using metabolic mapping (quantitative enzyme histochemistry). J. Histochem. Cytochem. 2014, 62, 813–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darmaun, D.; Matthews, D.E.; Bier, D.M. Glutamine and glutamate kinetics in humans. Am. J. Physiol. 1986, 251, E117–E126. [Google Scholar] [CrossRef]
- Duffy, T.E.; Vergara, F.; Plum, F. α-Ketoglutaramate in hepatic encephalopathy. Res. Publ. Assoc. Res. Nerv. Ment. Dis. 1974, 53, 39–52. [Google Scholar]
- Kuhara, T.; Inoue, Y.; Ohse, M.; Krasnikov, B.F.; Cooper, A.J.L. Urinary 2-hydroxy-5-oxoproline, the lactam form of α-ketoglutaramate, is markedly increased in urea cycle disorders. Anal. Bioanal. Chem. 2011, 400, 1843–1851. [Google Scholar] [CrossRef] [Green Version]
- Cooper, A.J.L.; Kuhara, T. α-Ketoglutaramate: An overlooked metabolite of glutamine and a biomarker for hepatic encephalopathy and inborn errors of the urea cycle. Metab. Brain Dis. 2014, 29, 991–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuhara, T.; Ohse, M.; Inoue, Y.; Cooper, A.J.L. A GC/MS-based metabolomic approach for diagnosing citrin deficiency. Anal. Bioanal. Chem. 2011, 400, 1881–1894. [Google Scholar] [CrossRef] [PubMed]
- Butterworth, R.F. hepatic encephalopathy in cirrhosis: Pathology and pathophysiology. Drugs 2019, 79 (Suppl. 1), 17–21. [Google Scholar] [CrossRef] [Green Version]
- Brusilow, S.W.; Koehler, R.C.; Traystman, R.J.; Cooper, A.J.L. Astrocyte glutamine synthetase: Importance in hyperammonemic syndromes and potential target for therapy. Neurotherapeutics 2010, 7, 452–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Refardt, J.; den Hoed, C.M.; Langendonk, J.; Zandee, W.; Charehbili, A.; Feelders, R.A.; de Herder, W.W.; Brabander, T.; Hofland, J. Prognostic significance of hyperammonemia in neuroendocrine neoplasm patients with liver metastases. Endocr. Relat. Cancer 2022, 29, 241–250. [Google Scholar] [CrossRef]
- Davis, A.; Clarke, S.; Ayesa, S.; Chan, D.L. Marked improvement in hyperammonaemic encephalopathy from multimodal treatment of metastatic neuroendocrine tumour. BMJ Case Rep. 2021, 14, e241191. [Google Scholar] [CrossRef]
- Okamoto, K.; Nozawa, H.; Hongo, K.; Iida, Y.; Kawai, K.; Sasaki, K.; Murono, K.; Kita, Y.; Ishihara, Y.; Takabayashi, N.; et al. Risk factors of mFOLFOX6-induced hyperammonemia in patients with colorectal cancer: An observational study. Int. J. Clin. Oncol. 2021, 26, 1477–1484. [Google Scholar] [CrossRef]
- Luo, C.; Shen, G.; Liu, N.; Gong, F.; Wei, X.; Yao, S.; Liu, D.; Teng, X.; Ye, N.; Zhang, N.; et al. Ammonia drives dendritic cells into dysfunction. J. Immunol. 2014, 193, 1080–1089. [Google Scholar] [CrossRef] [Green Version]
- Udupa, S.; Nguyen, S.; Hoang, G.; Nguyen, T.; Quinones, A.; Pham, K.; Asaka, R.; Nguyen, K.; Zhang, C.; Elgogary, A.; et al. Upregulation of the glutaminase II pathway contributes to glutamate production upon glutaminase 1 inhibition in pancreatic cancer. Proteomics 2019, 19, e1800451. [Google Scholar] [CrossRef] [Green Version]
- Robinson, M.M.; McBryant, S.J.; Tsukamoto, T.; Rojas, C.; Ferraris, D.V.; Hamilton, S.K.; Hansen, J.C.; Curthoys, N.P. Novel mechanism of inhibition of rat kidney-type glutaminase by bis-2-(5-phenylacetamido-1,2,4-thiadiazol-2-yl)ethyl sulfide (BPTES). Biochem. J. 2007, 406, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Stalnecker, C.A.; Ulrich, S.M.; Li, Y.; Ramachandran, S.; McBrayer, M.K.; DeBerardinis, R.J.; Cerione, R.A.; Erickson, J.W. Mechanism by which a recently discovered allosteric inhibitor blocks glutamine metabolism in transformed cells. Proc. Natl. Acad. Sci. USA 2015, 112, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Stalnecker, C.; Zhang, C.; McDermott, L.A.; Iyer, P.; O’Neill, J.; Reimer, S.; Cerione, R.A.; Katt, W.P. Characterization of the interactions of potent allosteric inhibitors with glutaminase C, a key enzyme in cancer cell glutamine metabolism. J. Biol. Chem. 2018, 293, 3535–3545. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Ramachandran, S.; Nguyen, T.T.; Stalnecker, C.A.; Cerione, R.A.; Erickson, J.W. The activation loop and substrate-binding cleft of glutaminase C are allosterically coupled. J. Biol. Chem. 2020, 295, 1328–1337. [Google Scholar] [CrossRef] [PubMed]
- Milano, S.K.; Huang, Q.; Nguyen, T.T.; Ramachandran, S.; Finke, A.; Kriksunov, I.; Schuller, D.J.; Szebenyi, D.M.; Arenholz, E.; McDermott, L.A.; et al. New insights into the molecular mechanisms of glutaminase C inhibitors in cancer cells using serial room temperature crystallography. J. Biol. Chem. 2022, 298, 101535. [Google Scholar] [CrossRef]
- Albers, E. Metabolic characteristics and importance of the universal methionine salvage pathway recycling methionine from 5′-methylthioadenosine. IUBMB Life 2009, 61, 1132–1142. [Google Scholar] [CrossRef] [PubMed]
- Sekowska, A.; Ashida, H.; Danchin, A. Revisiting the methionine salvage pathway and its paralogues. Microb. Biotechnol. 2019, 12, 77–97. [Google Scholar] [CrossRef] [Green Version]
- Wray, J.W.; Abeles, R.H. The methionine salvage pathway in Klebsiella pneumoniae and rat liver. Identification and characterization of two novel dioxygenases. J. Biol. Chem. 1995, 270, 3147–3153. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Wensink, P.C.; Abeles, R.H. One protein, two enzymes. J. Biol. Chem. 1999, 274, 1193–1195. [Google Scholar] [CrossRef] [Green Version]
- Dai, Y.; Pochapsky, T.C.; Abeles, R.H. Mechanistic studies of two dioxygenases in the methionine salvage pathway of Klebsiella pneumoniae. Biochemistry 2001, 40, 6379–6387. [Google Scholar] [CrossRef]
- Backlund, P.S., Jr.; Chang, C.P.; Smith, R.A. Identification of 2-keto-4-methylthiobutyrate as an intermediate compound in methionine synthesis from 5′-methylthioadenosine. J. Biol. Chem. 1982, 257, 4196–4202. [Google Scholar] [CrossRef]
- Ellens, K.W.; Richardson, L.G.; Frelin, O.; Collins, J.; Ribeiro, C.L.; Hsieh, Y.F.; Mullen, R.T.; Hanson, A.D. Evidence that glutamine transaminase and ω-amidase potentially act in tandem to close the methionine salvage cycle in bacteria and plants. Phytochemistry 2015, 113, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Basu, I.; Cordovano, G.; Das, I.; Belbin, T.J.; Guha, C.; Schramm, V.L. A transition state analogue of 5′-methylthioadenosine phosphorylase induces apoptosis in head and neck cancers. J. Biol. Chem. 2007, 282, 21477–21486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basu, I.; Locker, J.; Cassera, M.B.; Belbin, T.J.; Merino, E.F.; Dong, X.; Hemeon, I.; Evans, G.B.; Guha, C.; Schramm, V.L. Growth and metastases of human lung cancer are inhibited in mouse xenografts by a transition state analogue of 5′-methylthioadenosine phosphorylase. J. Biol. Chem. 2011, 286, 4902–4911. [Google Scholar] [CrossRef] [Green Version]
- Firestone, R.S.; Feng, M.; Basu, I.; Peregrina, K.; Augenlicht, L.H.; Schramm, V.L. Transition state analogue of MTAP extends lifespan of APCMin/+ mice. Sci. Rep. 2021, 11, 8844. [Google Scholar] [CrossRef] [PubMed]
- Affronti, H.C.; Rowsam, A.M.; Pellerite, A.J.; Rosario, S.R.; Long, M.D.; Jacobi, J.J.; Bianchi-Smiraglia, A.; Boerlin, C.S.; Gillard, B.M.; Karasik, E.; et al. Pharmacological polyamine catabolism upregulation with methionine salvage pathway inhibition as an effective prostate cancer therapy. Nat. Commun. 2020, 11, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Higuero, J.Á.; Betancor-Fernández, I.; Mesa-Torres, N.; Muga, A.; Salido, E.; Pey, A.L. Structural and functional insights on the roles of molecular chaperones in the mistargeting and aggregation phenotypes associated with primary hyperoxaluria type I. Adv. Protein Chem. Struct. Biol. 2019, 114, 119–152. [Google Scholar]
- Miller, E.; Litwack, G. Purification, properties, and identity of liver mitochondrial tyrosine aminotransferase. J. Biol. Chem. 1971, 246, 3234–3240. [Google Scholar] [CrossRef]
- Shrawder, E.; Martinez-Carrion, M. Evidence of phenylalanine transaminase activity in the isoenzymes of aspartate transaminase. J. Biol. Chem. 1972, 247, 2486–2492. [Google Scholar] [CrossRef]
- Caligiore, F.; Zangelmi, E.; Vetro, C.; Kentache, T.; Dewulf, J.P.; Veiga-da-Cunha, M.; Van Schaftingen, E.; Bommer, G.; Peracchi, A. Human cytosolic transaminases: Side activities and patterns of discrimination towards physiologically available alternative substrates. Cell Mol. Life Sci. 2022, 79, 421. [Google Scholar] [CrossRef]
- Vettore, L.; Westbrook, R.L.; Tennant, D.A. New aspects of amino acid metabolism in cancer. Br. J. Cancer 2020, 122, 150–156. [Google Scholar] [CrossRef]
- Chisari, A.; Golán, I.; Campisano, S.; Gélabert, C.; Moustakas, A.; Sancho, P.; Caja, L. Glucose and amino acid metabolic dependencies linked to stemness and metastasis in different aggressive cancer types. Front. Pharmacol. 2021, 12, 723798. [Google Scholar] [CrossRef]
- Yoo, H.C.; Han, J.M. Amino acid metabolism in cancer drug resistance. Cells 2022, 11, 140. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L.; Ginos, J.Z.; Meister, A. Synthesis and properties of the α-keto acids. Chem. Rev. 1983, 83, 321–358. [Google Scholar] [CrossRef]
- Liu, S.; He, L.; Yao, K. The antioxidative function of alpha-ketoglutarate and its applications. BioMed Res. Internat. 2018, 2018, 3408467. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wu, J.; Tang, W.; Zhou, X.; Lin, Q.; Luo, F.; Yin, Y.; Li, T. Prevention of oxidative stress by α-ketoglutarate via activation of CAR signaling and modulation of the expression of key antioxidant-associated targets in vivo and in vitro. J. Agric. Food Chem. 2018, 66, 11273–11283. [Google Scholar] [CrossRef]
- Kindrick, J.D.; Mole, D.R. Hypoxic regulation of gene transcription and chromatin: Cause and effect. Int. J. Mol. Sci. 2020, 21, 8320. [Google Scholar] [CrossRef]
- Losman, J.A.; Koivunen, P.; Kaelin, W.G., Jr. 2-Oxoglutarate-dependent dioxygenases in cancer. Nat. Rev. Cancer. 2020, 20, 710–726. [Google Scholar] [CrossRef]
- Eilertsen, M.; Andersen, S.; Al-Saad, S.; Kiselev, Y.; Donnem, T.; Stenvold, H.; Pettersen, I.; Al-Shibili, K.; Richardsen, E.; Busund, L.T.; et al. Monocarboxylate transporters 1-4 in NSCLC: MCT1 is an independent prognostic marker for survival. PLoS ONE 2014, 9, e105038. [Google Scholar] [CrossRef] [Green Version]
- Singh, M.; Afonso, J.; Sharma, D.; Gupta, R.; Kumar, V.; Rani, R.; Baltazar, F.; Kumar, V. Targeting monocarboxylate transporters (MCTs) in cancer: How close are we to the clinics? Semin. Cancer Biol. 2023, 90, 1–14. [Google Scholar] [CrossRef]
- Hirschey, M.D.; DeBerardinis, R.J.; Diehl, A.M.E.; Drew, J.E.; Frezza, C.; Green, M.F.; Jones, L.W.; Ko, Y.H.; Le, A.; Lea, M.A.; et al. Target Validation Team. Dysregulated metabolism contributes to oncogenesis. Semin. Cancer Biol. 2015, 35, S129–S150. [Google Scholar] [CrossRef] [Green Version]
- Ko, Y.H.; Niedźwiecka, K.; Casal, M.; Pedersen, P.L.; Ułaszewski, S. 3-Bromopyruvate as a potent anticancer therapy in honor and memory of the late Professor André Goffeau. Yeast 2019, 36, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Cui, H. Targeting glutamine induces apoptosis: A cancer therapy approach. Int. J. Mol. Sci. 2015, 16, 22830–22855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altman, B.J.; Stine, Z.E.; Dang, C.V. From Krebs to clinic: Glutamine metabolism to cancer therapy. Nat. Rev. Cancer 2016, 16, 619–6134. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Le, A. Glutamine metabolism in cancer. Adv. Exp. Med. Biol. 2018, 1063, 13–32. [Google Scholar]
- Bernfeld, E.; Foster, D.A. Glutamine as an essential amino acid for KRas-driven cancer cells. Trends Endocrinol. Metab. 2019, 30, 357–368. [Google Scholar] [CrossRef]
- Obara-Michleka, M.; Szeliga, M. Targeting glutamine addiction in gliomas. Cancers 2020, 12, 310. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Copeland, C.; Le, A. Glutamine metabolism in cancer. Adv. Exp. Med. Biol. 2021, 1311, 17–38. [Google Scholar]
- Zhou, Y.; Yu, H.; Cheng, S.; Chen, Y.; He, L.; Ren, J.; He, X.; Chen, J.; Zheng, L.; Li, F. Glutamate dehydrogenase 1 mediated glutaminolysis sustains HCC cells survival under glucose deprivation. J. Cancer 2022, 13, 1061–1072. [Google Scholar] [CrossRef]
- Li, M.; Li, C.H.; Allen, A.; Stanley, C.A.; Smith, T.J. Glutamate dehydrogenase: Structure, allosteric regulation, and role in insulin homeostasis. Neurochem. Res. 2014, 39, 433–445. [Google Scholar] [CrossRef] [PubMed]
- da Veiga Moreira, J.; Hamraz, M.; Abolhassani, M.; Bigan, E.; Pérès, S.; Paulevé, L.; Levy Nogueira, M.; Steyaert, J.-M.; Schwartz, L. The redox status of cancer cells supports mechanisms behind the Warburg effect. Metabolites 2016, 6, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katt, W.P.; Lukey, M.J.; Cerione, R.A. A tale of two glutaminases: Homologous enzymes with distinct roles in tumorigenesis. Future Med. Chem. 2017, 9, 223–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Zhang, C.; Lin, M.; Zhu, W.; Liang, Y.; Hong, X.; Zhao, Y.; Young, K.H.; Hu, W.; Feng, Z. Glutaminase 2 negatively regulates the PI3K/AKT signaling and shows tumor suppression activity in human hepatocellular carcinoma. Oncotarget 2014, 5, 2635–2647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, S.; Venkatesh, D.; Kanda, H.; Nakayama, A.; Hosokawa, H.; Lee, E.; Miki, T.; Stockwell, B.R.; Yokote, K.; Tanaka, T.; et al. GLS2 is a tumor suppressor and a regulator of ferroptosis in hepatocellular carcinoma. Cancer Res. 2022, 82, 3209–3222. [Google Scholar] [CrossRef] [PubMed]
- Levine, M.S.; Holland, A.J. The impact of mitotic errors on cell proliferation and tumorigenesis. Genes Dev. 2018, 32, 620–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felig, P.; Pozefsky, T.; Marliss, E.; Cahill, G.F., Jr. Alanine: Key role in gluconeogenesis. Science 1970, 167, 1003–1004. [Google Scholar] [CrossRef]
- Montal, E.; White, R.M. Zebrafish as a new model to study the crosstalk between tumor and host metabolism. Trends Cancer 2021, 7, 661–663. [Google Scholar] [CrossRef]
- Naser, F.J.; Jackstadt, M.M.; Fowle-Grider, R.; Spalding, J.L.; Cho, K.; Stancliffe, E.; Doonan, S.R.; Kramer, E.T.; Yao, L.; Krasnick, B.; et al. Isotope tracing in adult zebrafish reveals alanine cycling between melanoma and liver. Cell Metab. 2021, 33, 1493–1504.e5. [Google Scholar] [CrossRef]
- Yang, C.S.; Stampouloglou, E.; Kingston, N.M.; Zhang, L.; Monti, S.; Varelas, X. Glutamine-utilizing transaminases are a metabolic vulnerability of TAZ/YAP-activated cancer cells. EMBO Rep. 2018, 19, e43577. [Google Scholar] [CrossRef]
- Thornburg, J.M.; Nelson, K.K.; Clem, B.F.; Lane, A.N.; Arumugam, S.; Simmons, A.; Eaton, J.W.; Telang, S.; Chesney, J. Targeting aspartate aminotransferase in breast cancer. Breast Cancer Res. 2008, 10, R84. [Google Scholar] [CrossRef]
- Borst, P. The malate-aspartate shuttle (Borst cycle): How it started and developed into a major metabolic pathway. IUBMB Life 2020, 72, 2241–2259. [Google Scholar] [CrossRef]
- McKenna, M.C.; Waagepetersen, H.S.; Schousboe, A.; Sonnewald, U. Neuronal and astrocytic shuttle mechanisms for cytosolic-mitochondrial transfer of reducing equivalents: Current evidence and pharmacological tools. Biochem. Pharmacol. 2006, 71, 399–407. [Google Scholar] [CrossRef]
- Lee, J.S.; Choi, J.; Lee, S.H.; Kang, J.H.; Ha, J.S.; Kim, H.Y.; Jang, H.; Yook, J.I.; Kim, S.Y. Oxoglutarate carrier inhibition reduced melanoma growth and invasion by reducing ATP production. Pharmaceutics 2020, 12, 1128. [Google Scholar] [CrossRef] [PubMed]
- Sweatt, A.J.; Wood, M.; Suryawan, A.; Wallin, R.; Willingham, M.C.; Hutson, S.M. Branched-chain amino acid catabolism: Unique segregation of pathway enzymes in organ systems and peripheral nerves. Am. J. Physiol. Endocrinol. Metab. 2004, 286, E64–E76. [Google Scholar] [CrossRef] [PubMed]
- Hutson, S.M.; Sweatt, A.J.; Lanoue, K.F. Branched-chain amino acid metabolism: Implications for establishing safe intakes. J. Nutr. 2005, 135, 1557S–1564S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, M.E.; Hutson, S.M. BCAA Metabolism and NH3 homeostasis. Adv. Neurobiol. 2016, 13, 99–132. [Google Scholar]
- Cole, J.T.; Sweatt, A.J.; Hutson, S.M. Expression of mitochondrial branched-chain aminotransferase and α-keto-acid dehydrogenase in rat brain: Implications for neurotransmitter metabolism. Front. Neuroanat. 2012, 6, 18. [Google Scholar] [CrossRef] [Green Version]
- Jung, M.K.; Okekunle, A.P.; Lee, J.E.; Sung, M.K.; Lim, Y.J. Role of branched-chain amino acid metabolism in tumor development and progression. J. Cancer Prev. 2021, 26, 237–243. [Google Scholar] [CrossRef]
- Siess, E.A.; Kientsch-Engel, R.I.; Wieland, O.H. Concentration of free oxaloacetate in the mitochondrial compartment of isolated liver cells. Biochem. J. 1984, 218, 171–176. [Google Scholar] [CrossRef] [Green Version]
- Duffy, T.E.; Howse, D.C.; Plum, F. Cerebral energy metabolism during experimental status epilepticus. J. Neurochem. 1975, 24, 925–934. [Google Scholar] [CrossRef]
- Howse, D.C.; Duffy, T.E. Control of the redox state of the pyridine nucleotides in the rat cerebral cortex. Effect of electroshock-induced seizures. J. Neurochem. 1975, 24, 935–940. [Google Scholar] [CrossRef]
- Plaitakis, A.; Kalef-Ezra, E.; Kotzamani, D.; Zaganas, I.; Spanaki, C. The glutamate dehydrogenase pathway and its roles in cell and tissue biology in health and disease. Biology 2017, 6, 11. [Google Scholar] [CrossRef] [Green Version]
- McKenna, M.C.; Stridh, M.H.; McNair, L.F.; Sonnewald, U.; Waagepetersen, H.S.; Schousboe, A. Glutamate oxidation in astrocytes: Roles of glutamate dehydrogenase and aminotransferases. J. Neurosci. Res. 2016, 94, 1561–1571. [Google Scholar] [CrossRef]
- Pamiljans, V.; Krishnaswamy, P.R.; Dumville, G.; Meister, A. Studies on the mechanism of glutamine synthesis; isolation and properties of the enzyme from sheep brain. Biochemistry 1962, 1, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L.; Nieves, E.; Coleman, A.E.; Filc-DeRicco, S.; Gelbard, A.S. Short-term metabolic fate of [13N]ammonia in rat liver in vivo. J. Biol. Chem. 1987, 262, 1073–1080. [Google Scholar] [CrossRef]
- Brosnan, J.T. Glutamate, at the interface between amino acid and carbohydrate metabolism. J. Nutr. 2000, 130 (Suppl. 4S), 988S–990S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Li, D.; Alesi, G.N.; Fan, J.; Kang, H.B.; Lu, Z.; Boggon, T.J.; Jin, P.; Yi, H.; Wright, E.R.; et al. Glutamate dehydrogenase 1 signals through antioxidant glutathione peroxidase 1 to regulate redox homeostasis and tumor growth. Cancer Cell 2015, 27, 257–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Chun, J.; Pan, C.; Kumar, A.; Zhang, G.; Ha, Y.; Li, D.; Alesi, G.N.; Kang, Y.; Zhou, L.; et al. The PLAG1-GDH1 axis promotes anoikis resistance and tumor metastasis through CamKK2-AMPK signaling in LKB1-deficient lung cancer. Mol. Cell 2018, 69, 87–99.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eales, K.L.; Hollinshead, K.E.; Tennant, D.A. Hypoxia and metabolic adaptation of cancer cells. Oncogenesis 2016, 5, e190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, M.P. Redox modulation by reversal of the mitochondrial nicotinamide nucleotide transhydrogenase. Cell Metab. 2015, 22, 363–365. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Zhuang, Z.; Wu, T.; Lin, J.C.; Liu, Z.X.; Zhou, L.F.; Dai, T.; Lu, L.; Ju, H.Q. Nicotinamide nucleotide transhydrogenase-mediated redox homeostasis promotes tumor growth and metastasis in gastric cancer. Redox Biol. 2018, 18, 246–255. [Google Scholar] [CrossRef]
- Cooper, A.J.L.; Meister, A. An appreciation of Professor Alexander, E. Braunstein. The discovery and scope of enzymatic transamination. Biochimie 1989, 71, 387–404. [Google Scholar] [CrossRef] [PubMed]
- Snell, E.E. From bacterial nutrition to enzyme structure: A personal odyssey. Annu. Rev. Biochem. 1993, 62, 1–26. [Google Scholar] [CrossRef] [PubMed]
- Meister, A. Transamination. Adv. Enzymol. Relat. Subj. Biochem. 1955, 16, 185–246. [Google Scholar] [PubMed]
- Shen, D.; Kruger, L.; Deatherage, T.; Denton, T.T. Synthesis of α-ketoglutaramic acid. Anal. Biochem. 2020, 607, 113862. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.A.; Unkefer, P.J. Preparation of 2-hydroxy-5-oxoproline and Analogs Thereof. U.S. Patent 6,288,240 B1, 11 September 2001. [Google Scholar]
- Deng, L.; Zhou, Z.H. Spontaneous conversions of glutamine, histidine and arginine into α-hydroxycarboxylates with NH4VO3 or V2O5. Dalton Trans. 2020, 49, 11921–11930. [Google Scholar] [CrossRef] [PubMed]
- Denton, T.T.; Cooper, A.J.L. Chemistry, biochemistry and clinical relevance of the glutamine metabolite α-ketoglutaramate/2-hydroxy-5-oxoproline. Aus. J. Chem. 2023, in press. [Google Scholar] [CrossRef]
- Wise, D.R.; Thompson, C.B. Glutamine addiction: A new therapeutic target in cancer. Trends Biochem. Sci. 2010, 35, 427–433. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [Green Version]
- Márquez, J.; Alonso, F.; Matés, J.M.; Segura, J.A.; Martín-Rufián, M.; Campos-Sandoval, J.A. Glutamine addiction in gliomas. Neurochem. Res. 2017, 42, 1735–1746. [Google Scholar] [CrossRef]
- Martins, F.; Gonçalves, L.G.; Pojo, M.; Serpa, J. Take advantage of glutamine anaplerosis, the kernel of the metabolic rewiring in malignant gliomas. Biomolecules 2020, 10, 1370. [Google Scholar] [CrossRef]
- Louie, M.C.; Ton, J.; Brady, M.L.; Le, D.T.; Mar, J.N.; Lerner, C.A.; Gerencser, A.A.; Mookerjee, S.A. Total cellular ATP production changes with primary substrate in MCF7 breast cancer cells. Front. Oncol. 2020, 10, 1703. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xue, R.; Jiang, R.T.; Meng, Q.H. Characterization of metabolic landscape in hepatocellular carcinoma. World J. Gastrointest. Oncol. 2021, 13, 1144–1156. [Google Scholar] [CrossRef] [PubMed]
- Sniegowski, T.; Korac, K.; Bhutia, Y.D.; Ganapathy, V. SLC6A14 and SLC38A5 drive the glutaminolysis and serine-glycine-one-carbon pathways in cancer. Pharmaceuticals 2021, 14, 216. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Pan, H.; Ren, F.; Chen, H.; Ren, P. Targeting ASCT2-mediated glutamine metabolism inhibits proliferation and promotes apoptosis of pancreatic cancer cells. Biosci. Rep. 2022, 42, BSR20212171. [Google Scholar] [CrossRef]
- Myint, Z.W.; Sun, R.C.; Hensley, P.J.; James, A.C.; Wang, P.; Strup, S.E.; McDonald, R.J.; Yan, D.; St Clair, W.H.; Allison, D.B. Evaluation of glutaminase expression in prostate adenocarcinoma and correlation with clinicopathologic parameters. Cancers 2021, 13, 2157. [Google Scholar] [CrossRef]
- Halama, A.; Suhre, K. Advancing cancer treatment by targeting glutamine metabolism—A roadmap. Cancers 2022, 14, 553. [Google Scholar] [CrossRef]
- Choi, S.Y.C.; Ribeiro, C.F.; Wang, Y.; Loda, M.; Plymate, S.R.; Uo, T. Druggable Metabolic Vulnerabilities are exposed and masked during progression to castration resistant prostate cancer. Biomolecules 2022, 12, 1590. [Google Scholar] [CrossRef]
- Jiménez-Alonso, J.J.; López-Lázaro, M. Dietary manipulation of amino acids for cancer therapy. Nutrients 2023, 15, 2879. [Google Scholar] [CrossRef]
- Mardashev, S.R.; Lerman, M.I.; Benyumovich, M.S. Glutamine transaminase in brain tissue preparations and in cells of a human strain of dedifferentiated astrocytoma. Fed. Proc. Transl. 1963, 22, 976–977. [Google Scholar]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. Subcellular map of the human proteome. Science 2017, 356, 6340. [Google Scholar] [CrossRef]
- Ni, R.; Li, Z.; Li, L.; Peng, D.; Ming, Y.; Li, L.; Liu, Y. Rethinking glutamine metabolism and the regulation of glutamine addiction by oncogenes in cancer. Front. Oncol. 2023, 13, 1143798. [Google Scholar] [CrossRef] [PubMed]
- Peracchi, A.; Veiga-da-Cunha, M.; Kuhara, T.; Ellens, K.W.; Paczia, N.; Stroobant, V.; Seliga, A.K.; Marlaire, S.; Jaisson, S.; Bommer, G.T.; et al. Nit1 is a metabolite repair enzyme that hydrolyzes deaminated glutathione. Proc. Natl. Acad. Sci. USA 2017, 114, E3233–E3242. [Google Scholar] [CrossRef] [PubMed]
- Semba, S.; Han, S.Y.; Qin, H.R.; McCorkell, K.A.; Iliopoulos, D.; Pekarsky, Y.; Druck, T.; Trapasso, F.; Croce, C.M.; Huebner, K. Biological functions of mammalian Nit1, the counterpart of the invertebrate NitFhit Rosetta stone protein, a possible tumor suppressor. J. Biol. Chem. 2006, 281, 28244–28253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Okumura, H.; Yearsley, M.; Frankel, W.; Fong, L.Y.; Druck, T.; Huebner, K. Nit1 and Fhit tumor suppressor activities are additive. J. Cell Biochem. 2009, 107, 1097–1106. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.; Zhang, J.; Lu, Y.; Li, X.; Zhang, W.; Zhang, W.; Lin, W.; Zheng, L.; Li, X. NIT1 suppresses tumour proliferation by activating the TGFbeta1-Smad2/3 signalling pathway in colorectal cancer. Cell Death Dis. 2018, 9, 263. [Google Scholar] [CrossRef] [Green Version]
- Zheng, B.; Chai, R.; Yu, X. Downregulation of NIT2 inhibits colon cancer cell proliferation and induces cell cycle arrest through the caspase-3 and PARP pathways. Int. J. Mol. Med. 2015, 35, 1317–1322. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Wang, Z.; Feng, C. NIT2 overexpression predicts poor prognosis in tongue squamous cell carcinoma patients. Mol. Biol. Rep. 2020, 47, 1553–1561. [Google Scholar] [CrossRef] [Green Version]
- Duthie, S.J.; Mavrommatis, Y.; Rucklidge, G.; Reid, M.; Duncan, G.; Moyer, M.P.; Pirie, L.P.; Bestwick, C.S. The response of human colonocytes to folate deficiency in vitro: Functional and proteomic analyses. J. Proteome Res. 2008, 7, 3254–3266. [Google Scholar] [CrossRef]
- Wang, D.; Jensen, R.H.; Williams, K.E.; Pallavicini, M.G. Differential protein expression in MCF7 breast cancer cells transfected with ErbB2, neomycin resistance and luciferase plus yellow fluorescent protein. Proteomics 2004, 4, 2175–2183. [Google Scholar] [CrossRef]
- Pham, K.; Hanaford, A.R.; Poore, B.A.; Maxwell, M.J.; Sweeney, H.; Parthasarathy, A.; Alt, J.; Rais, R.; Slusher, B.S.; Eberhart, C.G.; et al. Comprehensive metabolic profiling of myc-amplified medulloblastoma tumors reveals key dependencies on amino acid, tricarboxylic acid and hexosamine pathways. Cancers 2022, 14, 1311. [Google Scholar] [CrossRef]
- Weil, A.G.; Wang, A.C.; Westwick, H.J.; Ibrahim, G.M.; Ariani, R.T.; Crevier, L.; Perreault, S.; Davidson, T.; Tseng, C.-H.; Fallah, A. Survival in pediatric medulloblastoma: A population-based observational study to improve prognostication. J. Neurooncol. 2016, 132, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Eberhart, C.G.; Kratz, J.; Wang, Y.; Summers, K.; Stearns, D.; Cohen, K.; Dang, C.V.; Burger, P.C. Histopathological and molecular prognostic markers in medulloblastoma. J. Neuropathol. Exp. Neurol. 2004, 63, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wise, D.R.; DeBerardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [PubMed]
- Gao, P.; Tchernyshyov, I.; Chang, T.-C.; Lee, Y.-S.; Kita, K.; Ochi, T.; Zeller, K.; De Marzo, A.M.; Van Eyk, J.E.; Mendell, J.T.; et al. c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 2009, 458, 762–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, Y.A.; Hong, J.; Asaka, R.; Asaka, S.; Hsu, F.C.; Suryo Rahmanto, Y.; Jung, J.G.; Chen, Y.W.; Yen, T.T.; Tomaszewski, A.; et al. Inhibition of the MYC-regulated glutaminase metabolic axis is an effective synthetic lethal approach for treating chemoresistant ovarian cancers. Cancer Res. 2020, 80, 4514–4526. [Google Scholar] [CrossRef]
- Pan, T.; Gao, L.; Wu, G.; Shen, G.; Xie, S.; Wen, H.; Yang, J.; Zhou, Y.; Tu, Z.; Qian, W. Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem. Biophys. Res. Commun. 2015, 456, 452–458. [Google Scholar] [CrossRef]
- Zhang, J.; Mao, S.Y.; Guo, Y.D.; Wu, Y.; Yao, X.D.; Huang, Y. Inhibition of GLS suppresses proliferation and promotes apoptosis in prostate cancer. Biosci. Rep. 2019, 39, 181826. [Google Scholar] [CrossRef] [Green Version]
- Kline, E.E.; Treat, E.G.; Averna, T.A.; Davis, M.S.; Smith, A.Y.; Sillerud, L.O. Citrate concentrations in human seminal fluid and expressed prostatic fluid determined via 1H nuclear magnetic resonance spectroscopy outperform prostate specific antigen in prostate cancer detection. J. Urol. 2006, 176, 2274–2279. [Google Scholar] [CrossRef]
- Franklin, R.B.; Feng, P.; Milon, B.; Desouki, M.M.; Singh, K.K.; Kajdacsy-Balla, A.; Bagasra, O.; Costello, L.C. hZIP1 zinc uptake transporter down regulation and zinc depletion in prostate cancer. Mol. Cancer 2005, 4, 32. [Google Scholar] [CrossRef] [Green Version]
- Costello, L.C.; Franklin, R.B. A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch. Biochem. Biophys. 2016, 611, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Costello, L.C.; Franklin, R.B.; Feng, P. Mitochondrial function, zinc, and intermediary metabolism relationships in normal prostate and prostate cancer. Mitochondrion 2005, 5, 143–153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eidelman, E.; Twum-Ampofo, J.; Ansari, J.; Siddiqui, M.M. The metabolic phenotype of prostate cancer. Front. Oncol. 2017, 7, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams-Ashman, H.G.; Pegg, A.E.; Lockwood, D.H. Mechanisms and regulation of polyamine and putrescine biosynthesis in male genital glands and other tissues of mammals. Adv. Enzyme Regul. 1969, 7, 291–323. [Google Scholar] [CrossRef]
- Pegg, A.E.; Lockwood, D.H.; Williams-Ashman, H.G. Concentrations of putrescine and polyamines and their enzymic synthesis during androgen-induced prostatic growth. Biochem. J. 1970, 117, 17–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purvis, K.; Magnus, O.; Mørkås, L.; Abyholm, T.; Rui, H. Ejaculate composition after masturbation and coitus in the human male. Int. J. Androl. 1986, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Lynch, M.J.; Nicholson, J.K. Proton MRS of human prostatic fluid: Correlations between citrate, spermine, and myo-inositol levels and changes with disease. Prostate 1997, 30, 248–255. [Google Scholar] [CrossRef]
- Nishio, T.; Sugino, K.; Yoshikawa, Y.; Matsumoto, M.; Oe, Y.; Sadakane, K.; Yoshikawa, K. K+ promotes the favorable effect of polyamine on gene expression better than Na. PLoS ONE 2020, 15, e0238447. [Google Scholar] [CrossRef]
- Stanbrough, M.; Bubley, G.J.; Ross, K.; Golub, T.R.; Rubin, M.A.; Penning, T.M.; Febbo, P.G.; Balk, S.P. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006, 66, 2815–2825. [Google Scholar] [CrossRef] [Green Version]
- Byrns, M.C.; Mindnich, R.; Duan, L.; Penning, T.M. Overexpression of aldo-keto reductase 1C3 (AKR1C3) in LNCaP cells diverts androgen metabolism towards testosterone resulting in resistance to the 5α-reductase inhibitor finasteride. J. Steroid Biochem. Mol. Biol. 2012, 130, 7–15. [Google Scholar] [CrossRef] [Green Version]
- Dorai, T.; Shah, A.; Summers, F.; Mathew, R.; Huang, J.; Hsieh, T.-C.; Wu, J.M. NRH:quinone oxidoreductase 2 (NQO2) and glutaminase (GLS) both play a role in large extracellular vesicles (LEV) formation in preclinical LNCaP-C4-2B prostate cancer model of progressive metastasis. Prostate 2018, 78, 1181–1195. [Google Scholar] [CrossRef] [Green Version]
- Minciacchi, V.R.; You, S.; Spinelli, C.; Morley, S.; Zandian, M.; Aspuria, P.J.; Cavallini, L.; Ciardiello, C.; Reis Sobreiro, M.; Morello, M.; et al. Large oncosomes contain distinct protein cargo and represent a separate functional class of tumor derived extracellular vesicles. Oncotarget 2015, 6, 11327–11341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wendler, F.; Stamp, G.W.; Chiamas, G. Tumor-stromal cell communication: Small vesicles signal big changes. Trends Cancer 2016, 2, 326–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagner, T.; Spinelli, C.; Minciacchi, V.R.; Balaj, L.; Zandian, M.; Conley, A.; Zijlstra, A.; Freeman, M.R.; Demichelis, F.; De, S.; et al. Large extracellular vesicles carry most of the tumour DNA circulating in prostate cancer patient plasma. J. Extracell. Vesicles 2018, 7, 1505403. [Google Scholar] [CrossRef] [Green Version]
- Ippolito, L.; Morandi, A.; Taddei, M.L.; Parri, M.; Comito, G.; Iscaro, A.; Raspollini, M.R.; Magherini, F.; Rapizzi, E.; Masquelier, J.; et al. Cancer-associated fibroblasts promote prostate cancer malignancy via metabolic rewiring and mitochondrial transfer. Oncogene 2019, 38, 5339–5355. [Google Scholar] [CrossRef]
- Wu, D.; Zhuo, L.; Wang, X. Metabolic reprogramming of carcinoma-associated fibroblasts and its impact on metabolic heterogeneity of tumors. Semin. Cell Dev. Biol. 2017, 64, 125–131. [Google Scholar] [CrossRef]
- von Ahrens, D.; Bhagat, T.D.; Nagrath, D.; Maitra, A.; Verma, A. The role of stromal cancer-associated fibroblasts in pancreatic cancer. J. Hematol. Oncol. 2017, 10, 76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Outschoorn, U.E.; Lisanti, M.P.; Sotgia, F. Catabolic cancer-associated fibroblasts transfer energy and biomass to anabolic cancer cells, fueling tumor growth. Semin. Cancer Biol. 2014, 25, 47–60. [Google Scholar] [CrossRef]
- Tajan, M.; Vousden, K.H. The quid pro quo of the tumor/stromal interaction. Cell Metab. 2016, 24, 645–646. [Google Scholar] [CrossRef] [Green Version]
- Avagliano, A.; Granato, G.; Ruocco, M.R.; Romano, V.; Belviso, I.; Carfora, A.; Montagnani, S.; Arcucci, A. Metabolic reprogramming of cancer associated fibroblasts: The slavery of stromal fibroblasts. Biomed. Res. Int. 2018, 2018, 6075403. [Google Scholar] [CrossRef] [Green Version]
- Harder, L.M.; Bunkenborg, J.; Andersen, J.S. Inducing autophagy: A comparative phosphoproteomic study of the cellular response to ammonia and rapamycin. Autophagy 2014, 10, 339–355. [Google Scholar] [CrossRef] [Green Version]
- Schiliro, C.; Firestein, B.L. Mechanisms of metabolic reprogramming in cancer cells supporting enhanced growth and proliferation. Cells. 2021, 10, 1056. [Google Scholar] [CrossRef] [PubMed]
- McGuirk, S.; Audet-Delage, Y.; St-Pierre, J. metabolic fitness and plasticity in cancer progression. Trends Cancer. 2020, 6, 49–61. [Google Scholar] [CrossRef] [PubMed]
- Hönigova, K.; Navratil, J.; Peltanova, B.; Polanska, H.H.; Raudenska, M.; Masarik, M. Metabolic tricks of cancer cells. Biochim. Biophys. Acta Rev. Cancer. 2022, 1877, 188705. [Google Scholar] [CrossRef]
- Sivanand, S.; Vander Heiden, M.G. Emerging roles for branched-chain amino acid metabolism in cancer. Cancer Cell. 2020, 37, 147–156. [Google Scholar] [CrossRef]
- Peng, H.; Wang, Y.; Luo, W. Multifaceted role of branched-chain amino acid metabolism in cancer. Oncogene 2020, 39, 6747–6756. [Google Scholar] [CrossRef] [PubMed]
- Ananieva, E.A.; Wilkinson, A.C. Branched chain amino acid metabolism in cancer. Curr. Opin. Clin. Nutr. Metab. Care 2018, 21, 64–70. [Google Scholar] [CrossRef] [Green Version]
- Paquette, M.; El-Houjeiri, L.; Pause, A. mTOR Pathways in cancer and autophagy. Cancers 2018, 10, 18. [Google Scholar] [CrossRef]
- Durán, R.V.; Hall, M.N. Glutaminolysis feeds mTORC1. Cell Cycle 2012, 11, 4107–4108. [Google Scholar] [CrossRef] [Green Version]
- Amaravadi, R.K.; Kimmelman, A.C.; Debnath, J. Targeting autophagy in cancer: Recent advances and future directions. Rev. Cancer Discov. 2019, 9, 1167–1181. [Google Scholar] [CrossRef]
- Otto, A.M. Metabolic constants and plasticity of cancer cells in a limiting glucose and glutamine microenvironment-A pyruvate perspective. Front. Oncol. 2020, 10, 596197. [Google Scholar] [CrossRef]
- Zhang, B.; Peng, H.; Zhou, M.; Bao, L.; Wang, C.; Cai, F.; Zhang, H.; Wang, J.E.; Niu, Y.; Chen, Y.; et al. Targeting BCAT1 combined with α-ketoglutarate triggers metabolic synthetic lethality in glioblastoma. Cancer Res. 2022, 82, 2388–2402. [Google Scholar] [CrossRef] [PubMed]
- Raffel, S.; Falcone, M.; Kneisel, N.; Hansson, J.; Wang, W.; Lutz, C.; Bullinger, L.; Poschet, G.; Nonnenmacher, Y.; Barnert, A.; et al. BCAT1 restricts αKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 2017, 551, 384–388. [Google Scholar] [CrossRef]
- Mosier, J.A.; Schwager, S.C.; Boyajian, D.A.; Reinhart-King, C.A. Cancer cell metabolic plasticity in migration and metastasis. Clin. Exp. Metastasis 2021, 38, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Lehuédé, C.; Dupuy, F.; Rabinovitch, R.; Jones, R.G.; Siegel, P.M. Metabolic plasticity as a determinant of tumor growth and metastasis. Cancer Res. 2016, 76, 5201–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knott, S.R.V.; Wagenblast, E.; Khan, S.; Kim, S.Y.; Soto, M.; Wagner, M.; Turgeon, M.O.; Fish, L.; Erard, N.; Gable, A.L.; et al. Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 2018, 554, 378–381. [Google Scholar] [CrossRef] [PubMed]
- Liao, J.; Liu, P.P.; Hou, G.; Shao, J.; Yang, J.; Liu, K.; Lu, W.; Wen, S.; Hu, Y.; Huang, P. Regulation of stem-like cancer cells by glutamine through β-catenin pathway mediated by redox signaling. Mol. Cancer 2017, 16, 51. [Google Scholar] [CrossRef] [Green Version]
- Shang, S.; Hua, F.; Hu, Z.-W. The regulation of β-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, N.N.; Hui, S.; Ghergurovich, J.M.; Fan, J.; Intlekofer, A.M.; White, R.M.; Rabinowitz, J.D.; Thompson, C.B.; Zhang, J. As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metab. 2018, 27, 428–438. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Fan, J.; Venneti, S.; Cross, J.R.; Takagi, T.; Bhinder, B.; Djaballah, H.; Kanai, M.; Cheng, E.H.; Judkins, A.R.; et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol. Cell 2014, 56, 205–218. [Google Scholar] [CrossRef] [Green Version]
- Krall, A.S.; Xu, S.; Graeber, T.G.; Braas, D.; Christofk, H.R. Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat. Commun. 2016, 7, 11457. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.; Rinaldi, S.; Scalbert, A.; Ferrari, P.; Achaintre, D.; Gunter, M.J.; Appleby, P.N.; Key, T.J.; Travis, R.C. Plasma concentrations and intakes of amino acids in male meat-eaters, fish-eaters, vegetarians and vegans: A cross-sectional analysis in the EPIC-Oxford cohort. Eur. J. Clin. Nutr. 2016, 70, 306–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooper, A.J.L.; Dorai, T.; Pinto, J.T.; Denton, T.T. The metabolic importance of the overlooked asparaginase II pathway. Anal. Biochem. 2022, 644, 114084. [Google Scholar] [CrossRef] [PubMed]
- Cooper, A.J.L. Asparagine transaminase from rat liver. J. Biol. Chem. 1977, 252, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wyckoff, J.; Wang, W.; Lin, E.Y.; Wang, Y.; Pixley, F.; Stanley, E.R.; Graf, T.; Pollard, J.W.; Segall, J.; Condeelis, J. A paracrine loop between tumor cells and macrophages is required for tumor cell migration in mammary tumors. Cancer Res. 2004, 64, 7022–7029. [Google Scholar] [CrossRef] [Green Version]
- Salem, A.F.; Whitaker-Menezes, D.; Lin, Z.; Martinez-Outschoorn, U.E.; Tanowitz, H.B.; Al-Zoubi, M.S.; Howell, A.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Two-compartment tumor metabolism: Autophagy in the tumor microenvironment and oxidative mitochondrial metabolism (OXPHOS) in cancer cells. Cell Cycle 2012, 11, 2545–2556. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Achreja, A.; Yeung, T.L.; Mangala, L.S.; Jiang, D.; Han, C.; Baddour, J.; Marini, J.C.; Ni, J.; Nakahara, R.; et al. Targeting stromal glutamine synthetase in tumors disrupts tumor microenvironment-regulated cancer cell growth. Cell Metab. 2016, 24, 685–700. [Google Scholar] [CrossRef] [Green Version]
- Ko, Y.-H.; Lin, Z.; Flomenberg, N.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P.; Martinez-Outschoorn, U.E. Glutamine fuels a vicious cycle of autophagy in the tumor stroma and oxidative mitochondrial metabolism in epithelial cancer cells. Cancer Biol. Ther. 2011, 12, 1085–1097. [Google Scholar] [CrossRef] [Green Version]
- Eng, C.H.; Abraham, R.T. Glutaminolysis yields a metabolic by-product that stimulates autophagy. Autophagy 2010, 6, 968–970. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.M.; Lee, Y.K.; Koo, J.S. Expression of glutamine metabolism-related proteins in thyroid cancer. Oncotarget 2016, 7, 53628–53641. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Kim, D.H.; Jung, W.-H.; Koo, J.S. Expression of glutamine metabolism-related proteins according to molecular subtype of breast cancer. Endocr. Relat. Cancer 2013, 20, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [Green Version]
- Vanhove, K.; Derveaux, E.; Graulus, G.J.; Mesotten, L.; Thomeer, M.; Noben, J.P.; Guedens, W.; Adriaensens, P. Glutamine addiction and therapeutic strategies in lung cancer. Int. J. Mol. Sci. 2019, 20, 252. [Google Scholar] [CrossRef] [Green Version]
- Magill, G.B.; Myers, W.P.; Reilly, H.C.; Putnam, R.C.; Magill, J.W.; Sykes, M.P.; Escher, G.C.; Karnofsky, D.A.; Bucgenal, J.H. Pharmacological and initial therapeutic observations on 6-diazo-5-oxo-l-norleucine (DON) in human neoplastic disease. Cancer 1957, 10, 1138–1150. [Google Scholar] [CrossRef] [PubMed]
- Livingston, R.B.; Venditti, J.M.; Cooney, D.A.; Carter, S.K. Glutamine antagonists in chemotherapy. Adv. Pharmacol. Chemother. 1970, 8, 57–120. [Google Scholar] [PubMed]
- Catane, R.; Von Hoff, D.D.; Glaubiger, D.L.; Muggia, F.M. Azaserine, DON, and azotomycin: Three diazo analogs of l-glutamine with clinical antitumor activity. Cancer Treat. Rep. 1979, 63, 1033–1038. [Google Scholar]
- Thangavelu, K.; Chong, Q.Y.; Low, B.C.; Sivaraman, J. Structural basis for the active site inhibition mechanism of human kidney-type glutaminase (KGA). Sci. Rep. 2014, 4, 3827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemberg, K.M.; Vornov, J.J.; Rais, R.; Slusher, B.S. We’re not “DON” yet: Optimal dosing and prodrug delivery of 6-diazo-5-oxo-l-norleucine. Mol. Cancer Ther. 2018, 17, 1824–1832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Earhart, R.H.; Amato, D.J.; Chang, A.Y.; Borden, E.C.; Shiraki, M.; Dowd, M.E.; Comis, R.L.; Davis, T.E.; Smith, T.J. Phase II trial of 6-diazo-5-oxo-l-norleucine versus aclacinomycin-A in advanced sarcomas and mesotheliomas. Investig. New Drugs 1990, 8, 113–119. [Google Scholar] [CrossRef]
- Lynch, G.; Kemeny, N.; Casper, E. Phase II evaluation of DON (6-diazo-5-oxo-l-norleucine) in patients with advanced colorectal carcinoma. Am. J. Clin. Oncol. 1982, 5, 541–543. [Google Scholar] [CrossRef]
- Rubin, J.; Sorensen, S.; Schutt, A.J.; van Hazel, G.A.; O’Connell, M.J.; Moertel, C.G. A phase II study of 6-diazo-5-oxo-l-norleucine (DON, NSC-7365) in advanced large bowel carcinoma. Am. J. Clin. Oncol. 1983, 6, 325–326. [Google Scholar] [CrossRef] [PubMed]
- Windmueller, H.G.; Spaeth, A.E. Respiratory fuels and nitrogen metabolism in vivo in small intestine of fed rats. Quantitative importance of glutamine, glutamate, and aspartate. J. Biol. Chem. 1980, 255, 107–112. [Google Scholar] [CrossRef]
- Hanaford, A.R.; Alt, J.; Rais, R.; Wang, S.Z.; Kaur, H.; Thorek, D.L.J.; Eberhart, C.G.; Slusher, B.S.; Martin, A.M.; Raabe, E.H. Orally bioavailable glutamine antagonist prodrug JHU-083 penetrates mouse brain and suppresses the growth of MYC-driven medulloblastoma. Transl. Oncol. 2019, 12, 1314–1322. [Google Scholar] [CrossRef]
- Pham, K.; Maxwell, M.J.; Sweeney, H.; Alt, J.; Rais, R.; Eberhart, C.G.; Slusher, B.S.; Raabe, E.H. Novel Glutamine antagonist JHU395 suppresses MYC-driven medulloblastoma growth and induces apoptosis. J. Neuropathol. Exp. Neurol. 2021, 80, 336–344. [Google Scholar] [CrossRef]
- Silva Teixeira, C.S.; Sousa, S.F.; Cerqueira, N.M.F.S.A. An unusual cys-glu-lys catalytic triad is responsible for the catalytic mechanism of the nitrilase superfamily: A QM/MM Study on Nit2. ChemPhysChem 2021, 22, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Elgogary, A.; Xu, Q.; Poore, B.; Alt, J.; Zimmermann, S.C.; Zhao, L.; Fu, J.; Chen, B.; Xia, S.; Liu, Y.; et al. Combination therapy with BPTES nanoparticles and metformin targets the metabolic heterogeneity of pancreatic cancer. Proc. Natl. Acad. Sci. USA 2016, 113, E5328–E5336. [Google Scholar] [CrossRef]
- Harding, J.J.; Telli, M.L.; Munster, P.N.; Le, M.H.; Molineaux, C.; Bennett, M.K.; Mittra, E.; Burris, H.A.; Clark, A.S.; Dunphy, M.; et al. Safety and tolerability of increasing doses of CB-839, a first-in-class, orally administered small molecule inhibitor of glutaminase, in solid tumors. J. Clin. Oncol. 2015, 33, 2512. [Google Scholar] [CrossRef]
- Soth, M.J.; Le, K.; di Francesco, M.E.; Hamilton, M.M.; Liu, G.; Burke, J.P.; Carroll, C.L.; Kovacs, J.J.; Bardenhagen, J.P.; Bristow, C.A.; et al. Discovery of IPN60090, a clinical stage selective glutaminase-1 (GLS-1) inhibitor with excellent pharmacokinetic and physicochemical properties. J. Med. Chem. 2020, 63, 12957–12977. [Google Scholar] [CrossRef] [PubMed]
- Momcilovic, M.; Bailey, S.T.; Lee, J.T.; Fishbein, M.C.; Magyar, C.; Braas, D.; Graeber, T.; Jackson, N.J.; Czernin, J.; Emberley, E.; et al. Targeted inhibition of EGFR and glutaminase induces metabolic crisis in EGFR mutant lung cancer. Cell Rep. 2017, 18, 601–610. [Google Scholar] [CrossRef]
- Jin, J.; Byun, J.K.; Choi, Y.K.; Park, K.G. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp. Mol. Med. 2023, 55, 706–715. [Google Scholar] [CrossRef]
- Shen, Y.A.; Chen, C.L.; Huang, Y.H.; Evans, E.E.; Cheng, C.C.; Chuang, Y.J.; Zhang, C.; Le, A. Inhibition of glutaminolysis in combination with other therapies to improve cancer treatment. Curr. Opin. Chem. Biol. 2021, 62, 64–81. [Google Scholar] [CrossRef] [PubMed]
- Ogier, G.; Chantepie, J.; Deshayes, C.; Chantegrel, B.; Charlot, C.; Doutheau, A.; Quash, G. Contribution of 4-methylthio-2-oxobutanoate and its transaminase to the growth of methionine dependent cells in culture: Effect of transaminase inhibitors. Biochem. Pharmacol. 1993, 45, 1631–1644. [Google Scholar] [CrossRef] [PubMed]
- Quash, G.; Roch, A.M.; Charlot, C.; Chantepie, J.; Thomas, V.; Hamedi-Sangsari, F.; Vila, J. 4-Methyl 2-oxobutanoate transaminase: A specific target for antiproliferative agents. Bull. Cancer 2004, 91, E61–E79. [Google Scholar] [PubMed]
- Wang, Q.; Hardie, R.A.; Hoy, A.J.; van Geldermalsen, M.; Gao, D.; Fazli, L.; Sadowski, M.C.; Balaban, S.; Schreuder, M.; Nagarajah, R.; et al. Targeting ASCT2-mediated glutamine uptake blocks prostate cancer growth and tumour development. J. Pathol. 2015, 236, 278–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, C.; Pereira, C.; Medeiros, R. ASCT2 and LAT1 Contribution to the hallmarks of cancer: From a molecular perspective to clinical translation. Cancers 2021, 13, 203. [Google Scholar] [CrossRef]
- Okano, N.; Naruge, D.; Kawai, K.; Kobayashi, T.; Nagashima, F.; Endou, H.; Furuse, J. First-in-human phase I study of JPH203, an l-type amino acid transporter 1 inhibitor, in patients with advanced solid tumors. Investig. New Drugs 2020, 38, 1495–1506. [Google Scholar] [CrossRef]
- Wang, Z.; Li, B.; Li, S.; Lin, W.; Wang, Z.; Wang, S.; Chen, W.; Shi, W.; Chen, T.; Zhou, H.; et al. Metabolic control of CD47 expression through LAT2-mediated amino acid uptake promotes tumor immune evasion. Nat. Commun. 2022, 13, 6308. [Google Scholar] [CrossRef]
- Zhao, X.; Jin, L.; Liu, Y.; Liu, Z.; Liu, Q. Bioinformatic analysis of the role of solute carrier-glutamine transporters in breast cancer. Ann. Transl. Med. 2022, 10, 777. [Google Scholar] [CrossRef]
- Wetzel, T.J.; Erfan, S.C.; Figueroa, L.D.; Wheeler, L.M.; Ananieva, E.A. Crosstalk between arginine, glutamine, and the branched chain amino acid metabolism in the tumor microenvironment. Front. Oncol. 2023, 13, 1186539. [Google Scholar] [CrossRef]
- Wetzel, T.J.; Erfan, S.C.; Ananieva, E.A. The emerging role of the branched chain aminotransferases, BCATc and BCATm, for anti-tumor T-cell immunity. Immunometabolism 2023, 5, e00014. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cooper, A.J.L.; Dorai, T.; Pinto, J.T.; Denton, T.T. Metabolic Heterogeneity, Plasticity, and Adaptation to “Glutamine Addiction” in Cancer Cells: The Role of Glutaminase and the GTωA [Glutamine Transaminase—ω-Amidase (Glutaminase II)] Pathway. Biology 2023, 12, 1131. https://doi.org/10.3390/biology12081131
Cooper AJL, Dorai T, Pinto JT, Denton TT. Metabolic Heterogeneity, Plasticity, and Adaptation to “Glutamine Addiction” in Cancer Cells: The Role of Glutaminase and the GTωA [Glutamine Transaminase—ω-Amidase (Glutaminase II)] Pathway. Biology. 2023; 12(8):1131. https://doi.org/10.3390/biology12081131
Chicago/Turabian StyleCooper, Arthur J. L., Thambi Dorai, John T. Pinto, and Travis T. Denton. 2023. "Metabolic Heterogeneity, Plasticity, and Adaptation to “Glutamine Addiction” in Cancer Cells: The Role of Glutaminase and the GTωA [Glutamine Transaminase—ω-Amidase (Glutaminase II)] Pathway" Biology 12, no. 8: 1131. https://doi.org/10.3390/biology12081131
APA StyleCooper, A. J. L., Dorai, T., Pinto, J. T., & Denton, T. T. (2023). Metabolic Heterogeneity, Plasticity, and Adaptation to “Glutamine Addiction” in Cancer Cells: The Role of Glutaminase and the GTωA [Glutamine Transaminase—ω-Amidase (Glutaminase II)] Pathway. Biology, 12(8), 1131. https://doi.org/10.3390/biology12081131