
EpiMCI: Predicting Multi-Way Chromatin Interactions from Epigenomic Signals

 , ,
, ,  and
and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Data Source
2.2. Data Preprocessing
2.3. Hypergraph Definition and Prediction Problem Statement
2.4. Hypergraph Construction
2.5. Vertex Feature Generation
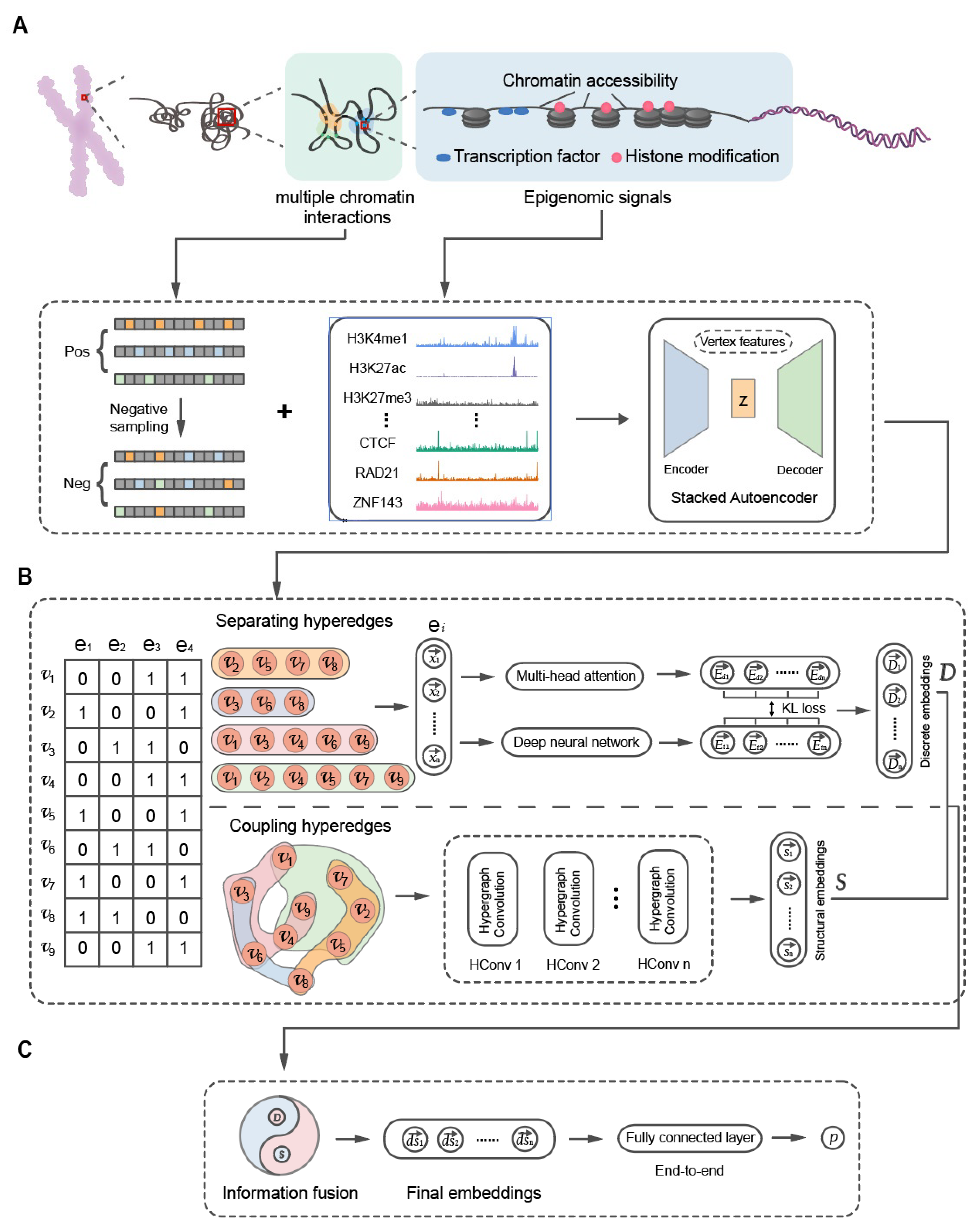
2.6. EpiMCI Model Architecture
2.7. Vertex Representation from Separating Hyperedges
2.8. Vertex Representation from Coupling Hyperedges
2.9. Experiment Setting and Evaluation Metrics
3. Results and Discussion
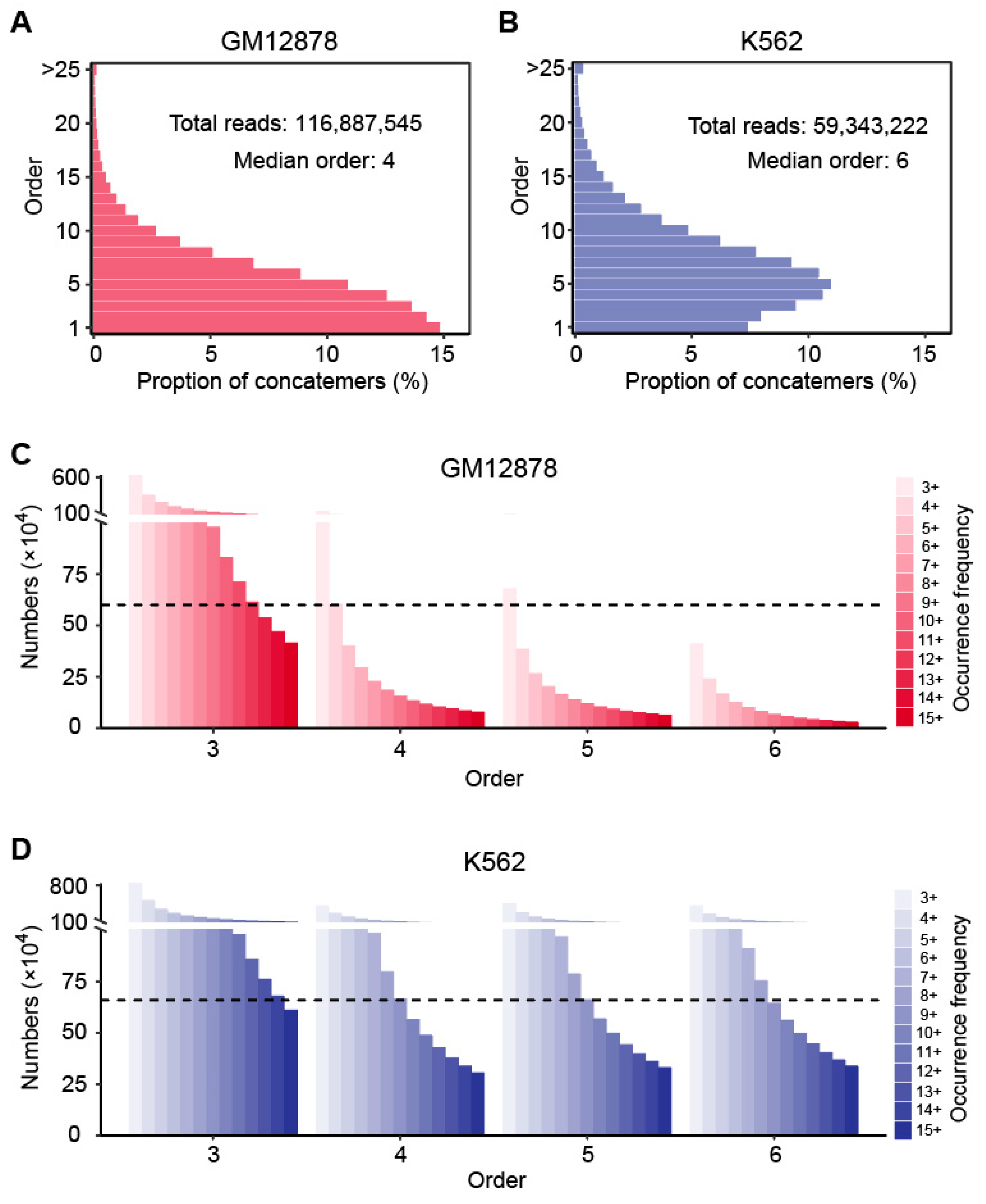
3.1. Hyperedge Generation
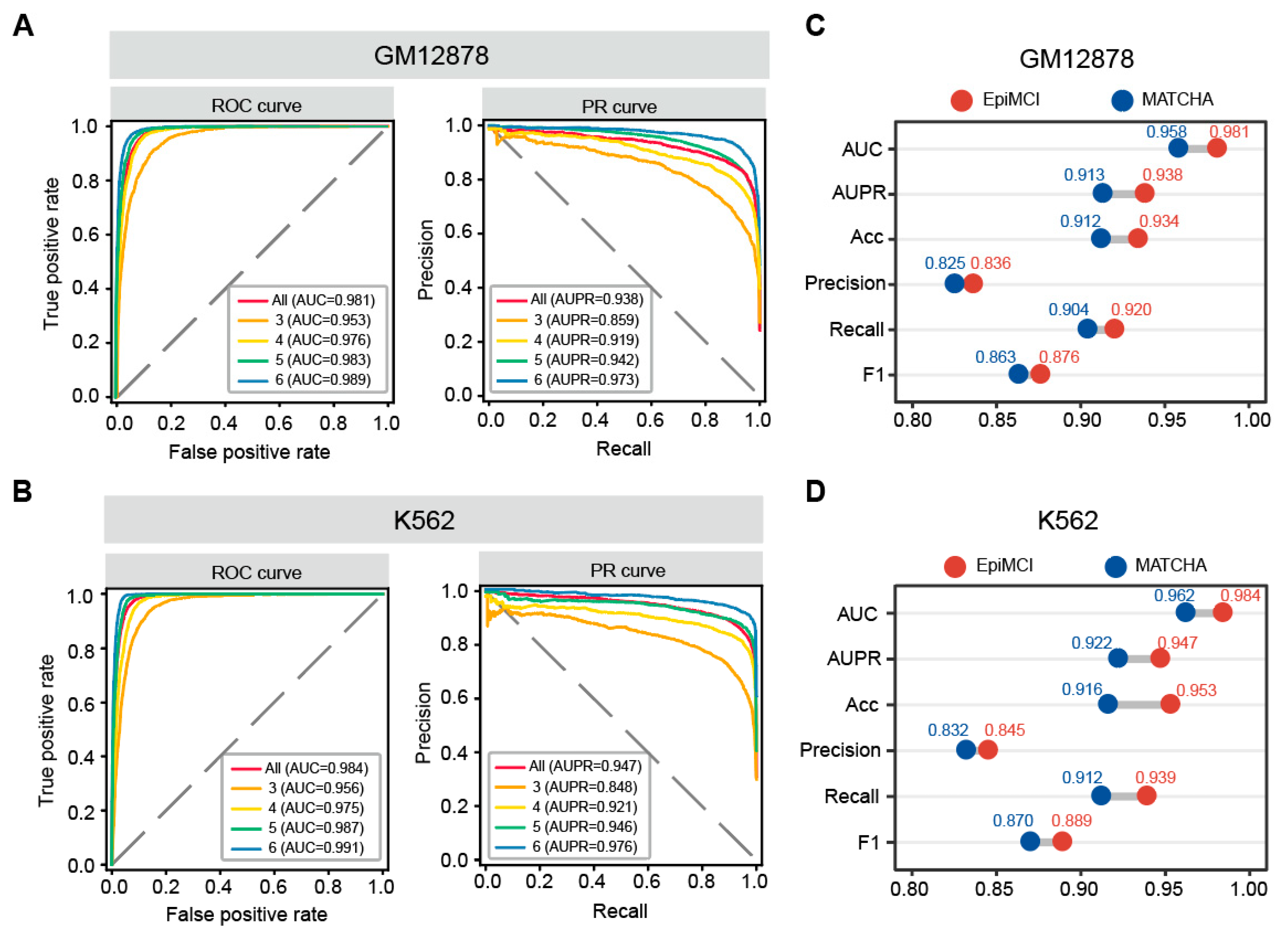
3.2. Multi-Way Chromatin Interaction Prediction
3.3. Model Performance Comparison
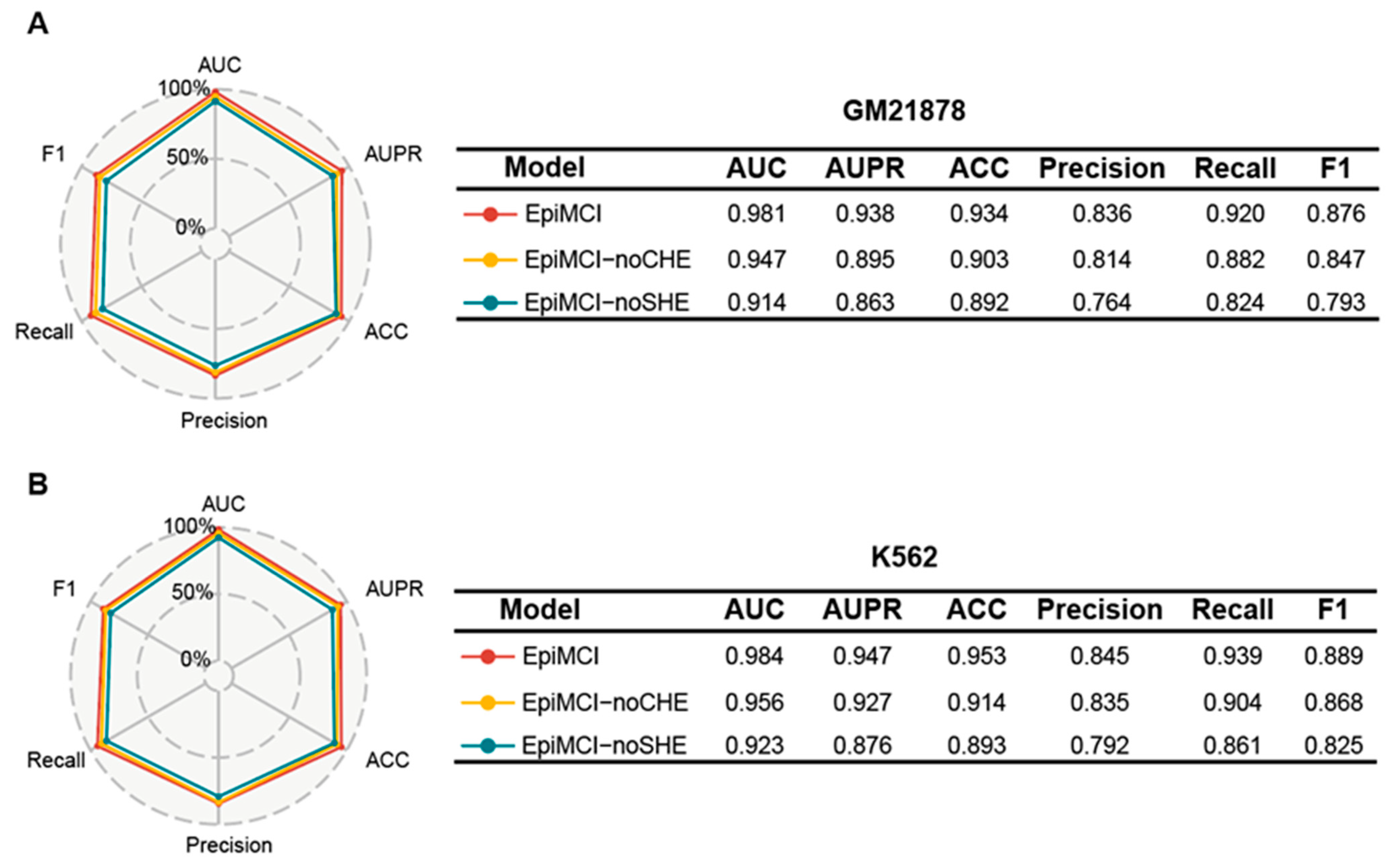
3.4. Ablation Experiment
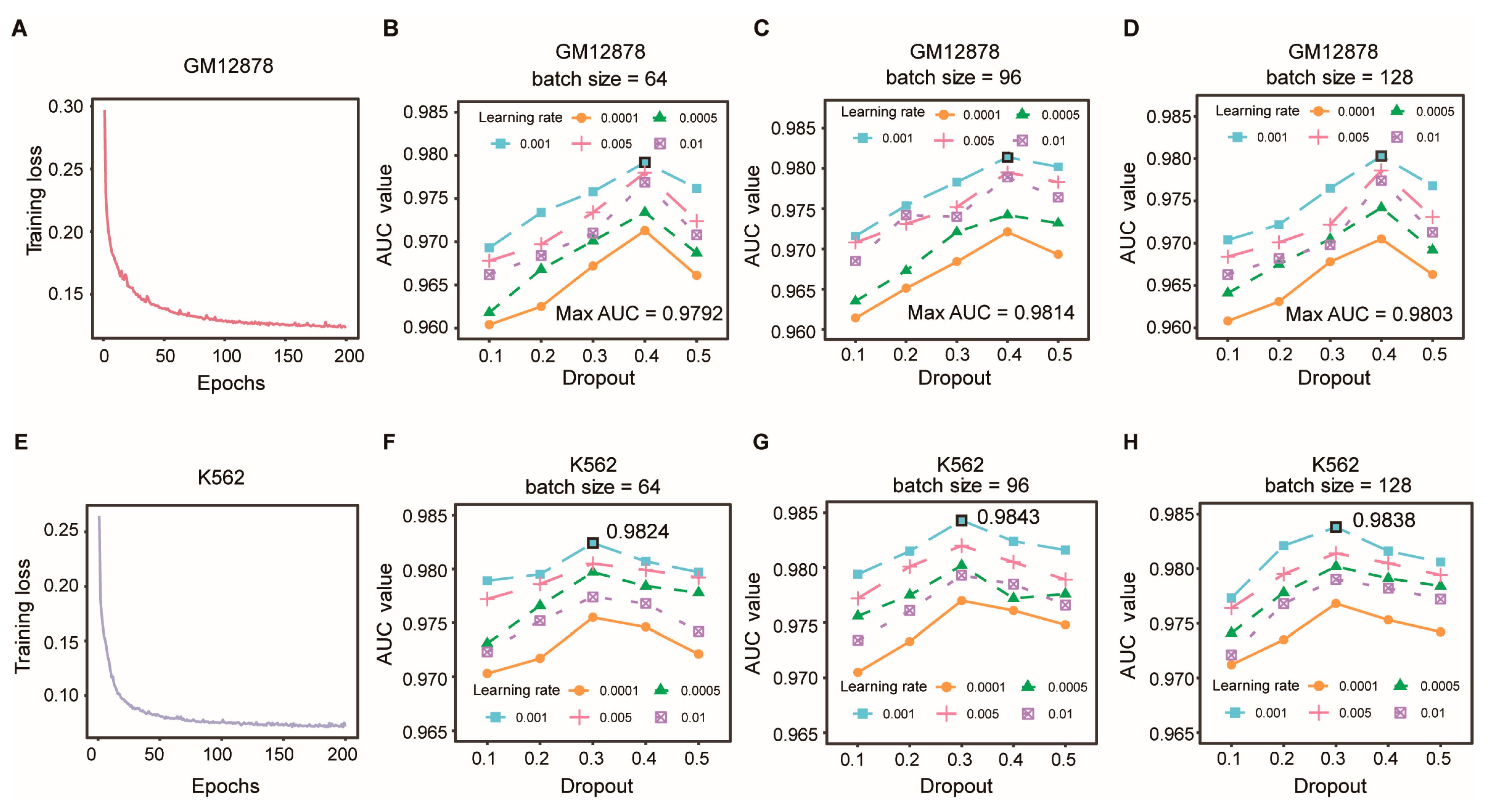
3.5. Optimization of Model Hyperparameters
3.6. Case Studies
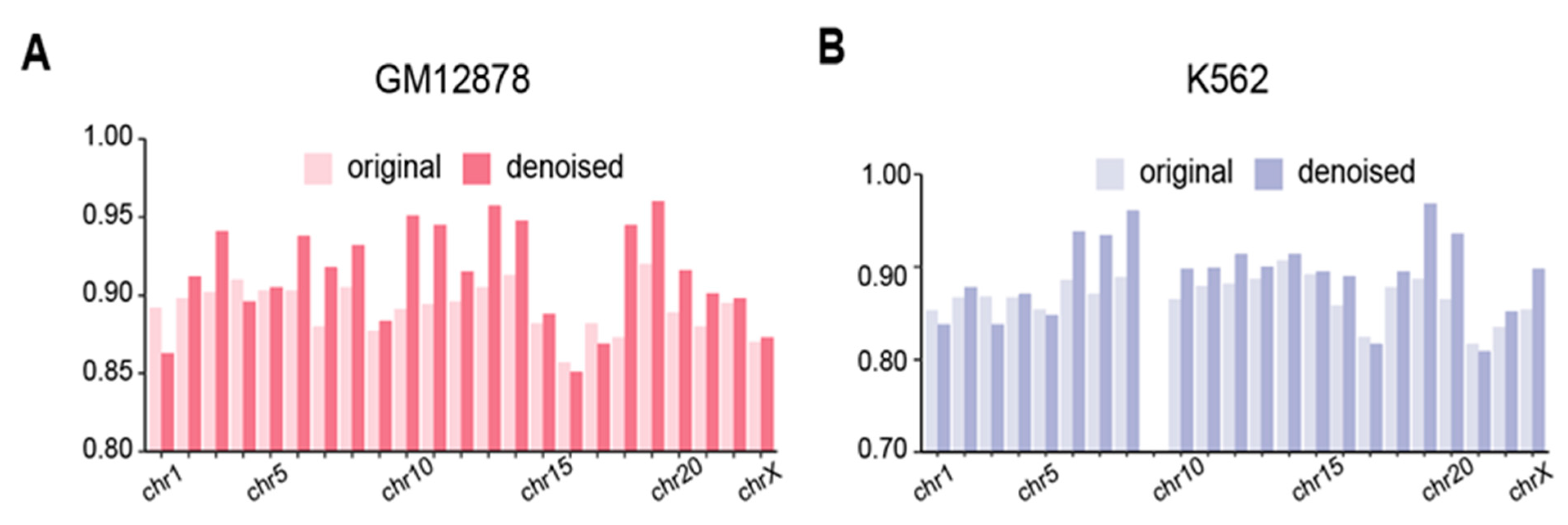
3.6.1. EpiMCI Improves HiPore-C Data Quality
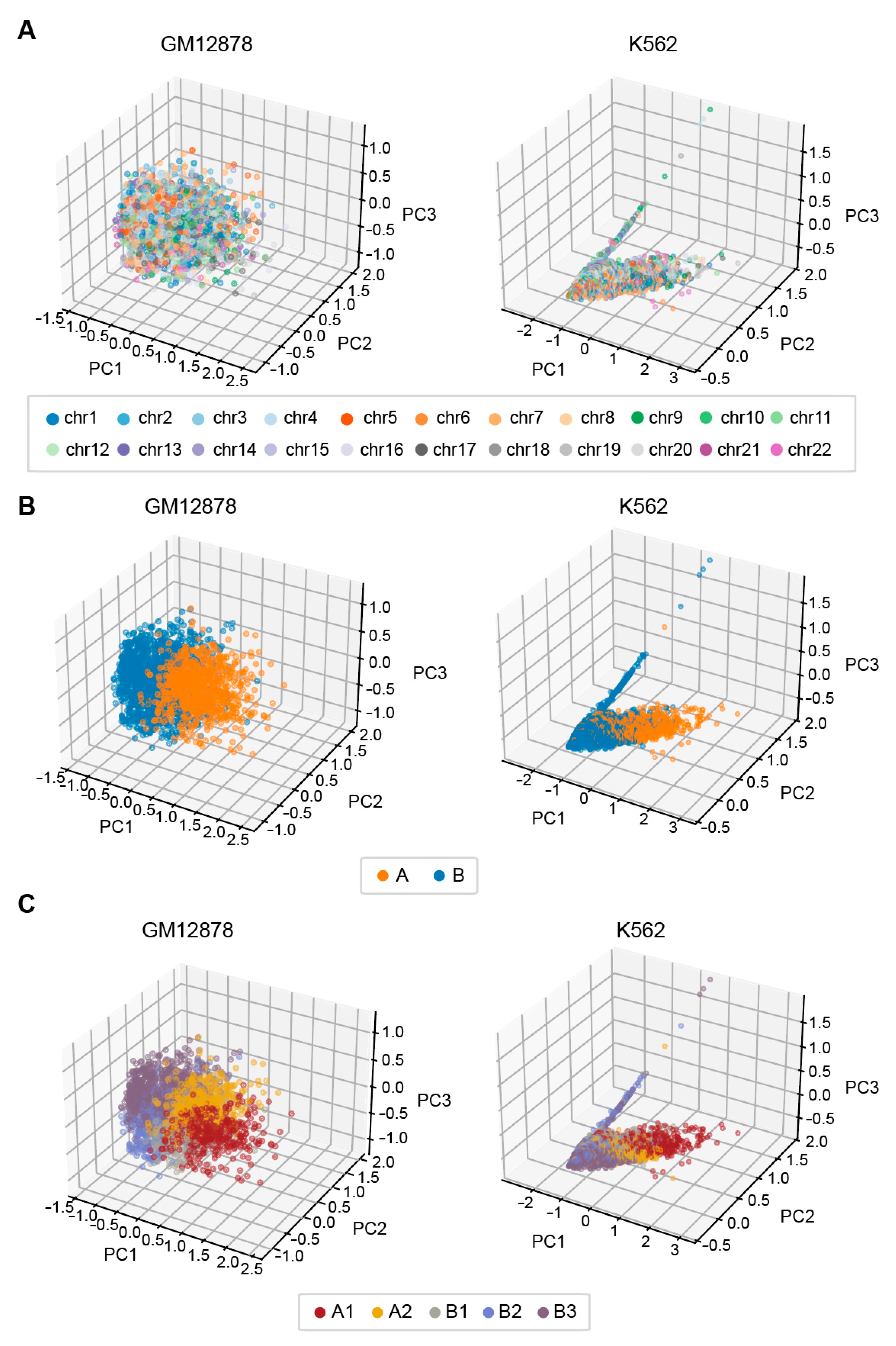
3.6.2. EpiMCI Reflects 3D Genome Global Positioning Patterns
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Misteli, T. The Self-Organizing Genome: Principles of Genome Architecture and Function. Cell 2020, 183, 28–45. [Google Scholar] [CrossRef] [PubMed]
- Oudelaar, A.M.; Higgs, D.R. The relationship between genome structure and function. Nat. Rev. Genet. 2021, 22, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Chen, C.; Teng, L.; Tan, K. Global view of enhancer-promoter interactome in human cells. Proc. Natl. Acad. Sci. USA 2014, 111, E2191–2199. [Google Scholar] [CrossRef] [PubMed]
- Fullwood, M.J.; Ruan, Y. ChIP-based methods for the identification of long-range chromatin interactions. J. Cell. Biochem. 2009, 107, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; Van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Huntley, M.H.; Durand, N.C.; Stamenova, E.K.; Bochkov, I.D.; Robinson, J.T.; Sanborn, A.L.; Machol, I.; Omer, A.D.; Lander, E.S.; et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell 2014, 159, 1665–1680. [Google Scholar] [CrossRef] [PubMed]
- Hou, C.; Li, L.; Qin, Z.S.; Corces, V.G. Gene density, transcription, and insulators contribute to the partition of the Drosophila genome into physical domains. Mol. Cell 2012, 48, 471–484. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Crane, E.; Bian, Q.; McCord, R.P.; Lajoie, B.R.; Wheeler, B.S.; Ralston, E.J.; Uzawa, S.; Dekker, J.; Meyer, B.J. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature 2015, 523, 240–244. [Google Scholar] [CrossRef]
- Guo, Y.; Xu, Q.; Canzio, D.; Shou, J.; Li, J.; Gorkin, D.U.; Jung, I.; Wu, H.; Zhai, Y.; Tang, Y.; et al. CRISPR Inversion of CTCF Sites Alters Genome Topology and Enhancer/Promoter Function. Cell 2015, 162, 900–910. [Google Scholar] [CrossRef] [PubMed]
- Jung, I.; Schmitt, A.; Diao, Y.; Lee, A.J.; Liu, T.; Yang, D.; Tan, C.; Eom, J.; Chan, M.; Chee, S.; et al. A compendium of promoter-centered long-range chromatin interactions in the human genome. Nat. Genet. 2019, 51, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Lupianez, D.G.; Kraft, K.; Heinrich, V.; Krawitz, P.; Brancati, F.; Klopocki, E.; Horn, D.; Kayserili, H.; Opitz, J.M.; Laxova, R.; et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015, 161, 1012–1025. [Google Scholar] [CrossRef]
- Kempfer, R.; Pombo, A. Methods for mapping 3D chromosome architecture. Nat. Rev. Genet. 2020, 21, 207–226. [Google Scholar] [CrossRef] [PubMed]
- Beagrie, R.A.; Scialdone, A.; Schueler, M.; Kraemer, D.C.; Chotalia, M.; Xie, S.Q.; Barbieri, M.; de Santiago, I.; Lavitas, L.M.; Branco, M.R.; et al. Complex multi-enhancer contacts captured by genome architecture mapping. Nature 2017, 543, 519–524. [Google Scholar] [CrossRef] [PubMed]
- Quinodoz, S.A.; Ollikainen, N.; Tabak, B.; Palla, A.; Schmidt, J.M.; Detmar, E.; Lai, M.M.; Shishkin, A.A.; Bhat, P.; Takei, Y.; et al. Higher-Order Inter-chromosomal Hubs Shape 3D Genome Organization in the Nucleus. Cell 2018, 174, 744–757.e724. [Google Scholar] [CrossRef] [PubMed]
- Oudelaar, A.M.; Davies, J.O.J.; Hanssen, L.L.P.; Telenius, J.M.; Schwessinger, R.; Liu, Y.; Brown, J.M.; Downes, D.J.; Chiariello, A.M.; Bianco, S.; et al. Single-allele chromatin interactions identify regulatory hubs in dynamic compartmentalized domains. Nat. Genet. 2018, 50, 1744–1751. [Google Scholar] [CrossRef]
- Allahyar, A.; Vermeulen, C.; Bouwman, B.A.M.; Krijger, P.H.L.; Verstegen, M.; Geeven, G.; van Kranenburg, M.; Pieterse, M.; Straver, R.; Haarhuis, J.H.I.; et al. Enhancer hubs and loop collisions identified from single-allele topologies. Nat. Genet. 2018, 50, 1151–1160. [Google Scholar] [CrossRef]
- Zheng, M.; Tian, S.Z.; Capurso, D.; Kim, M.; Maurya, R.; Lee, B.; Piecuch, E.; Gong, L.; Zhu, J.J.; Li, Z.; et al. Multiplex chromatin interactions with single-molecule precision. Nature 2019, 566, 558–562. [Google Scholar] [CrossRef]
- Deshpande, A.S.; Ulahannan, N.; Pendleton, M.; Dai, X.; Ly, L.; Behr, J.M.; Schwenk, S.; Liao, W.; Augello, M.A.; Tyer, C.; et al. Identifying synergistic high-order 3D chromatin conformations from genome-scale nanopore concatemer sequencing. Nat. Biotechnol. 2022, 40, 1488–1499. [Google Scholar] [CrossRef]
- Zhong, J.Y.; Niu, L.; Lin, Z.B.; Bai, X.; Chen, Y.; Luo, F.; Hou, C.; Xiao, C.L. High-throughput Pore-C reveals the single-allele topology and cell type-specificity of 3D genome folding. Nat. Commun. 2023, 14, 1250. [Google Scholar] [CrossRef] [PubMed]
- Whitaker, J.W.; Nguyen, T.T.; Zhu, Y.; Wildberg, A.; Wang, W. Computational schemes for the prediction and annotation of enhancers from epigenomic assays. Methods 2015, 72, 86–94. [Google Scholar] [CrossRef]
- Zhu, Y.; Chen, Z.; Zhang, K.; Wang, M.; Medovoy, D.; Whitaker, J.W.; Ding, B.; Li, N.; Zheng, L.; Wang, W. Constructing 3D interaction maps from 1D epigenomes. Nat. Commun. 2016, 7, 10812. [Google Scholar] [CrossRef] [PubMed]
- Cao, Q.; Anyansi, C.; Hu, X.; Xu, L.; Xiong, L.; Tang, W.; Mok, M.T.S.; Cheng, C.; Fan, X.; Gerstein, M.; et al. Reconstruction of enhancer-target networks in 935 samples of human primary cells, tissues and cell lines. Nat. Genet. 2017, 49, 1428–1436. [Google Scholar] [CrossRef]
- Roy, S.; Siahpirani, A.F.; Chasman, D.; Knaack, S.; Ay, F.; Stewart, R.; Wilson, M.; Sridharan, R. A predictive modeling approach for cell line-specific long-range regulatory interactions. Nucleic Acids Res. 2016, 44, 1977–1978. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Yang, Y.; Poczos, B.; Ma, J. Predicting enhancer-promoter interaction from genomic sequence with deep neural networks. Quant. Biol. 2019, 7, 122–137. [Google Scholar] [CrossRef]
- Whalen, S.; Truty, R.M.; Pollard, K.S. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat. Genet. 2016, 48, 488–496. [Google Scholar] [CrossRef]
- Di Pierro, M.; Cheng, R.R.; Lieberman Aiden, E.; Wolynes, P.G.; Onuchic, J.N. De novo prediction of human chromosome structures: Epigenetic marking patterns encode genome architecture. Proc. Natl. Acad. Sci. USA 2017, 114, 12126–12131. [Google Scholar] [CrossRef]
- Liu, T.; Wang, Z. DeepChIA-PET: Accurately predicting ChIA-PET from Hi-C and ChIP-seq with deep dilated networks. bioRxiv 2022. [Google Scholar] [CrossRef]
- Yang, R.; Das, A.; Gao, V.R.; Karbalayghareh, A.; Noble, W.S.; Bilmes, J.A.; Leslie, C.S. Epiphany: Predicting Hi-C contact maps from 1D epigenomic signals. Genome Biol. 2023, 24, 134. [Google Scholar] [CrossRef]
- Welling, M.; Kipf, T.N. Semi-supervised classification with graph convolutional networks. In Proceedings of the Journal International Conference on Learning Representations (ICLR 2017), Toulon, France, 24–26 April 2017. [Google Scholar]
- Defferrard, M.; Bresson, X.; Vandergheynst, P. Convolutional neural networks on graphs with fast localized spectral filtering. Adv. Neural Inf. Process. Syst. 2016, 29, 3844–3852. [Google Scholar]
- Feng, Y.; You, H.; Zhang, Z.; Ji, R.; Gao, Y. Hypergraph neural networks. In Proceedings of the AAAI Conference on Artificial Intelligence, Honolulu, HI, USA, 27 January–1 February 2019; pp. 3558–3565. [Google Scholar]
- Benson, A.R.; Gleich, D.F.; Leskovec, J. Higher-order organization of complex networks. Science 2016, 353, 163–166. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.M.; Klinvex, A.M.; Dunlavy, D.M. Advantages to modeling relational data using hypergraphs versus graphs. In Proceedings of the 2016 IEEE High Performance Extreme Computing Conference (HPEC), Waltham, MA USA, 13–15 September 2016; pp. 1–7. [Google Scholar]
- Dotson, G.A.; Chen, C.; Lindsly, S.; Cicalo, A.; Dilworth, S.; Ryan, C.; Jeyarajan, S.; Meixner, W.; Stansbury, C.; Pickard, J.; et al. Deciphering multi-way interactions in the human genome. Nat. Commun. 2022, 13, 5498. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Ma, J. MATCHA: Probing multi-way chromatin interaction with hypergraph representation learning. Cell Syst. 2020, 10, 397–407.e395. [Google Scholar] [CrossRef]
- Durand, N.C.; Shamim, M.S.; Machol, I.; Rao, S.S.; Huntley, M.H.; Lander, E.S.; Aiden, E.L. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. Cell Syst. 2016, 3, 95–98. [Google Scholar] [CrossRef]
- Ursu, O.; Boley, N.; Taranova, M.; Wang, Y.X.R.; Yardimci, G.G.; Stafford Noble, W.; Kundaje, A. GenomeDISCO: A concordance score for chromosome conformation capture experiments using random walks on contact map graphs. Bioinformatics 2018, 34, 2701–2707. [Google Scholar] [CrossRef] [PubMed]
- Busslinger, G.A.; Stocsits, R.R.; van der Lelij, P.; Axelsson, E.; Tedeschi, A.; Galjart, N.; Peters, J.M. Cohesin is positioned in mammalian genomes by transcription, CTCF and Wapl. Nature 2017, 544, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.P.; Huang, S.C.; Glenn St Hilaire, B.; Engreitz, J.M.; Perez, E.M.; Kieffer-Kwon, K.R.; Sanborn, A.L.; Johnstone, S.E.; Bascom, G.D.; Bochkov, I.D.; et al. Cohesin Loss Eliminates All Loop Domains. Cell 2017, 171, 305–320.e324. [Google Scholar] [CrossRef]
- Bengio, Y.; Lamblin, P.; Popovici, D.; Larochelle, H. Greedy layer-wise training of deep networks. Adv. Neural Inf. Process. Syst. 2006, 19, 153–160. [Google Scholar]
- Shin, H.-C.; Orton, M.R.; Collins, D.J.; Doran, S.J.; Leach, M.O. Stacked autoencoders for unsupervised feature learning and multiple organ detection in a pilot study using 4D patient data. IEEE Trans. Pattern Anal. Mach. Intell. 2012, 35, 1930–1943. [Google Scholar] [CrossRef]
- Yu, M.; Quan, T.; Peng, Q.; Yu, X.; Liu, L. A model-based collaborate filtering algorithm based on stacked AutoEncoder. Neural Comput. Appl. 2021, 34, 2503–2511. [Google Scholar] [CrossRef]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, Ł.; Polosukhin, I. Attention is all you need. Adv. Neural Inf. Process. Syst. 2017, 30, 6000–6010. [Google Scholar]
- Guo, S.; Wang, Y.; Yuan, H.; Huang, Z.; Chen, J.; Wang, X. TAERT: Triple-attentional explainable recommendation with temporal convolutional network. Inf. Sci. 2021, 567, 185–200. [Google Scholar] [CrossRef]
- Gao, Y.; Feng, Y.; Ji, S.; Ji, R. HGNN (+): General Hypergraph Neural Networks. IEEE Trans. Pattern Anal. Mach. Intell. 2022, 45, 3181–3199. [Google Scholar] [CrossRef]
- Maas, A.L.; Hannun, A.Y.; Ng, A.Y. Rectifier nonlinearities improve neural network acoustic models. In Proceedings of the ICML, Atlanta, GA, USA, 16–21 June 2013; p. 3. [Google Scholar]
- Han, J.; Moraga, C. The influence of the sigmoid function parameters on the speed of backpropagation learning. In Proceedings of the International Workshop on Artificial Neural Networks, Perth, WA, Australia, 27 November–1 December 1995; pp. 195–201. [Google Scholar]
- Daqi, G.; Yan, J. Classification methodologies of multilayer perceptrons with sigmoid activation functions. Pattern Recognit. 2005, 38, 1469–1482. [Google Scholar] [CrossRef]
- De Boer, P.-T.; Kroese, D.P.; Mannor, S.; Rubinstein, R.Y. A tutorial on the cross-entropy method. Ann. Oper. Res. 2005, 134, 19–67. [Google Scholar] [CrossRef]
- Lin, D.; Hong, P.; Zhang, S.; Xu, W.; Jamal, M.; Yan, K.; Lei, Y.; Li, L.; Ruan, Y.; Fu, Z.F. Digestion-ligation-only Hi-C is an efficient and cost-effective method for chromosome conformation capture. Nat. Genet. 2018, 50, 754–763. [Google Scholar] [CrossRef]
- Nagano, T.; Varnai, C.; Schoenfelder, S.; Javierre, B.M.; Wingett, S.W.; Fraser, P. Comparison of Hi-C results using in-solution versus in-nucleus ligation. Genome Biol. 2015, 16, 175. [Google Scholar] [CrossRef]
- Stilianoudakis, S.C.; Marshall, M.A.; Dozmorov, M.G. preciseTAD: A transfer learning framework for 3D domain boundary prediction at base-pair resolution. Bioinformatics 2021, 38, 621–630. [Google Scholar] [CrossRef]
- Stadhouders, R.; Filion, G.J.; Graf, T. Transcription factors and 3D genome conformation in cell-fate decisions. Nature 2019, 569, 345–354. [Google Scholar] [CrossRef]
- Xiong, K.; Ma, J. Revealing Hi-C subcompartments by imputing inter-chromosomal chromatin interactions. Nat. Commun. 2019, 10, 5069. [Google Scholar] [CrossRef] [PubMed]
- Ashoor, H.; Chen, X.; Rosikiewicz, W.; Wang, J.; Cheng, A.; Wang, P.; Ruan, Y.; Li, S. Graph embedding and unsupervised learning predict genomic sub-compartments from HiC chromatin interaction data. Nat. Commun. 2020, 11, 1173. [Google Scholar] [CrossRef] [PubMed]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, J.; Zhang, P.; Sun, W.; Zhang, J.; Zhang, W.; Hou, C.; Li, L. EpiMCI: Predicting Multi-Way Chromatin Interactions from Epigenomic Signals. Biology 2023, 12, 1203. https://doi.org/10.3390/biology12091203
Xu J, Zhang P, Sun W, Zhang J, Zhang W, Hou C, Li L. EpiMCI: Predicting Multi-Way Chromatin Interactions from Epigenomic Signals. Biology. 2023; 12(9):1203. https://doi.org/10.3390/biology12091203
Chicago/Turabian StyleXu, Jinsheng, Ping Zhang, Weicheng Sun, Junying Zhang, Wenxue Zhang, Chunhui Hou, and Li Li. 2023. "EpiMCI: Predicting Multi-Way Chromatin Interactions from Epigenomic Signals" Biology 12, no. 9: 1203. https://doi.org/10.3390/biology12091203
APA StyleXu, J., Zhang, P., Sun, W., Zhang, J., Zhang, W., Hou, C., & Li, L. (2023). EpiMCI: Predicting Multi-Way Chromatin Interactions from Epigenomic Signals. Biology, 12(9), 1203. https://doi.org/10.3390/biology12091203