
PMSeeker: A Scheme Based on the Greedy Algorithm and the Exhaustive Algorithm to Screen Low-Redundancy Marker Sets for Large-Scale Parentage Assignment with Full Parental Genotyping


Abstract
:Simple Summary
Abstract
1. Background
2. Materials and Methods
2.1. Algorithm and Scheme
2.1.1. Basic Definition
2.1.2. Exhaustive Algorithm
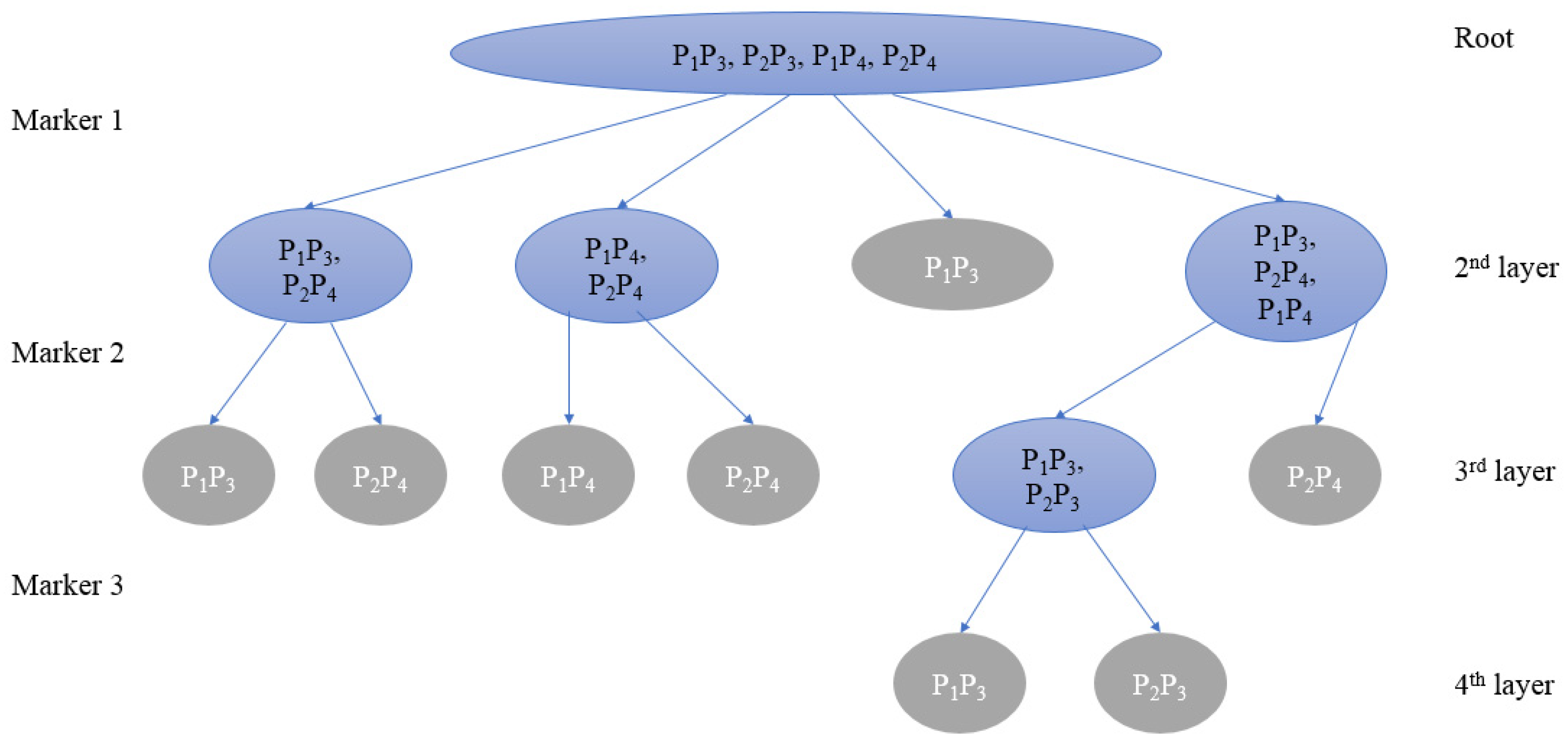
2.1.3. Multiway Tree-Based Greedy Algorithm
2.1.4. Scheme to Constitute a Low-Redundancy PMS
2.2. A Comparative Study on the Running Time of the Two Methods
2.3. PMS Screening Based on Real Molecular Markers
2.4. Application in a Real Case
3. Results
3.1. A Comparative Study on the Running Time of the Two Algorithms
3.2. PMS Screening Based on Real Molecular Markers
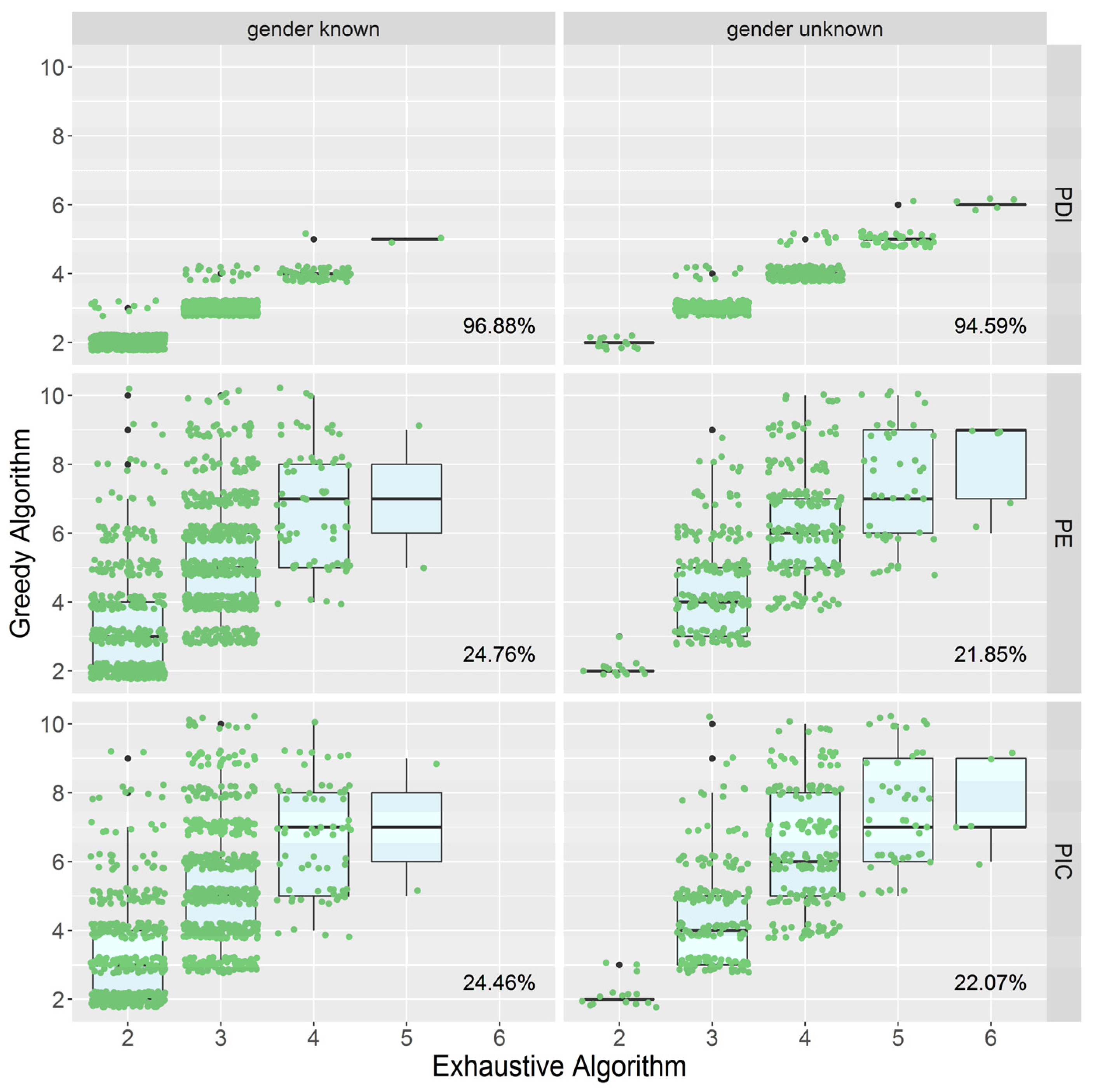
3.2.1. Summary of the Solution Acquisition Rate
3.2.2. Comparison of the Efficacy of PDI, PIC, and PE
3.3. Application in a Real Case
3.4. The PMSeeker Online Tool for PMS Screening
4. Discussion
4.1. The Advantages and Disadvantages of the Two Algorithms and the Efficacy of Their Combination
4.2. Comparison of the Statistics Used in PMS Screening
4.3. Effectiveness, Problems, and Solutions in Practical Applications
4.3.1. The Efficacy of the Low-Redundancy Scheme
4.3.2. The Different Impacts of Two Types of Problematic Markers
4.3.3. The Different Efficacies of SSR and SNP
4.3.4. About the Software Tools for Parentage Assignment
4.4. Scalability of the Scheme
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vandeputte, M.; Rossignol, M.-N.; Pincent, C. From theory to practice: Empirical evaluation of the assignment power of marker sets for pedigree analysis in fish breeding. Aquaculture 2011, 314, 80–86. [Google Scholar] [CrossRef]
- Yue, G.H.; Xia, J.H. Practical Considerations of Molecular Parentage Analysis in Fish. J. World Aquac. Soc. 2014, 45, 89–103. [Google Scholar] [CrossRef]
- Flanagan, S.P.; Jones, A.G. The future of parentage analysis: From microsatellites to SNPs and beyond. Mol. Ecol. 2019, 28, 544–567. [Google Scholar] [CrossRef]
- Vandeputte, M.; Haffray, P. Parentage assignment with genomic markers: A major advance for understanding and exploiting genetic variation of quantitative traits in farmed aquatic animals. Front. Genet. 2014, 5, 432. [Google Scholar] [CrossRef]
- Tortereau, F.; Moreno, C.R.; Tosser-Klopp, G.; Servin, B.; Raoul, J. Development of a SNP panel dedicated to parentage assignment in French sheep populations. BMC Genet. 2017, 18, 50. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, R.; Meagher, T.R.; Smouse, P.E. Parentage analysis with genetic markers in natural populations. I. The expected proportion of offspring with unambiguous paternity. Genetics 1988, 118, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Jamieson, A.; Taylor, S.C.S. Comparisons of three probability formulae for parentage exclusion. Anim. Genet. 1997, 28, 397–400. [Google Scholar] [CrossRef]
- Zhang, T.; Guo, L.; Shi, M.; Xu, L.; Chen, Y.; Zhang, L.; Gao, H.; Li, J.; Gao, X. Selection and effectiveness of informative SNPs for paternity in Chinese Simmental cattle based on a high-density SNP array. Gene 2018, 673, 211–216. [Google Scholar] [CrossRef] [PubMed]
- Vandeputte, M. An accurate formula to calculate exclusion power of marker sets in parentage assignment. Genet. Sel. Evol. 2012, 44, 36. [Google Scholar] [CrossRef] [PubMed]
- Matson, S.E.; Camara, M.D.; Elchert, W.; Banks, M.A. P-LOCI: A computer program for choosing the most efficient set of loci for parentage assignment. Mol. Ecol. Resour. 2008, 8, 765–768. [Google Scholar] [CrossRef]
- Jones, A.G.; Small, C.M.; Paczolt, K.A.; Ratterman, N.L. A practical guide to methods of parentage analysis. Mol. Ecol. Resour. 2010, 10, 6–30. [Google Scholar] [CrossRef] [PubMed]
- Zhivotovsky, L.A.; Underhill, P.A.; Cinnioğlu, C.; Kayser, M.; Morar, B.; Kivisild, T.; Scozzari, R.; Cruciani, F.; Destro-Bisol, G.; Spedini, G.; et al. The Effective Mutation Rate at Y Chromosome Short Tandem Repeats, with Application to Human Population-Divergence Time. Am. J. Hum. Genet. 2004, 74, 50–61. [Google Scholar] [CrossRef] [PubMed]
- Städele, V.; Vigilant, L. Strategies for determining kinship in wild populations using genetic data. Ecol. Evol. 2016, 6, 6107–6120. [Google Scholar] [CrossRef] [PubMed]
- Fisher, P.; Malthus, B.; Walker, M.C.; Corbett, G.; Spelman, R.J. The number of single nucleotide polymorphisms and on-farm data required for whole-herd parentage testing in dairy cattle herds. J. Dairy Sci. 2009, 92, 369–374. [Google Scholar] [CrossRef] [PubMed]
- Conrad, D.F.; Keebler, J.E.M.; DePristo, M.A.; Lindsay, S.J.; Zhang, Y.; Casals, F.; Idaghdour, Y.; Hartl, C.L.; Torroja, C.; Garimella, K.V.; et al. Variation in genome-wide mutation rates within and between human families. Nat. Genet. 2011, 43, 712–714. [Google Scholar] [CrossRef] [PubMed]
- Kidd, K.K.; Pakstis, A.J.; Speed, W.C.; Lagacé, R.; Chang, J.; Wootton, S.; Haigh, E.; Kidd, J.R. Current sequencing technology makes microhaplotypes a powerful new type of genetic marker for forensics. Forensic Sci. Int. Genet. 2014, 12, 215–224. [Google Scholar] [CrossRef]
- Xia, L.; Shi, M.; Zhang, W.; Duan, Y.; Cheng, Y.-Y.; Wu, N.; Xia, X.-Q. A method for paternity testing of grass carp (Ctenopharyngodon idellus) using microhaplotypes. Acta Hydrobiol. Sin. 2020, 44, 509–517. [Google Scholar] [CrossRef]
- Wang, Y.; Lu, Y.; Zhang, Y.; Ning, Z.; Li, Y.; Zhao, Q.; Lu, H.; Huang, R.; Xia, X.; Feng, Q.; et al. The draft genome of the grass carp (Ctenopharyngodon idellus) provides insights into its evolution and vegetarian adaptation. Nat. Genet. 2015, 47, 625–631. [Google Scholar] [CrossRef]
- Andrews, K.R.; Adams, J.R.; Cassirer, E.F.; Plowright, R.K.; Gardner, C.; Dwire, M.; Hohenlohe, P.A.; Waits, L.P. A bioinformatic pipeline for identifying informative SNP panels for parentage assignment from RADseq data. Mol. Ecol. Resour. 2018, 18, 1263–1281. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L.; Marshall, T.C. Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol. Ecol. 2007, 16, 1099–1106. [Google Scholar] [CrossRef]
- Jones, O.R.; Wang, J. COLONY: A program for parentage and sibship inference from multilocus genotype data. Mol. Ecol. Resour. 2010, 10, 551–555. [Google Scholar] [CrossRef]
- Botstein, D.; White, R.L.; Skolnick, M.; Davis, R.W. Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am. J. Hum. Genet. 1980, 32, 314–331. [Google Scholar] [PubMed]
- Serrote, C.M.L.; Reiniger, L.R.S.; Silva, K.B.; Rabaiolli, S.M.d.S.; Stefanel, C.M. Determining the Polymorphism Information Content of a molecular marker. Gene 2020, 726, 144175. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Sun, D.; Zhang, Y. Establishment of paternity testing system using microsatellite markers in Chinese Holstein. J. Genet. Genom. 2008, 35, 279–284. [Google Scholar] [CrossRef] [PubMed]
- Heaton, M.P.; Leymaster, K.A.; Kalbfleisch, T.S.; Kijas, J.W.; Clarke, S.M.; McEwan, J.; Maddox, J.F.; Basnayake, V.; Petrik, D.T.; Simpson, B.; et al. SNPs for Parentage Testing and Traceability in Globally Diverse Breeds of Sheep. PLoS ONE 2014, 9, e94851. [Google Scholar] [CrossRef]
- Yang, W.; Zheng, J.; Jia, B.; Wei, H.; Wang, G.; Yang, F. Isolation of novel microsatellite markers and their application for genetic diversity and parentage analyses in sika deer. Gene 2018, 643, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Langdon, K.S.; King, G.J.; Nock, C.J. DNA paternity testing indicates unexpectedly high levels of self-fertilisation in macadamia. Tree Genet. Genomes 2019, 15, 29. [Google Scholar] [CrossRef]
- Tan, L.-Q.; Liu, Q.-L.; Zhou, B.; Yang, C.-J.; Zou, X.; Yu, Y.-Y.; Wang, Y.; Hu, J.-H.; Zou, Y.; Chen, S.-X.; et al. Paternity analysis using SSR markers reveals that the anthocyanin-rich tea cultivar ‘Ziyan’ is self-compatible. Sci. Hortic. 2019, 245, 258–262. [Google Scholar] [CrossRef]
- Morón, J.A.; Veli, E.A.; Membrillo, A.; Paredes, M.M.; Gutiérrez, G.A. Genetic diversity and validation of a microsatellite panel for parentage testing for alpacas (Vicugna pacos) on three Peruvian farms. Small Rumin. Res. 2020, 193, 106246. [Google Scholar] [CrossRef]
- Thongda, W.; Zhao, H.; Zhang, D.; Jescovitch, L.N.; Liu, M.; Guo, X.; Schrandt, M.; Powers, S.P.; Peatman, E. Development of SNP Panels as a New Tool to Assess the Genetic Diversity, Population Structure, and Parentage Analysis of the Eastern Oyster (Crassostrea virginica). Mar. Biotechnol. 2018, 20, 385–395. [Google Scholar] [CrossRef]
- Zhao, H.; Li, C.; Hargrove, J.S.; Bowen, B.R.; Thongda, W.; Zhang, D.; Mohammed, H.; Beck, B.H.; Austin, J.D.; Peatman, E. SNP marker panels for parentage assignment and traceability in the Florida bass (Micropterus floridanus). Aquaculture 2018, 485, 30–38. [Google Scholar] [CrossRef]
- Hu, L.R.; Li, D.; Chu, Q.; Wang, Y.C.; Zhou, L.; Yu, Y.; Zhang, Y.; Zhang, S.L.; Usman, T.; Xie, Z.Q.; et al. Selection and implementation of single nucleotide polymorphism markers for parentage analysis in crossbred cattle population. Animal 2021, 15, 100066. [Google Scholar] [CrossRef]
- Cooke, T.; Yee, M.-C.; Muzzio, M.; Sockell, A.; Bell, R.; Cornejo, O.; Kelley, J.; Bailliet, G.; Bravi, C.; Bustamante, C.; et al. GBStools: A Statistical Method for Estimating Allelic Dropout in Reduced Representation Sequencing Data. PLoS Genet. 2016, 12, e1005631. [Google Scholar] [CrossRef]
- Rocheta, M.; Miguel Dionísio, F.; Fonseca, L.; Pires, A.M. Paternity analysis in Excel. Comput. Methods Programs Biomed. 2007, 88, 234–238. [Google Scholar] [CrossRef]
- Huisman, J. Pedigree reconstruction from SNP data: Parentage assignment, sibship clustering and beyond. Mol. Ecol. Resour. 2017, 17, 1009–1024. [Google Scholar] [CrossRef]
- Taggart, J.B. FAP: An exclusion-based parental assignment program with enhanced predictive functions. Mol. Ecol. Notes 2007, 7, 412–415. [Google Scholar] [CrossRef]
- Foote, A.; Simma, D.; Khatkar, M.; Raadsma, H.; Guppy, J.; Coman, G.; Giardina, E.; Jerry, D.; Zenger, K.; Wade, N. Considerations for Maintaining Family Diversity in Commercially Mass-Spawned Penaeid Shrimp: A Case Study on Penaeus monodon. Front. Genet. 2019, 10, 1127. [Google Scholar] [CrossRef]
- Warner, P.A.; Willis, B.L.; van Oppen, M.J.H. Sperm dispersal distances estimated by parentage analysis in a brooding scleractinian coral. Mol. Ecol. 2016, 25, 1398–1415. [Google Scholar] [CrossRef]
- Karaket, T.; Poompuang, S. CERVUS vs. COLONY for successful parentage and sibship determinations in freshwater prawn Macrobrachium rosenbergii de Man. Aquaculture 2012, 324–325, 307–311. [Google Scholar] [CrossRef]
- Clarke, S.M.; Henry, H.M.; Dodds, K.G.; Jowett, T.W.D.; Manley, T.R.; Anderson, R.M.; McEwan, J.C. A High Throughput Single Nucleotide Polymorphism Multiplex Assay for Parentage Assignment in New Zealand Sheep. PLoS ONE 2014, 9, e93392. [Google Scholar] [CrossRef]
- Heaton, M.P.; Harhay, G.P.; Bennett, G.L.; Stone, R.T.; Grosse, W.M.; Casas, E.; Keele, J.W.; Smith, T.P.; Chitko-McKown, C.G.; Laegreid, W.W. Selection and use of SNP markers for animal identification and paternity analysis in U.S. beef cattle. Mamm. Genome Off. J. Int. Mamm. Genome Soc. 2002, 13, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Weller, J.I.; Feldmesser, E.; Golik, M.; Tager-Cohen, I.; Domochovsky, R.; Alus, O.; Ezra, E.; Ron, M. Factors affecting incorrect paternity assignment in the Israeli Holstein population. J. Dairy Sci. 2004, 87, 2627–2640. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Gender | Group | Both Algorithms | Greedy Algorithm | |||||
|---|---|---|---|---|---|---|---|---|
| ResS Number | ResS Rate (%) | OS Number | OS Rate (%) | RedS Number | Rate of OS and RedS (%) | IS Number | ||
| Known | 1 | 326 | 32.6 | 319 | 97.85 | 4 | 99.08 | 3 |
| 2 | 349 | 34.9 | 334 | 95.7 | 15 | 100 | 0 | |
| 3 | 351 | 35.1 | 341 | 97.15 | 10 | 100 | 0 | |
| Sum | 1026 | 34.2 | 994 | 96.88 | 29 | 99.71 | 3 | |
| Unknown | 1 | 146 | 14.6 | 142 | 97.26 | 4 | 100 | 0 |
| 2 | 145 | 14.5 | 139 | 95.86 | 5 | 98.62 | 1 | |
| 3 | 153 | 15.3 | 139 | 90.85 | 14 | 100 | 0 | |
| Sum | 444 | 14.8 | 420 | 94.59 | 23 | 99.77 | 1 | |
| r (Gender Known) | r (Gender Unknown) | |
|---|---|---|
| PIC vs. PE | 0.9886 | 0.9886 |
| PDI vs. PIC | −0.7783 | −0.8738 |
| PDI vs. PE | −0.7913 | −0.8766 |
| Indicators | Gender Known | Gender Unknown | ||
|---|---|---|---|---|
| Exhaustive | Greedy | Exhaustive | Greedy | |
| Marker number | 2.6686 ± 0.6051 | 2.6998 ± 0.6321 | 3.6306 ± 0.7593 | 3.6847 ± 0.7920 |
| Average PIC | 0.6012 ± 0.0844 | 0.6141 ± 0.0823 | 0.6138 ± 0.0739 | 0.6231 ± 0.0711 |
| CPE | 0.9078 ± 0.0507 | 0.9143 ± 0.0478 | 0.9665 ± 0.0214 | 0.9698 ± 0.0180 |
| PMS † | Number/Candidate Number | Average PIC | CPE | CERVUS Accuracy Rate (%) | COLONY Accuracy Rate (%) | Failures on Genotype Error ‡ | Failures on Missing Genotype § | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Offspring | CERVUS | COLONY | Offspring | CERVUS | COLONY | ||||||
| S1-1 | 3/22 | 0.669 | 0.9611 | 16 (88.89) | 18 (100.00) | 2 | 2 | 0 | 0 | - | - |
| S1-2 | 4/22 | 0.6754 | 0.9871 | 18 (100.00) | 18 (100.00) | 1 | 0 | 0 | 1 | 0 | 0 |
| S1-1+2 | 5/22 | 0.6817 | 0.9962 | 18 (100.00) | 18 (100.00) | 2 | 0 | 0 | 1 | 0 | 0 |
| 1-1 | 4/999 | 0.3023 | 0.6613 | 14 (82.35) | 13 (76.47) | 3 | 3 | 3 | 3 | 0 | 1 |
| 1-2 | 5/999 | 0.3309 | 0.7705 | 17 (100.00) | 17 (100.00) | 0 | - | - | 2 | 0 | 0 |
| 1-1+2 | 9/999 | 0.3182 | 0.9223 | 16 (94.12) | 17 (100.00) | 3 | 1 | 0 | 5 | 0 | 0 |
| 4-1 | 6/326 | 0.3682 | 0.8573 | 15 (88.24) | 14 (82.35) | 1 | 1 | 1 | 5 (3) ¶ | 1 (3) | 2 (3) |
| 4-2 | 5/326 | 0.3652 | 0.8000 | 17 (100.00) | 17 (100.00) | 0 | - | - | 3 | 0 | 0 |
| 4-1+2 | 11/326 | 0.3668 | 0.9714 | 17 (100.00) | 17 (100.00) | 1 | 0 | 0 | 6 (4, 2) | 0 | 0 |
| 45-1 | 6/201 | 0.3690 | 0.8587 | 14 (82.35) | 15 (88.24) | 1 | 1 | 1 | 5 (3) | 2 (3) | 1 (3) |
| 45-2 | 6/201 | 0.3733 | 0.8609 | 15 (88.24) | 16 (94.12) | 1 | 1 | 0 | 4 (3) | 1 (3) | 1 (3) |
| 45-1+2 | 12/201 | 0.3712 | 0.9802 | 16 (94.12) | 16 (94.12) | 2 | 0 | 0 | 6 (6, 2, 2) | 1 (6) | 1 (6) |
| 475-1 | 6/111 | 0.3662 | 0.8557 | 16 (94.12) | 16 (94.12) | 1 | 1 | 1 | 4 (3) | 0 | 0 |
| 475-2 | 6/111 | 0.3705 | 0.8589 | 17 (100.00) | 17 (100.00) | 0 | - | - | 4 (3) | 0 | 0 |
| 475-1+2 | 12/111 | 0.3719 | 0.9273 | 17 (100.00) | 17 (100.00) | 1 | 0 | 0 | 6 (4, 3, 2) | 0 | 0 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xia, L.; Shi, M.; Li, H.; Zhang, W.; Cheng, Y.; Xia, X.-Q. PMSeeker: A Scheme Based on the Greedy Algorithm and the Exhaustive Algorithm to Screen Low-Redundancy Marker Sets for Large-Scale Parentage Assignment with Full Parental Genotyping. Biology 2024, 13, 100. https://doi.org/10.3390/biology13020100
Xia L, Shi M, Li H, Zhang W, Cheng Y, Xia X-Q. PMSeeker: A Scheme Based on the Greedy Algorithm and the Exhaustive Algorithm to Screen Low-Redundancy Marker Sets for Large-Scale Parentage Assignment with Full Parental Genotyping. Biology. 2024; 13(2):100. https://doi.org/10.3390/biology13020100
Chicago/Turabian StyleXia, Lei, Mijuan Shi, Heng Li, Wanting Zhang, Yingyin Cheng, and Xiao-Qin Xia. 2024. "PMSeeker: A Scheme Based on the Greedy Algorithm and the Exhaustive Algorithm to Screen Low-Redundancy Marker Sets for Large-Scale Parentage Assignment with Full Parental Genotyping" Biology 13, no. 2: 100. https://doi.org/10.3390/biology13020100
APA StyleXia, L., Shi, M., Li, H., Zhang, W., Cheng, Y., & Xia, X. -Q. (2024). PMSeeker: A Scheme Based on the Greedy Algorithm and the Exhaustive Algorithm to Screen Low-Redundancy Marker Sets for Large-Scale Parentage Assignment with Full Parental Genotyping. Biology, 13(2), 100. https://doi.org/10.3390/biology13020100