
Current Applications and Challenges of Next-Generation Sequencing in Plasma Circulating Tumour DNA of Ovarian Cancer
 , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
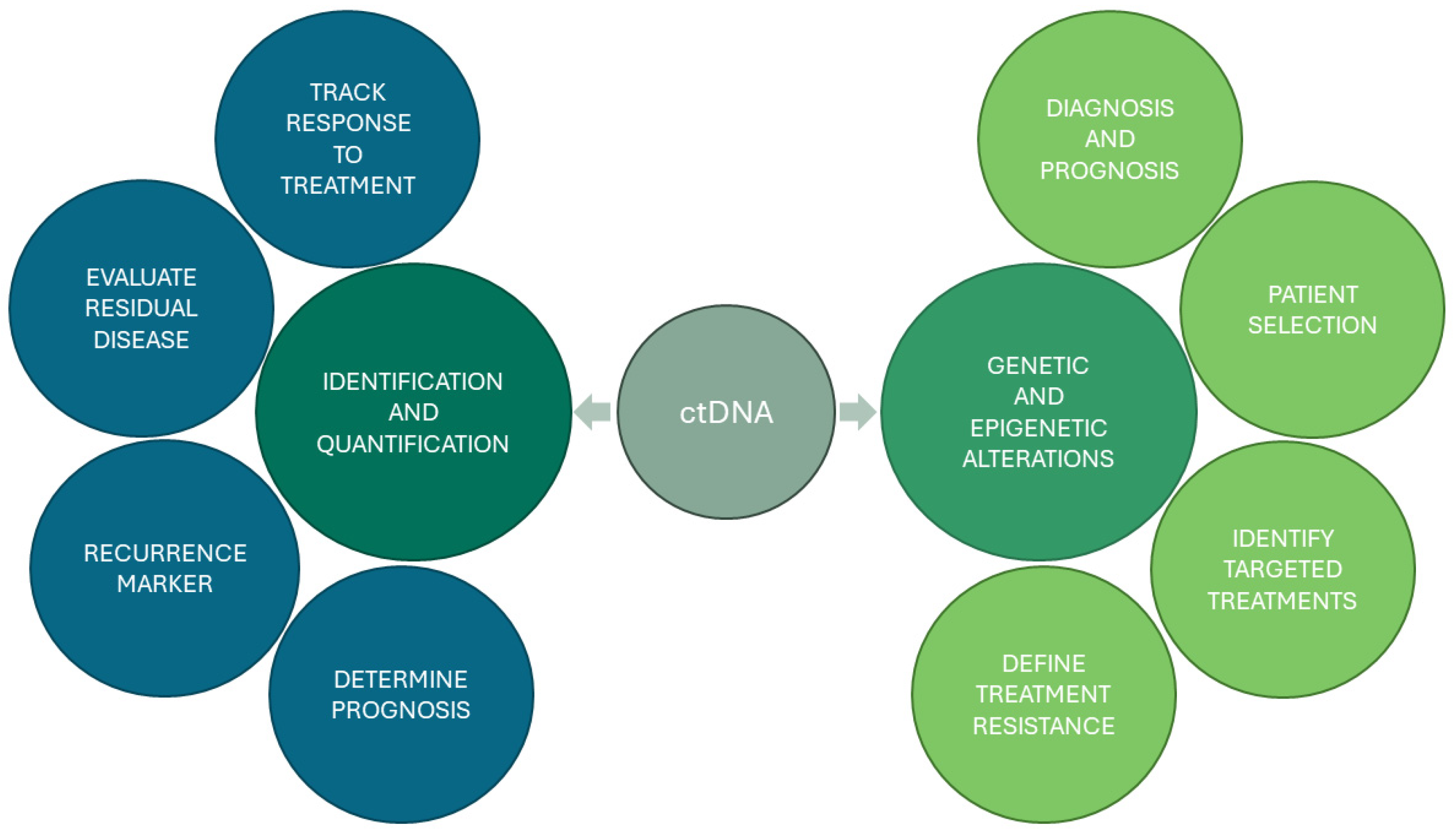
2. Choosing NGS for ctDNA Detection and Quantification
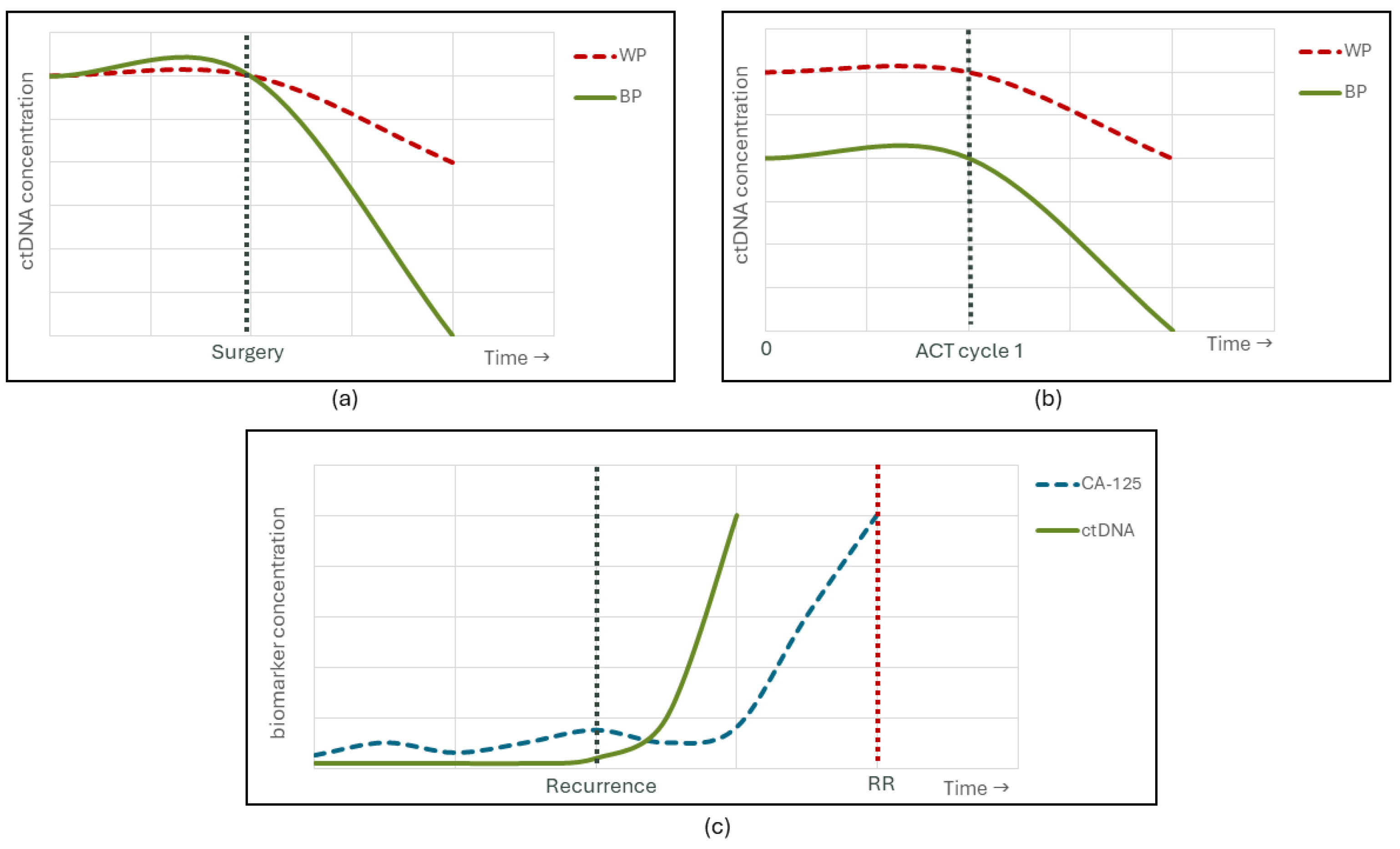
3. NGS-Based Prognostication and Prediction of Treatment Response
4. The Challenge of Tumour Clonality
5. Methylation Patterns in ctDNA
6. ctDNA in Non-High-Grade Epithelial Ovarian Cancers and Rare Subtypes
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Abigél, B.; Jong, B.; Orsolya, B. The Application of Circulating Tumor Cell and Cell-Free DNA Liquid Biopsies in Ovarian Cancer. Mol. Cell Probes 2022, 66, 101871. [Google Scholar]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2022. CA Cancer J Clin 2022, 72, 7–33. [Google Scholar] [CrossRef]
- De Leo, A.; Santini, D.; Ceccarelli, C.; Santandrea, G.; Palicelli, A.; Acquaviva, G.; Chiarucci, F.; Rosini, F.; Ravegnini, G.; Pession, A.; et al. What Is New on Ovarian Carcinoma: Integrated Morphologic and Molecular Analysis Following the New 2020 World Health Organization Classification of Female Genital Tumors. Diagnostics 2021, 11, 697. [Google Scholar] [CrossRef] [PubMed]
- Du-Bois, A.; Leslie, C.; Leslie, Z.; Tarek, M.M.; Elin, S.G. Liquid Biopsy in Ovarian Cancer Using Circulating Tumor DNA and Cells: Ready for Prime Time? Cancer Lett. 2020, 468, 59–71. [Google Scholar]
- Colombo, N.; Sessa, C.; du Bois, A.; Ledermann, J.; McCluggage, W.G.; McNeish, I.; Morice, P.; Pignata, S.; Ray-Coquard, I.; Vergote, I.; et al. ESMO-ESGO Consensus Conference Recommendations on Ovarian Cancer: Pathology and Molecular Biology, Early and Advanced Stages, Borderline Tumours and Recurrent Disease. Ann. Oncol. 2019, 30, 672–705. [Google Scholar] [CrossRef]
- Charkhchi, P.; Cybulski, C.; Gronwald, J.; Wong, F.O.; Narod, S.A.; Akbari, M.R. CA125 and Ovarian Cancer: A Comprehensive Review. Cancers 2020, 12, 3730. [Google Scholar] [CrossRef] [PubMed]
- Widschwendter, M.; Zikan, M.; Wahl, B.; Lempiäinen, H.; Paprotka, T.; Evans, I.; Jones, A.; Ghazali, S.; Reisel, D.; Eichner, J.; et al. The Potential of Circulating Tumor DNA Methylation Analysis for the Early Detection and Management of Ovarian Cancer. Genome Med. 2017, 9, 116. [Google Scholar] [CrossRef] [PubMed]
- Meinusha, G.; Christoph, W.; Matthew, W.; Marcus, Q.B.; Thomas, K. High-Throughput Approaches for Precision Medicine in High-Grade Serous Ovarian Cancer. J. Hematol. Oncol. 2020, 13, 134. [Google Scholar]
- Carolina, M.S.; Innocenza, P.; Serena, M.B.; Giuseppe, C.; Giorgia, P.; Federica, T.; Violante, D.D.; Angela, M.; Ludovico, M. Role of Circulating Biomarkers in Platinum-Resistant Ovarian Cancer. Int. J. Mol. Sci. 2021, 22, 13650. [Google Scholar]
- Jie, W.Z.; Parsa, C.; Mohammad, R.A. Potential Clinical Utility of Liquid Biopsies in Ovarian Cancer. Mol. Cancer 2022, 21, 114. [Google Scholar]
- Sánchez-Herrero, E.; Serna-Blasco, R.; Robado de Lope, L.; González-Rumayor, V.; Romero, A.; Provencio, M. Circulating Tumor DNA as a Cancer Biomarker: An Overview of Biological Features and Factors That May Impact on CtDNA Analysis. Front. Oncol. 2022, 12, 943253. [Google Scholar] [CrossRef] [PubMed]
- Fang, Y.; Jun, T.; Zihao, Z.; Chunling, Z.; Yuancai, X. Circulating Tumor DNA: A Noninvasive Biomarker for Tracking Ovarian Cancer. Reprod. Biol. Endocrinol. 2021, 19, 178. [Google Scholar]
- Ting, X.; Chenyan, F.; Yaqing, C. Advances in Application of Circulating Tumor DNA in Ovarian Cancer. Funct. Integr. Genom. 2023, 23, 250. [Google Scholar]
- Parkinson, C.A.; Gale, D.; Piskorz, A.M.; Biggs, H.; Hodgkin, C.; Addley, H.; Freeman, S.; Moyle, P.; Sala, E.; Sayal, K.; et al. Exploratory Analysis of TP53 Mutations in Circulating Tumour DNA as Biomarkers of Treatment Response for Patients with Relapsed High-Grade Serous Ovarian Carcinoma: A Retrospective Study. PLoS Med. 2016, 13, e1002198. [Google Scholar] [CrossRef]
- Sullivan, B.G.; Lo, A.; Yu, J.; Gonda, A.; Dehkordi-Vakil, F.; Dayyani, F.; Senthil, M. Circulating Tumor DNA Is Unreliable to Detect Somatic Gene Alterations in Gastrointestinal Peritoneal Carcinomatosis. Ann. Surg. Oncol. 2023, 30, 278–284. [Google Scholar] [CrossRef] [PubMed]
- Zill, O.A.; Banks, K.C.; Fairclough, S.R.; Mortimer, S.A.; Vowles, J.V.; Mokhtari, R.; Gandara, D.R.; Mack, P.C.; Odegaard, J.I.; Nagy, R.J.; et al. The Landscape of Actionable Genomic Alterations in Cell-Free Circulating Tumor DNA from 21,807 Advanced Cancer Patients. Clin. Cancer Res. 2018, 24, 3528–3538. [Google Scholar] [CrossRef] [PubMed]
- Ana, B.; Ana, P.; Pedro, P.; Manuela, P.; Manuel, R.T. Potential Clinical Applications of Circulating Cell-Free DNA in Ovarian Cancer Patients. Expert. Rev. Mol. Med. 2018, 20, e6. [Google Scholar]
- Lam, V.K.; Zhang, J.; Wu, C.C.; Tran, H.T.; Li, L.; Diao, L.; Wang, J.; Rinsurongkawong, W.; Raymond, V.M.; Lanman, R.B.; et al. Genotype-Specific Differences in Circulating Tumor DNA Levels in Advanced NSCLC. J. Thorac. Oncol. 2021, 16, 601–609. [Google Scholar] [CrossRef]
- Mouliere, F.; Chandrananda, D.; Piskorz, A.M.; Moore, E.K.; Morris, J.; Ahlborn, L.B.; Mair, R.; Goranova, T.; Marass, F.; Heider, K.; et al. Enhanced Detection of Circulating Tumor DNA by Fragment Size Analysis. Sci. Transl. Med. 2018, 10, eaat4921. [Google Scholar] [CrossRef]
- Havell, M.; Dineika, C.; Elizabeth, M.; Florent, M.; James, M.; James, D.B.; Christopher, G.S.; Nitzan, R. Refined Characterization of Circulating Tumor DNA through Biological Feature Integration. Sci. Rep. 2022, 12, 1928. [Google Scholar]
- Hasenleithner, S.O.; Speicher, M.R. A Clinician’s Handbook for Using CtDNA throughout the Patient Journey. Mol. Cancer 2022, 21, 81. [Google Scholar] [CrossRef]
- Barbosa, A.; Pinto, P.; Peixoto, A.; Guerra, J.; Pinto, C.; Santos, C.; Pinheiro, M.; Escudeiro, C.; Bartosch, C.; Silva, J.; et al. Gene Panel Tumor Testing in Ovarian Cancer Patients Significantly Increases the Yield of Clinically Actionable Germline Variants beyond BRCA1/BRCA2. Cancers 2020, 12, 2834. [Google Scholar] [CrossRef]
- Singh, R.R. Target Enrichment Approaches for Next-Generation Sequencing Applications in Oncology. Diagnostics 2022, 12, 1539. [Google Scholar] [CrossRef]
- Silvia, R.V.; Floris, H.G.; Ronald, V.M.; Corine, M.B.; Jean, C.H.; Hendrikus, J.D.; Winand, N.M.D.; Patricia, C.E.G.; Ramon, S.; Helena, C.V.D.; et al. TP53 Mutations in Serum Circulating Cell-Free Tumor DNA As Longitudinal Biomarker for High-Grade Serous Ovarian Cancer. Biomolecules 2020, 10, 415. [Google Scholar]
- Newman, A.M.; Bratman, S.V.; To, J.; Wynne, J.F.; Eclov, N.C.W.; Modlin, L.A.; Liu, C.L.; Neal, J.W.; Wakelee, H.A.; Merritt, R.E.; et al. An Ultrasensitive Method for Quantitating Circulating Tumor DNA with Broad Patient Coverage. Nat. Med. 2014, 20, 548–554. [Google Scholar] [CrossRef]
- Pereira, E.; Camacho-Vanegas, O.; Anand, S.; Sebra, R.; Camacho, S.C.; Garnar-Wortzel, L.; Nair, N.; Moshier, E.; Wooten, M.; Uzilov, A.; et al. Personalized Circulating Tumor DNA Biomarkers Dynamically Predict Treatment Response and Survival in Gynecologic Cancers. PLoS ONE 2015, 10, e0145754. [Google Scholar] [CrossRef] [PubMed]
- Zviran, A.; Schulman, R.C.; Shah, M.; Hill, S.T.K.; Deochand, S.; Khamnei, C.C.; Maloney, D.; Patel, K.; Liao, W.; Widman, A.J.; et al. Genome-Wide Cell-Free DNA Mutational Integration Enables Ultra-Sensitive Cancer Monitoring. Nat. Med. 2020, 26, 1114–1124. [Google Scholar] [CrossRef] [PubMed]
- Phallen, J.; Sausen, M.; Adleff, V.; Leal, A.; Hruban, C.; White, J.; Anagnostou, V.; Fiksel, J.; Cristiano, S.; Papp, E.; et al. Direct Detection of Early-Stage Cancers Using Circulating Tumor DNA. Sci. Transl. Med. 2017, 9, eaan2415. [Google Scholar] [CrossRef]
- Cohen, J.D.; Li, L.; Wang, Y.; Thoburn, C.; Afsari, B.; Danilova, L.; Douville, C.; Javed, A.A.; Wong, F.; Mattox, A.; et al. Detection and Localization of Surgically Resectable Cancers with a Multi-Analyte Blood Test. Science 2018, 359, 926–930. [Google Scholar] [CrossRef]
- Salk, J.J.; Schmitt, M.W.; Loeb, L.A. Enhancing the Accuracy of Next-Generation Sequencing for Detecting Rare and Subclonal Mutations. Nat. Rev. Genet. 2018, 19, 269–285. [Google Scholar] [CrossRef] [PubMed]
- Leslie, C.; Tindaro, G.; Aaron, B.B.; Anna, L.R.; Colin, S.; Benhur, A.; Tarek, M.M.; Elin, S.G. Identification of TP53 Mutations in Circulating Tumour DNA in High Grade Serous Ovarian Carcinoma Using next Generation Sequencing Technologies. Sci. Rep. 2023, 13, 278. [Google Scholar]
- Widman, A.J.; Shah, M.; Øgaard, N.; Khamnei, C.C.; Frydendahl, A.; Deshpande, A.; Arora, A.; Zhang, M.; Halmos, D.; Bass, J.; et al. Machine Learning Guided Signal Enrichment for Ultrasensitive Plasma Tumor Burden Monitoring. bioRxiv 2022. [Google Scholar] [CrossRef]
- Poplin, R.; Chang, P.-C.; Alexander, D.; Schwartz, S.; Colthurst, T.; Ku, A.; Newburger, D.; Dijamco, J.; Nguyen, N.; Afshar, P.T.; et al. A Universal SNP and Small-Indel Variant Caller Using Deep Neural Networks. Nat. Biotechnol. 2018, 36, 983–987. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Sedlazeck, F.J.; Lam, T.-W.; Schatz, M.C. A Multi-Task Convolutional Deep Neural Network for Variant Calling in Single Molecule Sequencing. Nat. Commun. 2019, 10, 998. [Google Scholar] [CrossRef]
- Abdelwahab, O.; Belzile, F.; Torkamaneh, D. Performance Analysis of Conventional and AI-Based Variant Callers Using Short and Long Reads. BMC Bioinform. 2023, 24, 472. [Google Scholar] [CrossRef]
- Nakabayashi, M.; Kawashima, A.; Yasuhara, R.; Hayakawa, Y.; Miyamoto, S.; Iizuka, C.; Sekizawa, A. Massively Parallel Sequencing of Cell-Free DNA in Plasma for Detecting Gynaecological Tumour-Associated Copy Number Alteration. Sci. Rep. 2018, 8, 11205. [Google Scholar] [CrossRef]
- Sharbatoghli, M.; Fattahi, F.; Aboulkheyr, E.H.; Akbari, A.; Akhavan, S.; Ebrahimi, M.; Asadi-Lari, M.; Totonchi, M.; Madjd, Z. Copy Number Variation of Circulating Tumor DNA (CtDNA) Detected Using NIPT in Neoadjuvant Chemotherapy-Treated Ovarian Cancer Patients. Front. Genet. 2022, 13, 938985. [Google Scholar] [CrossRef] [PubMed]
- Oikkonen, J.; Zhang, K.; Salminen, L.; Schulman, I.; Lavikka, K.; Andersson, N. Prospective Longitudinal ctDNA Workflow Reveals Clinically Actionable Alterations in Ovarian Cancer. JCO Precis Oncol. 2019, 3, 1–12. [Google Scholar] [CrossRef]
- Iwahashi, N.; Sakai, K.; Noguchi, T.; Yahata, T.; Matsukawa, H.; Toujima, S.; Nishio, K.; Ino, K. Liquid Biopsy-Based Comprehensive Gene Mutation Profiling for Gynecological Cancer Using CAncer Personalized Profiling by Deep Sequencing. Sci. Rep. 2019, 9, 10426. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Iwahashi, N.; Sakai, K.; Matsuda, K.; Matsukawa, H.; Toujima, S.; Nishio, K.; Ino, K. Comprehensive Gene Mutation Profiling of Circulating Tumor DNA in Ovarian Cancer: Its Pathological and Prognostic Impact. Cancers 2020, 12, 3382. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Franco, A.; Darlington, E.; Carter, L.; Cook, N.; Graham, D.M.; Thistlethwaite, F.; Roxburgh, P.; Young, R.J.; Symeonides, S.N.; Basu, B.; et al. TARGET National: A U.K.-Wide Liquid-Based Molecular Profiling Program to Enhance Recruitment to Early-Phase Trials. J. Clin. Oncol. 2022, 40, TPS3163. [Google Scholar] [CrossRef]
- Rothwell, D.G.; Ayub, M.; Cook, N.; Thistlethwaite, F.; Carter, L.; Dean, E.; Smith, N.; Villa, S.; Dransfield, J.; Clipson, A.; et al. Utility of CtDNA to Support Patient Selection for Early Phase Clinical Trials: The TARGET Study. Nat. Med. 2019, 25, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Charo, L.M.; Eskander, R.N.; Okamura, R.; Patel, S.P.; Nikanjam, M.; Lanman, R.B.; Piccioni, D.E.; Kato, S.; McHale, M.T.; Kurzrock, R. Clinical Implications of Plasma Circulating Tumor DNA in Gynecologic Cancer Patients. Mol. Oncol. 2021, 15, 67–79. [Google Scholar] [CrossRef]
- Bertucci, F.; Gonçalves, A.; Guille, A.; Adelaïde, J.; Garnier, S.; Carbuccia, N.; Billon, E.; Finetti, P.; Sfumato, P.; Monneur, A.; et al. Prospective High-Throughput Genome Profiling of Advanced Cancers: Results of the PERMED-01 Clinical Trial. Genome Med. 2021, 13, 87. [Google Scholar] [CrossRef]
- Renaud, S.; Séverine, G.; Arnaud, G.; Nadine, C.; Jihane, P.; José, A.; Magali, P.; Maria, C.; Frédérique, R.; Max, C.; et al. Whole-Genome/Exome Analysis of Circulating Tumor DNA and Comparison to Tumor Genomics from Patients with Heavily Pre-Treated Ovarian Cancer: Subset Analysis of the PERMED-01 Trial. Front. Oncol. 2022, 12, 946257. [Google Scholar]
- Kevin, K.L.; Maria, I.H.; Amit, M.O.; Ana, O.; Isabelle, R.C.; Anna, V.T.; Elena, H.; Marc, R.R.; Carmen, S.; Lan-Thanh, V.; et al. BRCA Reversion Mutations in Circulating Tumor DNA Predict Primary and Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2019, 9, 210–219. [Google Scholar]
- Kim, Y.N.; Shim, Y.; Seo, J.; Choi, Z.; Lee, Y.J.; Shin, S.; Kim, S.W.; Kim, S.; Choi, J.R.; Lee, J.Y.; et al. Investigation of PARP Inhibitor Resistance Based on Serially Collected Circulating Tumor DNA in Patients With BRCA-Mutated Ovarian Cancer. Clin. Cancer Res. 2023, 29, 2725–2734. [Google Scholar] [CrossRef] [PubMed]
- Weigelt, B.; Comino-Méndez, I.; De Bruijn, I.; Tian, L.; Meisel, J.L.; García-Murillas, I.; Fribbens, C.; Cutts, R.; Martelotto, L.G.; Ng, C.K.Y.; et al. Diverse BRCA1 and BRCA2 Reversion Mutations in Circulating Cell-Free DNA of Therapy-Resistant Breast or Ovarian Cancer. Clin. Cancer Res. 2017, 23, 6708–6720. [Google Scholar] [CrossRef] [PubMed]
- Christie, E.L.; Fereday, S.; Doig, K.; Pattnaik, S.; Dawson, S.J.; Bowtell, D.D.L. Reversion of BRCA1/2 Germline Mutations Detected in Circulating Tumor DNA from Patients with High-Grade Serous Ovarian Cancer. J. Clin. Oncol. 2017, 35, 1274–1280. [Google Scholar] [CrossRef] [PubMed]
- Kondrashova, O.; Nguyen, M.; Shield-Artin, K.; Tinker, A.V.; Teng, N.N.H.; Harrell, M.I.; Kuiper, M.J.; Ho, G.Y.; Barker, H.; Jasin, M.; et al. Secondary Somatic Mutations Restoring RAD51C and RAD51D Associated with Acquired Resistance to the PARP Inhibitor Rucaparib in High-Grade Ovarian Carcinoma. Cancer Discov. 2017, 7, 984–998. [Google Scholar] [CrossRef] [PubMed]
- Paracchini, L.; Beltrame, L.; Grassi, T.; Inglesi, A.; Fruscio, R.; Landoni, F.; Ippolito, D.; Marchette, M.D.; Paderno, M.; Adorni, M.; et al. Genome-Wide Copy-Number Alterations in Circulating Tumor DNA as a Novel Biomarker for Patients with High-Grade Serous Ovarian Cancer. Clin. Cancer Res. 2021, 27, 2549–2559. [Google Scholar] [CrossRef]
- Hou, J.Y.; Chapman, J.S.; Kalashnikova, E.; Pierson, W.; Smith-McCune, K.; Pineda, G.; Vattakalam, R.M.; Ross, A.; Mills, M.; Suarez, C.J.; et al. Circulating Tumor DNA Monitoring for Early Recurrence Detection in Epithelial Ovarian Cancer. Gynecol. Oncol. 2022, 167, 334–341. [Google Scholar] [CrossRef]
- Chao, A.; Chen, S.J.; Chen, H.C.; Tan, K.T.; Hsiao, W.; Jung, S.M.; Yang, L.-Y.; Huang, K.-G.; Chou, H.-H.; Huang, H.-J.; et al. Mutations in Circulating Tumor DNA Detected in the Postoperative Period Predict Poor Survival in Patients with Ovarian Cancer. Biomed. J. 2022, 46, 100563. [Google Scholar] [CrossRef]
- Zhu, J.W.; Wong, F.; Szymiczek, A.; Ene, G.E.V.; Zhang, S.; May, T.; Narod, S.A.; Kotsopoulos, J.; Akbari, M.R. Evaluating the Utility of CtDNA in Detecting Residual Cancer and Predicting Recurrence in Patients with Serous Ovarian Cancer. Int. J. Mol. Sci. 2023, 24, 14388. [Google Scholar] [CrossRef]
- Marian, C.A.; Fernando, L.A.F.; Alayne, M.T.; Barros, L.A.D.R.; Lopes, A.; Silva, L.C.F.F.; Luz, A.S.; Cruz, F.J.S.M.; Del Giglio, A. Increased Circulating Tumor DNA as a Noninvasive Biomarker of Early Treatment Response in Patients with Metastatic Ovarian Carcinoma: A Pilot Study. Tumour Biol. 2020, 42, 1010428320919198. [Google Scholar]
- Alves, M.C.; Fonseca, F.L.A.; Yamada, A.M.T.D. Evaluation of Circulating Tumor DNA in Patients with Ovarian Cancer Harboring Somatic PIK3CA or KRAS Mutations. Cancer Res. Treat. 2020, 52, 1219–1228. [Google Scholar]
- Steffensen, K.D.; Madsen, C.V.; Andersen, R.F.; Waldstrøm, M.; Adimi, P.; Jakobsen, A. Prognostic Importance of Cell-Free DNA in Chemotherapy Resistant Ovarian Cancer Treated with Bevacizumab. Eur. J. Cancer 2014, 50, 2611–2618. [Google Scholar] [CrossRef] [PubMed]
- Lara, P.; Laura, M.; Luca, B.; Landoni, F.; Fruscio, R.; Grassi, T.; Dalessandro, M.L.; D’Incalci, M.; Marchini, S. Targeted Mutational Analysis of Circulating Tumor DNA to Decipher Temporal Heterogeneity of High-Grade Serous Ovarian Cancer. Cancers 2022, 14, 3697. [Google Scholar]
- Han, M.R.; Lee, S.H.; Park, J.Y.; Hong, H.; Ho, J.Y.; Hur, S.Y.; Choi, Y.J. Clinical Implications of Circulating Tumor DNA from Ascites and Serial Plasma in Ovarian Cancer. Cancer Res. Treat. 2020, 52, 779–788. [Google Scholar] [CrossRef] [PubMed]
- Lara, P.; Maurizio, D.; Sergio, M. Liquid Biopsy in the Clinical Management of High-Grade Serous Epithelial Ovarian Cancer-Current Use and Future Opportunities. Cancers 2021, 13, 2386. [Google Scholar]
- Barbosa, A.; Pinto, P.; Peixoto, A.; Guerra, J.; Pinheiro, M.; Santos, C.; Pinto, C.; Escudeiro, C.; Bartosch, C.; Santos, R.; et al. Next Generation Sequencing of Tumor and Matched Plasma Samples: Identification of Somatic Variants in CtDNA From Ovarian Cancer Patients. Front. Oncol. 2021, 11, 754094. [Google Scholar] [CrossRef]
- Gerratana, L.; Movarek, M.; Wehbe, F.; Katam, N.; Mahalingam, D.; Donahue, J.; Shah, A.; Chae, Y.K.; Mulcahy, M.; Tsarwhas, D.; et al. Genomic Landscape of Advanced Solid Tumors in Circulating Tumor DNA and Correlation With Tissue Sequencing: A Single Institution’s Experience. JCO Precis. Oncol. 2022, 6, e2100289. [Google Scholar] [CrossRef]
- Simone, K.T.; Malene, P.S.; Karen, D.; Inge, S.P. Early Diagnosis of Ovarian Cancer Based on Methylation Profiles in Peripheral Blood Cell-Free DNA: A Systematic Review. Clin. Epigenet. 2023, 15, 24. [Google Scholar]
- Sandeep, S.K.; Swamy, S.N.; Premalatha, C.S.; Pallavi, V.R.; Gawari, R. Aberrant Promoter Hypermethylation of RASSF1a and BRCA1 in Circulating Cell-Free Tumor DNA Serves as a Biomarker of Ovarian Carcinoma. Asian Pac. J. Cancer Prev. 2019, 20, 3001–3005. [Google Scholar]
- Trinidad, C.V.; Tetlow, A.L.; Bantis, L.E.; Godwin, A.K. Reducing Ovarian Cancer Mortality Through Early Detection: Approaches Using Circulating Biomarkers. Cancer Prev. Res. 2020, 13, 241–252. [Google Scholar] [CrossRef]
- Liggett, T.E.; Melnikov, A.; Yi, Q.; Replogle, C.; Hu, W.; Rotmensch, J.; Kamat, A.; Sood, A.K.; Levenson, V. Distinctive DNA Methylation Patterns of Cell-Free Plasma DNA in Women with Malignant Ovarian Tumors. Gynecol. Oncol. 2011, 120, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Alka, S.; Sameer, G.; Jaydeep, A.B.; Manisha, S. Detection of Aberrant Methylation of HOXA9 and HIC1 through Multiplex MethyLight Assay in Serum DNA for the Early Detection of Epithelial Ovarian Cancer. Int. J. Cancer 2020, 147, 1740–1752. [Google Scholar]
- Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Swanton, C.; Seiden, M.V.; Liu, M.C.; Oxnard, G.R.; Klein, E.A.; Smith, D.; Richards, D.; et al. Sensitive and Specific Multi-Cancer Detection and Localization Using Methylation Signatures in Cell-Free DNA. Ann. Oncol. 2020, 31, 745–759. [Google Scholar] [CrossRef] [PubMed]
- Dana, D.; Dušan, B.; Bálint, N.; Grendár, M.; Poka, R.; Soltész, B.; Jagelková, M.; Zelinová, K.; Lasabová, Z.; Zubor, P.; et al. Aberrant Methylation Status of Tumour Suppressor Genes in Ovarian Cancer Tissue and Paired Plasma Samples. Int. J. Mol. Sci. 2019, 20, 4119. [Google Scholar]
- Flanagan, J.M.; Wilhelm-Benartzi, C.S.; Metcalf, M.; Kaye, S.B.; Brown, R. Association of Somatic DNA Methylation Variability with Progression-Free Survival and Toxicity in Ovarian Cancer Patients. Ann. Oncol. 2013, 24, 2813–2818. [Google Scholar] [CrossRef] [PubMed]
- Flanagan, J.M.; Wilson, A.; Koo, C.; Masrour, N.; Gallon, J.; Loomis, E.; Flower, K.; Wilhelm-Benartzi, C.; Hergovich, A.; Cunnea, P.; et al. Platinum-Based Chemotherapy Induces Methylation Changes in Blood DNA Associated with Overall Survival in Patients with Ovarian Cancer. Clin. Cancer Res. 2017, 23, 2213–2222. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Zhang, Y.; Li, C.; Liao, Y.; Wang, G.; Xu, J.; Li, Y.; Yuan, G.; Sun, Y.; Zhang, R. Plasma CfDNA Methylation Markers for the Detection and Prognosis of Ovarian Cancer. EBioMedicine 2022, 83, 104222. [Google Scholar] [CrossRef] [PubMed]
- Tomar, T.; Alkema, N.G.; Schreuder, L.; Meersma, G.J.; de Meyer, T.; van Criekinge, W.; Klip, H.G.; Fiegl, H.; van Nieuwenhuysen, E.; Vergote, I.; et al. Methylome Analysis of Extreme Chemoresponsive Patients Identifies Novel Markers of Platinum Sensitivity in High-Grade Serous Ovarian Cancer. BMC Med. 2017, 15, 116. [Google Scholar] [CrossRef] [PubMed]
- Houshdaran, S.; Hawley, S.; Palmer, C.; Campan, M.; Olsen, M.N.; Ventura, A.P.; Knudsen, B.S.; Drescher, C.W.; Urban, N.D.; Brown, P.O.; et al. DNA Methylation Profiles of Ovarian Epithelial Carcinoma Tumors and Cell Lines. PLoS ONE 2010, 5, e9359. [Google Scholar] [CrossRef] [PubMed]
- Rodger, E.J.; Almomani, S.N.; Ludgate, J.L.; Stockwell, P.A.; Baguley, B.C.; Eccles, M.R.; Chatterjee, A. Comparison of Global DNA Methylation Patterns in Human Melanoma Tissues and Their Derivative Cell Lines. Cancers 2021, 13, 2123. [Google Scholar] [CrossRef]
- Ohnmacht, A.J.; Rajamani, A.; Avar, G.; Kutkaite, G.; Gonçalves, E.; Saur, D.; Menden, M.P. The Pharmacoepigenomic Landscape of Cancer Cell Lines Reveals the Epigenetic Component of Drug Sensitivity. Commun. Biol. 2023, 6, 825. [Google Scholar] [CrossRef] [PubMed]
- Balgkouranidou, I.; Chimonidou, M.; Milaki, G.; Tsarouxa, E.G.; Kakolyris, S.; Welch, D.R.; Georgoulias, V.; Lianidou, E.S. Breast Cancer Metastasis Suppressor-1 Promoter Methylation in Cell-Free DNA Provides Prognostic Information in Non-Small Cell Lung Cancer. Br. J. Cancer 2014, 110, 2054–2062. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.A.; Vikramdeo, K.S.; Sudan, S.K.; Singh, S.; Wilhite, A.; Dasgupta, S.; Rocconi, R.P.; Singh, A.P. Platinum-Resistant Ovarian Cancer: From Drug Resistance Mechanisms to Liquid Biopsy-Based Biomarkers for Disease Management. Semin. Cancer Biol. 2021, 77, 99–109. [Google Scholar] [CrossRef]
- Maha, E.; Katharina, P.; Lan, K.; Oliveira-Ferrer, L.; Peine, S.; Müller, V.; Woelber, L.; Schmalfeldt, B.; Pantel, K.; Joosse, S.A. BRCA1 Promoter Hypermethylation on Circulating Tumor DNA Correlates with Improved Survival of Patients with Ovarian Cancer. Mol. Oncol. 2021, 15, 3615–3625. [Google Scholar]
- Kalachand, R.D.; O’Riain, C.; Toomey, S.; Carr, A.; Timms, K.M.; O’Toole, S.; Madden, S.; Bates, M.; O’Leary, J.J.; Gleeson, N.; et al. Prevalence of Tumor BRCA1 and BRCA2 Dysfunction in Unselected Patients with Ovarian Cancer. Obs. Gynecol. Sci. 2020, 63, 643–654. [Google Scholar] [CrossRef]
- Olga, K.; Monique, T.; Ksenija, N.; Lieschke, E.; Ho, G.-Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 28, 3970. [Google Scholar]
- Maria, R.; Rikke, F.A.; Anders, J.; Karina, D.S. Circulating HOXA9-Methylated Tumour DNA: A Novel Biomarker of Response to Poly (ADP-Ribose) Polymerase Inhibition in BRCA-Mutated Epithelial Ovarian Cancer. Eur. J. Cancer 2020, 125, 121–129. [Google Scholar]
- Pandya, D.; Camacho, S.C.; Padron, M.M.; Camacho-Vanegas, O.; Billaud, J.-N.; Beddoe, A.-M.; Irish, J.; Yoxtheimer, L.; Kalir, T.; RoseFigura, J.; et al. Rapid Development and Use of Patient-Specific CtDNA Biomarkers to Avoid a “Rash Decision” in an Ovarian Cancer Patient. Mol. Case Stud. 2019, 5, a004648. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| CA-125 | ctDNA |
|---|---|
| Non-invasive | |
| Can be altered by other coexisting physiological and pathological conditions | |
| Inexpensive and highly available | Expensive and restricted to specialist centres |
| Simple methodology | Complex methodology |
| Results in minutes-hours | Results in days and weeks |
| Quantitative marker | Quantitative and qualitative markers |
| One continuous variable | Can generate multiple continuous and discrete variables |
| Only informative regarding the presence/absence of treatment response and recurrence | Yields more information regarding treatment response and tumour recurrence, like resistance mechanisms and targetable genetic alterations |
| Directly interpreted by the clinician | Requires specialised interpretation |
| Easily detected in blood and urine | Low concentrations in biological fluids |
| The utility is limited to producing tumours (mainly restricted to HGSOC) | Theoretically applicable to all histological subtypes |
| Established and recognised clinical utility in trials | Clinical utility is debatable and requires confirmation in prospective trials |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roque, R.; Ribeiro, I.P.; Figueiredo-Dias, M.; Gourley, C.; Carreira, I.M. Current Applications and Challenges of Next-Generation Sequencing in Plasma Circulating Tumour DNA of Ovarian Cancer. Biology 2024, 13, 88. https://doi.org/10.3390/biology13020088
Roque R, Ribeiro IP, Figueiredo-Dias M, Gourley C, Carreira IM. Current Applications and Challenges of Next-Generation Sequencing in Plasma Circulating Tumour DNA of Ovarian Cancer. Biology. 2024; 13(2):88. https://doi.org/10.3390/biology13020088
Chicago/Turabian StyleRoque, Ricardo, Ilda Patrícia Ribeiro, Margarida Figueiredo-Dias, Charlie Gourley, and Isabel Marques Carreira. 2024. "Current Applications and Challenges of Next-Generation Sequencing in Plasma Circulating Tumour DNA of Ovarian Cancer" Biology 13, no. 2: 88. https://doi.org/10.3390/biology13020088
APA StyleRoque, R., Ribeiro, I. P., Figueiredo-Dias, M., Gourley, C., & Carreira, I. M. (2024). Current Applications and Challenges of Next-Generation Sequencing in Plasma Circulating Tumour DNA of Ovarian Cancer. Biology, 13(2), 88. https://doi.org/10.3390/biology13020088