
Personalized Driver Gene Prediction Using Graph Convolutional Networks with Conditional Random Fields


Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Datasets
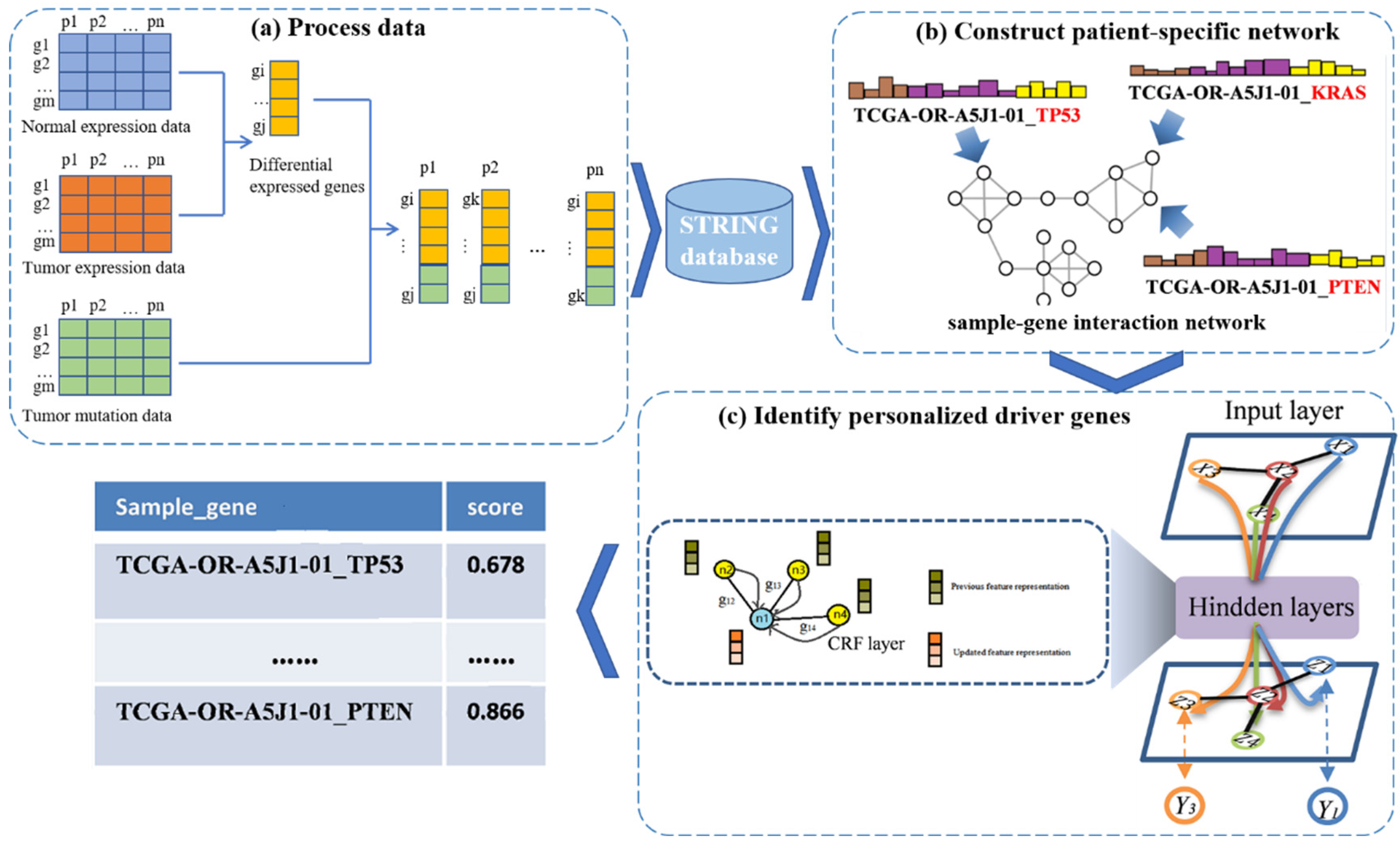
2.2. PDGCN
2.2.1. Construction of Personalized Networks
2.2.2. Graph Convolutional Network for Node Embedding
2.2.3. CRF Layer for Embedding Update
2.2.4. Overall Loss and Optimization
3. Results
3.1. Experimental Setup
3.2. Evaluation Metrics
3.3. Effect of Node2vec Dimensions
3.4. Ablation Experiments
3.5. Effects of Different Weights in Loss Function
3.6. Analysis at the Population Level
3.7. Analysis at the Individual Level
3.8. Analysis of Identifying Rare Drivers
3.9. Survival Analysis
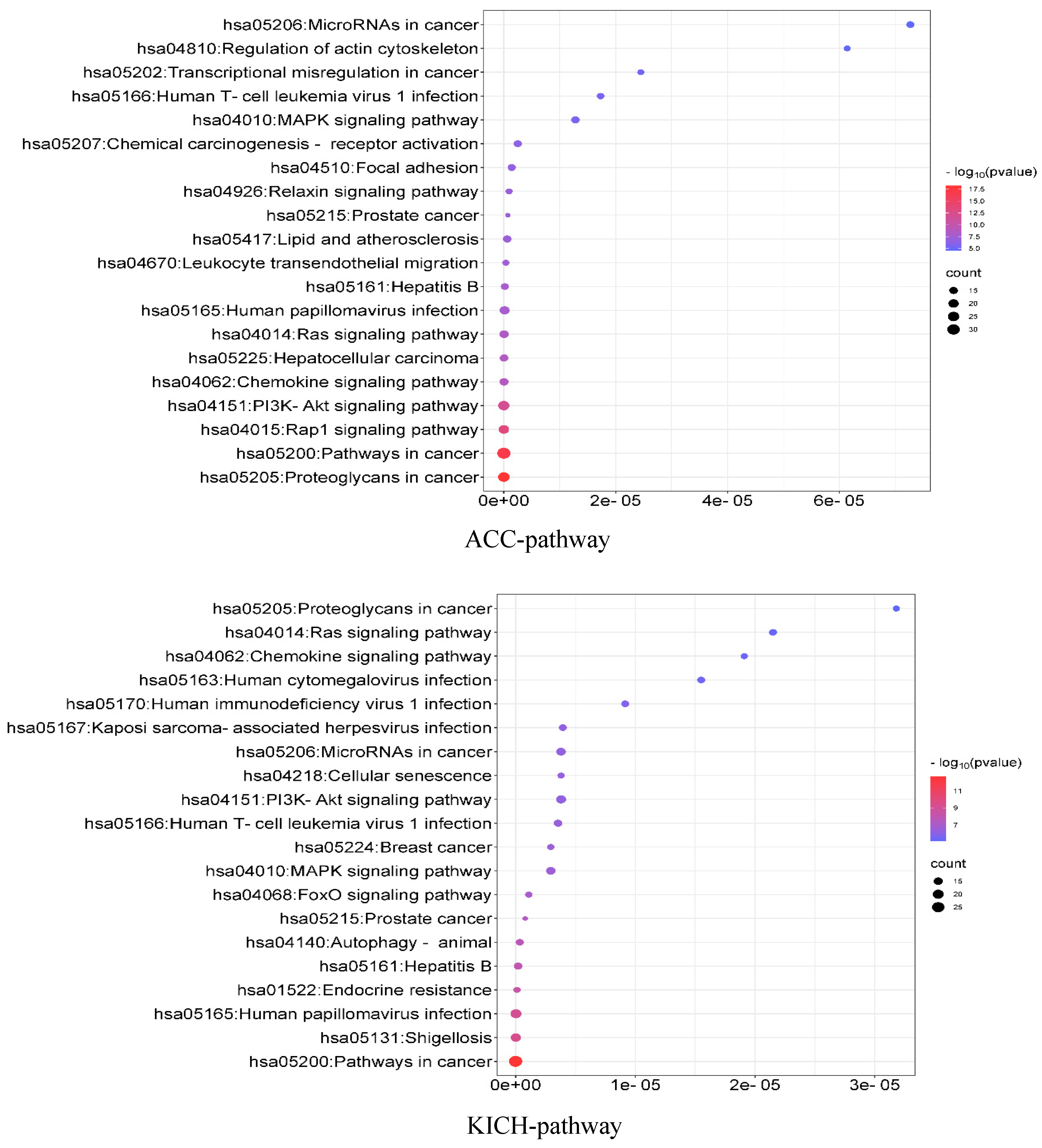
3.10. Enrichment Analysis
4. Conclusions and Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef]
- Loomans-Kropp, H.A.; Umar, A. Cancer prevention and screening: The next step in the era of precision medicine. NPJ Precis. Oncol. 2019, 3, 3. [Google Scholar] [CrossRef] [PubMed]
- Anandakrishnan, R.; Varghese, R.T.; Kinney, N.A.; Garner, H.R. Estimating the number of genetic mutations (hits) required for carcinogenesis based on the distribution of somatic mutations. PLoS Comput. Biol. 2019, 15, e1006881. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Martínez-Jiménez, F.; Muiños, F.; Sentís, I.; Deu-Pons, J.; Reyes-Salazar, I.; Arnedo-Pac, C.; Mularoni, L.; Pich, O.; Bonet, J.; Kranas, H. A compendium of mutational cancer driver genes. Nat. Rev. Cancer 2020, 20, 555–572. [Google Scholar] [CrossRef]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar] [CrossRef]
- Banerji, S.; Cibulskis, K.; Rangel-Escareno, C.; Brown, K.K.; Carter, S.L.; Frederick, A.M.; Lawrence, M.S.; Sivachenko, A.Y.; Sougnez, C.; Zou, L.; et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012, 486, 405–409. [Google Scholar] [CrossRef]
- Dees, N.D.; Zhang, Q.; Kandoth, C.; Wendl, M.C.; Schierding, W.; Koboldt, D.C.; Mooney, T.B.; Callaway, M.B.; Dooling, D.; Mardis, E.R.; et al. MuSiC: Identifying mutational significance in cancer genomes. Genome Res. 2012, 22, 1589–1598. [Google Scholar] [CrossRef]
- Tamborero, D.; Gonzalez-Perez, A.; Lopez-Bigas, N. OncodriveCLUST: Exploiting the positional clustering of somatic mutations to identify cancer genes. Bioinformatics 2013, 29, 2238–2244. [Google Scholar] [CrossRef]
- Wood, L.D.; Parsons, D.W.; Jones, S.; Lin, J.; Sjoblom, T.; Leary, R.J.; Shen, D.; Boca, S.M.; Barber, T.; Ptak, J.; et al. The genomic landscapes of human breast and colorectal cancers. Science 2007, 318, 1108–1113. [Google Scholar] [CrossRef]
- Bashashati, A.; Haffari, G.; Ding, J.; Ha, G.; Lui, K.; Rosner, J.; Huntsman, D.G.; Caldas, C.; Aparicio, S.A.; Shah, S.P. DriverNet: Uncovering the impact of somatic driver mutations on transcriptional networks in cancer. Genome Biol. 2012, 13, 1–14. [Google Scholar] [CrossRef]
- Leiserson, M.D.; Vandin, F.; Wu, H.-T.; Dobson, J.R.; Eldridge, J.V.; Thomas, J.L.; Papoutsaki, A.; Kim, Y.; Niu, B.; McLellan, M. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat. Genet. 2015, 47, 106–114. [Google Scholar] [CrossRef]
- Schulte-Sasse, R.; Budach, S.; Hnisz, D.; Marsico, A. Integration of multiomics data with graph convolutional networks to identify new cancer genes and their associated molecular mechanisms. Nat. Mach. Intell. 2021, 3, 513–526. [Google Scholar] [CrossRef]
- Peng, W.; Tang, Q.; Dai, W.; Chen, T. Improving cancer driver gene identification using multi-task learning on graph convolutional network. Brief. Bioinform. 2022, 23, bbab432. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, D.; Chng, K.R.; Sherbaf, F.G.; Kiesel, A.; Chia, B.K.; Sia, Y.Y.; Huang, S.K.; Hoon, D.S.; Liu, E.T.; Hillmer, A. Patient-specific driver gene prediction and risk assessment through integrated network analysis of cancer omics profiles. Nucleic Acids Res. 2015, 43, e44. [Google Scholar] [CrossRef]
- Hou, J.P.; Ma, J. DawnRank: Discovering personalized driver genes in cancer. Genome Med. 2014, 6, 56. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, Y.; Ji, H.; Aihara, K.; Chen, L. Personalized characterization of diseases using sample-specific networks. Nucleic Acids Res. 2016, 44, e164. [Google Scholar] [CrossRef]
- Guo, W.-F.; Zhang, S.-W.; Liu, L.-L.; Liu, F.; Shi, Q.-Q.; Zhang, L.; Tang, Y.; Zeng, T.; Chen, L. Discovering personalized driver mutation profiles of single samples in cancer by network control strategy. Bioinformatics 2018, 34, 1893–1903. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhang, S.-W.; Zeng, T.; Li, Y.; Gao, J.; Chen, L. A novel network control model for identifying personalized driver genes in cancer. PLoS Comput. Biol. 2019, 15, e1007520. [Google Scholar] [CrossRef]
- Pham, V.V.H.; Liu, L.; Bracken, C.P.; Nguyen, T.; Goodall, G.J.; Li, J.; Le, T.D. pDriver: A novel method for unravelling personalized coding and miRNA cancer drivers. Bioinformatics 2021, 37, 3285–3292. [Google Scholar] [CrossRef]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Canc Genome Atlas Res, N. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef]
- Goldman, M.J.; Craft, B.; Hastie, M.; Repecka, K.; McDade, F.; Kamath, A.; Banerjee, A.; Luo, Y.; Rogers, D.; Brooks, A.N.; et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat. Biotechnol. 2020, 38, 675–678. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING Database in 2021: Customizable Protein-Protein Networks, and Functional Characterization of User-Uploaded Gene/Measurement Sets. Nucleic Acids Res. 2021, 49, D605–D612. Available online: https://cn.string-db.org/ (accessed on 1 July 2022). [CrossRef] [PubMed]
- Repana, D.; Nulsen, J.; Dressler, L.; Bortolomeazzi, M.; Venkata, S.K.; Tourna, A.; Yakovleva, A.; Palmieri, T.; Ciccarelli, F.D. The Network of Cancer Genes (NCG): A comprehensive catalogue of known and candidate cancer genes from cancer sequencing screens. Genome Biol. 2019, 20, 1. [Google Scholar] [CrossRef] [PubMed]
- Futreal, P.A.; Coin, L.; Marshall, M.; Down, T.; Hubbard, T.; Wooster, R.; Rahman, N.; Stratton, M.R. A census of human cancer genes. Nat. Rev. Cancer 2004, 4, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B. Comprehensive characterization of cancer driver genes and mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [PubMed]
- Nulsen, J.; Misetic, H.; Yau, C.; Ciccarelli, F.D. Pan-cancer detection of driver genes at the single-patient resolution. Genome Med. 2021, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Grover, A.; Leskovec, J.; Assoc Comp, M. node2vec: Scalable Feature Learning for Networks. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD), San Francisco, CA, USA, 13–17 August 2016; pp. 855–864. [Google Scholar]
- Perozzi, B.; Al-Rfou, R.; Skiena, S. DeepWalk: Online Learning of Social Representations. In Proceedings of the 20th ACM SIGKDD International Conference on Knowledge Discovery and Data Mining (KDD), New York, NY, USA, 24–27 August 2014; ACM: New York, NY, USA, 2014; pp. 701–710. [Google Scholar]
- Niepert, M.; Ahmed, M.; Kutzkov, K. Learning Convolutional Neural Networks for Graphs. In Proceedings of the 33rd International Conference on Machine Learning, New York, NY, USA, 20–22 June 2016; pp. 2014–2023. [Google Scholar]
- Lafferty, J.; McCallum, A.; Pereira, F.C. Conditional random fields: Probabilistic models for segmenting and labeling sequence data. In ICML; Williams College: Williamstown, MA, USA, 2001; pp. 282–289. [Google Scholar]
- Long, Y.; Wu, M.; Kwoh, C.K.; Luo, J.; Li, X. Predicting human microbe-drug associations via graph convolutional network with conditional random field. Bioinformatics 2020, 36, 4918–4927. [Google Scholar] [CrossRef] [PubMed]
- Vaswani, A.; Shazeer, N.; Parmar, N.; Uszkoreit, J.; Jones, L.; Gomez, A.N.; Kaiser, L.; Polosukhin, I. Attention Is All You Need. In Proceedings of the 31st Annual Conference on Neural Information Processing Systems (NIPS), Long Beach, CA, USA, 4–9 December 2017; pp. 5998–6008. [Google Scholar]
- Zhao, W.; Gu, X.; Chen, S.; Wu, J.; Zhou, Z. MODIG: Integrating multi-omics and multi-dimensional gene network for cancer driver gene identification based on graph attention network model. Bioinformatics 2022, 38, 4901–4907. [Google Scholar] [CrossRef]
- Zhao, S.; Liu, J.; Nanga, P.; Liu, Y.; Cicek, A.E.; Knoblauch, N.; He, C.; Stephens, M.; He, X. Detailed modeling of positive selection improves detection of cancer driver genes. Nat. Commun. 2019, 10, 3399. [Google Scholar] [CrossRef]
- Shi, X.; Teng, H.; Shi, L.; Bi, W.; Wei, W.; Mao, F.; Sun, Z. Comprehensive evaluation of computational methods for predicting cancer driver genes. Brief. Bioinform. 2022, 23, bbab548. [Google Scholar] [CrossRef]
- Tang, Z.; Kang, B.; Li, C.; Chen, T.; Zhang, Z. GEPIA2: An enhanced web server for large-scale expression profiling and interactive analysis. Nucleic Acids Res. 2019, 47, W556–W560. [Google Scholar] [CrossRef]
- Zhang, S.-W.; Wang, Z.-N.; Li, Y.; Guo, W.-F. Prioritization of cancer driver gene with prize-collecting steiner tree by introducing an edge weighted strategy in the personalized gene interaction network. Bmc Bioinform. 2022, 23, 341. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID Bioinformatics Resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Huang, V.; Zheng, J.; Qi, Z.; Wang, J.; Place, R.F.; Yu, J.; Li, H.; Li, L.-C. Ago1 Interacts with RNA Polymerase II and Binds to the Promoters of Actively Transcribed Genes in Human Cancer Cells. PLoS Genet. 2013, 9, e1003821. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, D.; Grondal, S.; Rydstrom, J.; DePierre, J.W. Levels of cytochrome P-450, steroidogenesis and microsomal and cytosolic epoxide hydrolases in normal human adrenal tissue and corresponding tumors. Cancer Biochem. Biophys. 1992, 12, 283–291. [Google Scholar] [PubMed]
- Patalano, A.; Brancato, V.; Mantero, F. Adrenocortical Cancer Treatment. Horm. Res. 2009, 71, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Gomperts, B.D.; Tatham, P.E.; Kramer, I.M. Signal Transduction; Gulf Professional Publishing: Oxford, UK, 2002. [Google Scholar]
- Akhtar, M.; Kardar, H.; Linjawi, T.; McClintock, J.; Ali, M.A. Chromophobe cell carcinoma of the kidney. A clinicopathologic study of 21 cases. Am. J. Surg. Pathol. 1995, 19, 1245–1256. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, G.; Pea, M.; Chilosi, M.; Brunelli, M.; Scarpa, A.; Colato, C.; Tardanico, R.; Zamboni, G.; Bonetti, F. Parvalbumin is constantly expressed in chromophobe renal carcinoma. Mod. Pathol. 2001, 14, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Lin, Z.; Ye, Y.; Luo, R.; Zeng, L. ARRB2 promotes colorectal cancer growth through triggering WTAP. Acta Biochim. Et Biophys. Sin. 2021, 53, 85–93. [Google Scholar] [CrossRef]
- Jiang, T.; Yu, J.T.; Wang, Y.L.; Wang, H.F.; Zhang, W.; Hu, N.; Tan, L.; Sun, L.; Tan, M.S.; Zhu, X.C. The genetic variation of ARRB2 is associated with late-onset Alzheimer’s disease in Han Chinese. Curr. Alzheimer Res. 2014, 11, 408–412. [Google Scholar] [CrossRef]
- Zhou, B.; Song, H.; Xu, W.; Zhang, Y.; Liu, Y.; Qi, W. The Comprehensive Analysis of Hub Gene ARRB2 in Prostate Cancer. Dis. Markers 2022, 2022, 8518378. [Google Scholar] [CrossRef]
- Ma, T.; Zhao, Y.; Wei, K.; Yao, G.; Pan, C.; Liu, B.; Xia, Y.; He, Z.; Qi, X.; Li, Z. MicroRNA-124 functions as a tumor suppressor by regulating CDH2 and epithelial-mesenchymal transition in non-small cell lung cancer. Cell. Physiol. Biochem. 2016, 38, 1563–1574. [Google Scholar] [CrossRef]
- Miao, J.; Wang, W.; Wu, S.; Zang, X.; Li, Y.; Wang, J.; Zhan, R.; Gao, M.; Hu, M.; Li, J. miR-194 suppresses proliferation and migration and promotes apoptosis of osteosarcoma cells by targeting CDH2. Cell. Physiol. Biochem. 2018, 45, 1966–1974. [Google Scholar] [CrossRef]
- Chen, Q.; Cai, J.; Jiang, C. CDH2 expression is of prognostic significance in glioma and predicts the efficacy of temozolomide therapy in patients with glioblastοma. Oncol. Lett. 2018, 15, 7415–7422. [Google Scholar] [PubMed]
- Cao, L.; Cheng, H.; Jiang, Q.; Li, H.; Wu, Z. APEX1 is a novel diagnostic and prognostic biomarker for hepatocellular carcinoma. Aging 2020, 12, 4573. [Google Scholar] [CrossRef] [PubMed]
- Tummanatsakun, D.; Proungvitaya, T.; Roytrakul, S.; Limpaiboon, T.; Wongkham, S.; Wongkham, C.; Silsirivanit, A.; Somintara, O.; Sangkhamanon, S.; Proungvitaya, S. Serum apurinic/apyrimidinic endodeoxyribonuclease 1 (APEX1) level as a potential biomarker of cholangiocarcinoma. Biomolecules 2019, 9, 413. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Yang, D.; Cogdell, D.; Du, X.; Li, H.; Pang, Y.; Sun, Y.; Hu, L.; Sun, B.; Trent, J. APEX1 gene amplification and its protein overexpression in osteosarcoma: Correlation with recurrence, metastasis, and survival. Technol. Cancer Res. Treat. 2010, 9, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Feng, Q.; Qi, Y.; Cui, G.; Zhao, S. PPARGC1A is upregulated and facilitates lung cancer metastasis. Exp. Cell Res. 2017, 359, 356–360. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Wang, W.; Li, Y.; Yang, D.; Li, X.; Shen, C.; Liu, Y.; Ke, X.; Guo, S.; Guo, Z. HSP90AA1-mediated autophagy promotes drug resistance in osteosarcoma. J. Exp. Clin. Cancer Res. 2018, 37, 201. [Google Scholar] [CrossRef] [PubMed]
- Okino, K.; Nagai, H.; Hatta, M.; Nagahata, T.; Yoneyama, K.; Ohta, Y.; Jin, E.; Kawanami, O.; Araki, T.; Emi, M. Up-regulation and overproduction of DVL-1, the human counterpart of the Drosophila dishevelled gene, in cervical squamous cell carcinoma. Oncol. Rep. 2003, 10, 1219–1223. [Google Scholar] [CrossRef] [PubMed]
- Nagahata, T.; Shimada, T.; Harada, A.; Nagai, H.; Onda, M.; Yokoyama, S.; Shiba, T.; Jin, E.; Kawanami, O.; Emi, M. Amplification, up-regulation and over-expression of DVL-1, the human counterpart of the Drosophila disheveled gene, in primary breast cancers. Cancer Sci. 2003, 94, 515–518. [Google Scholar] [CrossRef]
- Smith, M.J.; O’Sullivan, J.; Bhaskar, S.S.; Hadfield, K.D.; Poke, G.; Caird, J.; Sharif, S.; Eccles, D.; Fitzpatrick, D.; Rawluk, D. Loss-of-function mutations in SMARCE1 cause an inherited disorder of multiple spinal meningiomas. Nat. Genet. 2013, 45, 295–298. [Google Scholar] [CrossRef]
- Wang, P.; Xie, M.; Yang, D.; Wang, F.; Chen, E. Integrative multi-omics analysis reveals the landscape of Cyclin-Dependent Kinase (CDK) family genes in pan-cancer. Res. Sq. 2022. [Google Scholar] [CrossRef]
- Caliskan, A.; Andac, A.C.; Arga, K.Y. Novel molecular signatures and potential therapeutics in renal cell carcinomas: Insights from a comparative analysis of subtypes. Genomics 2020, 112, 3166–3178. [Google Scholar] [CrossRef]
- Zhou, L.; Yin, B.; Liu, Y.; Hong, Y.; Zhang, C.; Fan, J. Mechanism and function of decreased FOXO1 in renal cell carcinoma. J. Surg. Oncol. 2012, 105, 841–847. [Google Scholar] [CrossRef]
- Kojima, T.; Shimazui, T.; Horie, R.; Hinotsu, S.; Oikawa, T.; Kawai, K.; Suzuki, H.; Meno, K.; Akaza, H.; Uchida, K. FOXO1 and TCF7L2 genes involved in metastasis and poor prognosis in clear cell renal cell carcinoma. Genes Chromosomes Cancer 2010, 49, 379–389. [Google Scholar] [CrossRef]
- Xu, J.; Perecman, A.; Wiggins, A.; Kalantzakos, T.; Das, S.; Sullivan, T.; Rieger-Christ, K. MetastamiRs in Renal Cell Carcinoma: An Overview of MicroRNA Implicated in Metastatic Kidney Cancer. Exon Publ. 2022, 71–93. [Google Scholar]
- Erdem, M.; Erdem, S.; Sanli, O.; Sak, H.; Kilicaslan, I.; Sahin, F.; Telci, D. Up-regulation of TGM2 with ITGB1 and SDC4 is important in the development and metastasis of renal cell carcinoma. In Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2014; Volume 32, pp. 25.e13–25.e20. [Google Scholar]
- Bruder, E.; Moch, H.; Ehrlich, D.; Leuschner, I.; Harms, D.; Argani, P.; Briner, J.; Graf, N.; Selle, B.; Rufle, A. Wnt signaling pathway analysis in renal cell carcinoma in young patients. Mod. Pathol. 2007, 20, 1217–1229. [Google Scholar] [CrossRef] [PubMed]
- Cui, J.; Yuan, Y.; Shanmugam, M.K.; Anbalagan, D.; Tan, T.Z.; Sethi, G.; Kumar, A.P.; Lim, L.H.K. MicroRNA-196a promotes renal cancer cell migration and invasion by targeting BRAM1 to regulate SMAD and MAPK signaling pathways. Int. J. Biol. Sci. 2021, 17, 4254. [Google Scholar] [CrossRef] [PubMed]
- Dirim, A.; Haberal, A.N.; Goren, M.R.; Tekin, M.I.; Peskircioglu, L.; Demirhan, B.; Ozkardes, H. VEGF, COX-2, and PCNA expression in renal cell carcinoma subtypes and their prognostic value. Int. Urol. Nephrol. 2008, 40, 861–868. [Google Scholar] [CrossRef] [PubMed]
- Altintas, E.; Kaynar, M.; Celik, Z.E.; Celik, M.; Kilic, O.; Akand, M.; Goktas, S. Expression of Ring Box-1 protein and its relationship with Fuhrman grade and other clinical-pathological parameters in renal cell cancer. In Urologic Oncology: Seminars and Original Investigations; Elsevier: Amsterdam, The Netherlands, 2020; Volume 38, pp. 6.e17–6.e22. [Google Scholar]
- Chen, C.; Chi, H.; Min, L.; Junhua, Z. Downregulation of guanine nucleotide-binding protein beta 1 (GNB1) is associated with worsened prognosis of clearcell renal cell carcinoma and is related to VEGF signaling pathway. JBUON 2017, 22, 1441–1446. [Google Scholar]
- Gara, S.K.; Wang, Y.; Patel, D.; Liu-Chittenden, Y.; Jain, M.; Boufraqech, M.; Zhang, L.; Meltzer, P.S.; Kebebew, E. Integrated genome-wide analysis of genomic changes and gene regulation in human adrenocortical tissue samples. Nucleic Acids Res. 2015, 43, 9327–9339. [Google Scholar] [CrossRef]
- Kaidi, A.; Jackson, S.P. KAT5 tyrosine phosphorylation couples chromatin sensing to ATM signalling. Nature 2013, 498, 70–74. [Google Scholar] [CrossRef]
- Mouat, I.C.; Omata, K.; McDaniel, A.S.; Hattangady, N.G.; Talapatra, D.; Cani, A.K.; Hovelson, D.H.; Tomlins, S.A.; Rainey, W.E.; Hammer, G.D. Somatic mutations in adrenocortical carcinoma with primary aldosteronism or hyperreninemic hyperaldosteronism. Endocr.-Relat. Cancer 2019, 26, 217–225. [Google Scholar] [CrossRef]
- Lin, S.; Qiu, L.; Liang, K.; Zhang, H.; Xian, M.; Chen, Z.; Wei, J.; Fu, S.; Gong, X.; Ding, K. KAT2A/E2F1 Promotes Cell Proliferation and Migration via Upregulating the Expression of UBE2C in Pan-Cancer. Genes 2022, 13, 1817. [Google Scholar] [CrossRef]
- Altieri, B.; Ronchi, C.L.; Kroiss, M.; Fassnacht, M. Next-generation therapies for adrenocortical carcinoma. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101434. [Google Scholar] [CrossRef]
- Shaikh, L.H.; Zhou, J.; Teo, A.E.D.; Garg, S.; Neogi, S.G.; Figg, N.; Yeo, G.S.; Yu, H.; Maguire, J.J.; Zhao, W. LGR5 activates noncanonical Wnt signaling and inhibits aldosterone production in the human adrenal. J. Clin. Endocrinol. Metab. 2015, 100, E836–E844. [Google Scholar] [CrossRef]
- Ruggiero, C.; Lalli, E. VAV2: A novel prognostic marker and a druggable target for adrenocortical carcinoma. Oncotarget 2017, 8, 88257. [Google Scholar] [CrossRef]
- Ruggiero, C.; Doghman-Bouguerra, M.; Sbiera, S.; Sbiera, I.; Parsons, M.; Ragazzon, B.; Morin, A.; Robidel, E.; Favier, J.; Bertherat, J.; et al. Dosage-dependent regulation of VAV2 expression by steroidogenic factor-1 drives adrenocortical carcinoma cell invasion. Sci. Signal. 2017, 10, eaal2464. [Google Scholar] [CrossRef]
- Parviainen, H.; Schrade, A.; Kiiveri, S.; Prunskaite-Hyyrylainen, R.; Haglund, C.; Vainio, S.; Wilson, D.B.; Arola, J.; Heikinheimo, M. Expression of Wnt and TGF-β pathway components and key adrenal transcription factors in adrenocortical tumors: Association to carcinoma aggressiveness. Pathol.-Res. Pract. 2013, 209, 503–509. [Google Scholar] [CrossRef]
- Zhu, Y.; Wang, M.; Zhao, X.; Zhang, L.; Wu, Y.; Wang, B.; Hu, W. Rottlerin as a novel chemotherapy agent for adrenocortical carcinoma. Oncotarget 2017, 8, 22825. [Google Scholar] [CrossRef]
- Gayarre, J.; Kamieniak, M.M.; Cazorla-Jiménez, A.; Muñoz-Repeto, I.; Borrego, S.; García-Donas, J.; Hernando, S.; Robles-Díaz, L.; García-Bueno, J.M.; y Cajal, T.R. The NER-related gene GTF2H5 predicts survival in high-grade serous ovarian cancer patients. J. Gynecol. Oncol. 2016, 27. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, M.; Fogarty, E.; Janga, M.; Surendran, K. Notch signaling in kidney development, maintenance, and disease. Biomolecules 2019, 9, 692. [Google Scholar] [CrossRef]
- Peri, S.; Devarajan, K.; Yang, D.H.; Knudson, A.G.; Balachandran, S. Meta-analysis identifies NF-κB as a therapeutic target in renal cancer. PLoS ONE 2013, 8, e76746. [Google Scholar] [CrossRef]
- Lind, G.E.; Kleivi, K.; Meling, G.I.; Teixeira, M.R.; Thiis-Evensen, E.; Rognum, T.O.; Lothe, R.A. ADAMTS1, CRABP1, and NR3C1 identified as epigenetically deregulated genes in colorectal tumorigenesis. Anal. Cell. Pathol. 2006, 28, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Song, L.; Xu, Y.; Xu, Y.; Zheng, M.; Zhang, P.; Wang, Q. Midkine promotes breast cancer cell proliferation and migration by upregulating NR3C1 expression and activating the NF-κB pathway. Mol. Biol. Rep. 2022, 49, 2953–2961. [Google Scholar] [CrossRef] [PubMed]
- Jakob, J.A.; Bassett, R.L., Jr.; Ng, C.S.; Curry, J.L.; Joseph, R.W.; Alvarado, G.C.; Rohlfs, M.L.; Richard, J.; Gershenwald, J.E.; Kim, K.B. NRAS mutation status is an independent prognostic factor in metastatic melanoma. Cancer 2012, 118, 4014–4023. [Google Scholar] [CrossRef] [PubMed]
- Therkildsen, C.; Bergmann, T.K.; Henrichsen-Schnack, T.; Ladelund, S.; Nilbert, M. The predictive value of KRAS, NRAS, BRAF, PIK3CA and PTEN for anti-EGFR treatment in metastatic colorectal cancer: A systematic review and meta-analysis. Acta Oncol. 2014, 53, 852–864. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.; Cao, Y. Differentially expressed genes SNRPC and PRPF38A are potential biomarkers candidates for osteosarcoma. Res. Sq. 2020. [Google Scholar] [CrossRef]
- Liu, Y.; Ni, R.; Zhang, H.; Miao, L.; Wang, J.; Jia, W.; Wang, Y. Identification of feature genes for smoking-related lung adenocarcinoma based on gene expression profile data. OncoTargets Ther. 2016, 9, 7397. [Google Scholar] [CrossRef]
- Sathe, A.; Nawroth, R. Targeting the PI3K/AKT/mTOR Pathway in Bladder Cancer. Urothelial Carcinoma Methods Protoc. 2018, 1665, 335–350. [Google Scholar]
- Schiffman, M.; Doorbar, J.; Wentzensen, N.; De Sanjosé, S.; Fakhry, C.; Monk, B.J.; Stanley, M.A.; Franceschi, S. Carcinogenic human papillomavirus infection. Nat. Rev. Dis. Primers 2016, 2, 16086. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.K.; Choi, E.J. Pathological roles of MAPK signaling pathways in human diseases. Biochim. Et Biophys. Acta (BBA)-Mol. Basis Dis. 2010, 1802, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Di Leva, G.; Garofalo, M.; Croce, C.M. MicroRNAs in cancer. Annu. Rev. Pathol. 2014, 9, 287. [Google Scholar] [CrossRef]
- Mazal, P.R.; Exner, M.; Haitel, A.; Krieger, S.; Thomson, R.B.; Aronson, P.S.; Susani, M. Expression of kidney-specific cadherin distinguishes chromophobe renal cell carcinoma from renal oncocytoma. Hum. Pathol. 2005, 36, 22–28. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ACC Data | KICH Data | |||||
|---|---|---|---|---|---|---|
| Acc | Aupr | Auc | Acc | Aupr | Auc | |
| GCN-CRF-Node10 | 0.849 | 0.813 | 0.890 | 0.865 | 0.810 | 0.912 |
| GCN-CRF-Node20 | 0.855 | 0.802 | 0.898 | 0.858 | 0.814 | 0.919 |
| GCN-CRF-Node30 | 0.833 | 0.758 | 0.884 | 0.866 | 0.803 | 0.900 |
| ACC Data | KICH Data | |||||
|---|---|---|---|---|---|---|
| Acc | Aupr | Auc | Acc | Aupr | Auc | |
| GCN | 0.821 | 0.772 | 0.879 | 0.804 | 0.807 | 0.904 |
| GCN-CRF | 0.850 | 0.796 | 0.892 | 0.821 | 0.813 | 0.902 |
| GCN-CRF-Node20 | 0.855 | 0.802 | 0.898 | 0.858 | 0.814 | 0.919 |
| ACC Data | KICH Data | |||||
|---|---|---|---|---|---|---|
| Acc | Aupr | Auc | Acc | Aupr | Auc | |
| Weight of 1 time | 0.855 | 0.802 | 0.898 | 0.858 | 0.814 | 0.919 |
| Weight of 2 times | 0.861 | 0.812 | 0.912 | 0.848 | 0.816 | 0.929 |
| Weight of 3 times | 0.851 | 0.813 | 0.928 | 0.830 | 0.803 | 0.916 |
| Weight of 4 times | 0.857 | 0.811 | 0.920 | 0.784 | 0.778 | 0.920 |
| Weight of 5 times | 0.846 | 0.790 | 0.920 | 0.767 | 0.795 | 0.897 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, P.-J.; Zhu, A.-D.; Cao, R.; Zheng, C. Personalized Driver Gene Prediction Using Graph Convolutional Networks with Conditional Random Fields. Biology 2024, 13, 184. https://doi.org/10.3390/biology13030184
Wei P-J, Zhu A-D, Cao R, Zheng C. Personalized Driver Gene Prediction Using Graph Convolutional Networks with Conditional Random Fields. Biology. 2024; 13(3):184. https://doi.org/10.3390/biology13030184
Chicago/Turabian StyleWei, Pi-Jing, An-Dong Zhu, Ruifen Cao, and Chunhou Zheng. 2024. "Personalized Driver Gene Prediction Using Graph Convolutional Networks with Conditional Random Fields" Biology 13, no. 3: 184. https://doi.org/10.3390/biology13030184
APA StyleWei, P. -J., Zhu, A. -D., Cao, R., & Zheng, C. (2024). Personalized Driver Gene Prediction Using Graph Convolutional Networks with Conditional Random Fields. Biology, 13(3), 184. https://doi.org/10.3390/biology13030184