
Metformin: From Diabetes to Cancer—Unveiling Molecular Mechanisms and Therapeutic Strategies

 ,
,  , , ,
, , ,  and
and 
Abstract
:Simple Summary
Abstract
1. Introduction
Pharmacokinetics
2. Mechanisms of Action
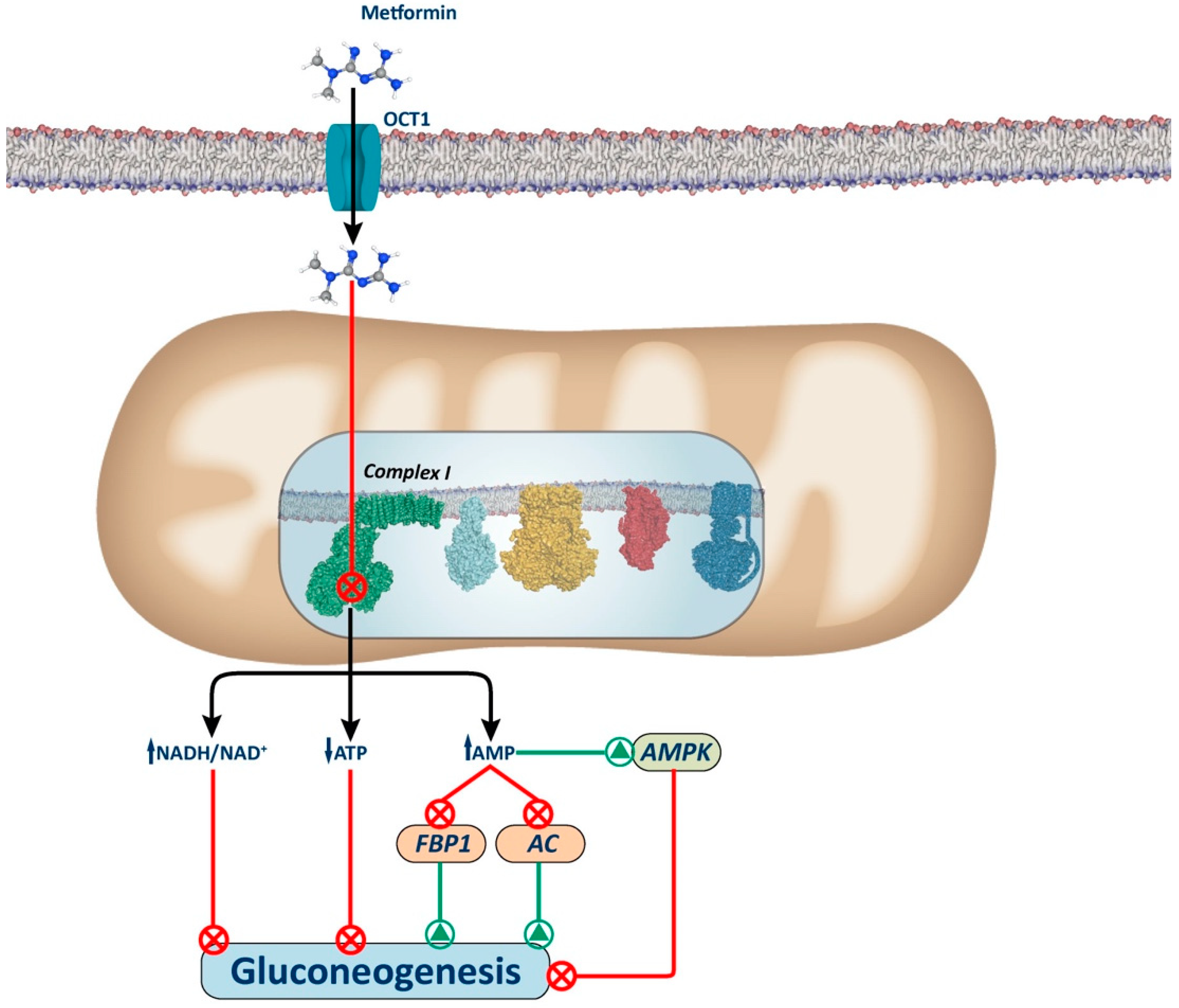
2.1. Complex I Inhibition
2.2. Adenosine Monophosphate-Activated Protein Kinase (AMPK)-Dependent Mechanisms
Adenosine Monophosphate-Activated Protein Kinase (AMPK)-Dependent Lysosomal Pathway
2.3. Adenosine Monophosphate-Activated Protein Kinase (AMPK)-Independent Mechanisms
2.4. Complex IV Inhibition
2.5. Epigenetic Effects of Metformin
2.5.1. Effects of Metformin on the Acetylation Profile
2.5.2. DNA Methylation
2.6. Effects of Metformin on microRNAs
2.7. Effects of Metformin on the Microbiota
2.8. Effects of Metformin as an Anticancer Agent
3. Metformin in Cancer Risk and Treatment
3.1. Breast Cancer
3.1.1. Clinical Studies
3.1.2. Animal Studies
3.1.3. In Vitro Studies
3.2. Colorectal Cancer
3.2.1. Clinical Studies
3.2.2. Animal Studies
3.2.3. In Vitro Studies
4. Future Perspectives and Improvements in Cancer Therapy
5. Concluding Remarks
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
List of Abbreviations
| 5-FU | 5-fluorouracil |
| ACC | Acetyl-CoA carboxylase |
| ADP | Adenosine diphosphate |
| AMP | Adenosine monophosphate |
| AKT | Protein kinase B |
| AMPK | AMP-activated protein kinase |
| APC | Adenomatous polyposis coli |
| ATP | Adenosine triphosphate |
| AXIN | Axin 1 |
| BMI | Body Mass Index |
| CAMK-beta | Calcium/calmodulin protein kinase 2 |
| CBP | CREB-binding protein |
| CD133 | Cluster of differentiation 133 |
| cAMP | Cyclic adenosine monophosphate |
| cGPDH | Cytosolic glycerol-3-phosphate dehydrogenase |
| CpG | Cytosine–phosphate–guanine |
| CPT1 | Carnitine palmitoyltransferase 1 |
| CRC | Colorectal cancer |
| CREB | cAMP response element-binding protein |
| CRTC2 | CREB-regulated transcription co-activator 2 |
| CSC | Cancer stem cells |
| CTL | Cytotoxic T lymphocyte |
| CTLA4 | Cytotoxic T lymphocyte antigen 4 |
| DICER1 | Dicer 1, Ribonuclease III |
| DFS | Disease-free survival |
| DFX | Deferasirox |
| DHAP | Dihydroxyacetone phosphate |
| DMH | Dimethylhydrazine |
| DXR | Dexrazoxane |
| EGF | Epidermal growth factor |
| EMT | Epithelial–mesenchymal transition |
| ER | Endoplasmic reticulum |
| ERAD | ER-associated degradation |
| ERK | Extracellular-Signal-Regulated Kinase |
| FASN | Fatty acid synthase |
| FXR | Farnesoid X receptor |
| G3P | Glycerol-3-phosphate |
| GDF15 | Growth/differentiation factor-15 |
| GLOBOCAN | Global Cancer Observatory |
| GLP1 | Glucagon-like peptide 1 |
| GLUT1 | Glucose transport protein 1 |
| GUDCA | Glycoursodeoxycholic acid |
| H2S | Hydrogen sulfide |
| HAT | Histone acetyltransferase |
| HDAC | Histone deacetylase |
| HNF-4α | hepatocyte nuclear factor-4α |
| HER2- | Human epidermal growth factor receptor 2-negative |
| HOMA-IR | Homeostatic model assessment for insulin resistance |
| HR+ | Hormone receptor-positive |
| IARC | International Agency for Research on Cancer |
| IGF-1 | Insulin-like growth factor 1 |
| ITC | Isothiocyanate |
| KLF5 | Krüppel-like factor 5 |
| Ki-67 | Antigen Kiel 67 |
| LKB1 | Liver kinase B1 |
| M-MDSCs | Monocytic myeloid-derived suppressor cells+B14 |
| MAPK | Mitogen-activated protein kinases |
| MATE | Multidrug and toxin extruders |
| mGPDH | Mitochondrial glycerol-3-phosphate dehydrogenase |
| MICB | MHC Class I Polypeptide-Related Sequence B |
| miRNA | microRNA |
| MMP-x | Matrix metalloproteinase-x |
| mTOR | Mammalian target of rapamycin |
| mTORC1 | Mammalian target of rapamycin complex 1 |
| NADH | Nicotinamide adenine dinucleotide |
| NAMPT | Nicotinamide phosphoribosyltransferase |
| NDH1 | NADH–menaquinone oxidoreductase |
| NDI1 | NADH–ubiquinone reductase (H(+)-translocating) |
| NDUFS7 | NADH dehydrogenase [ubiquinone] iron-sulfur protein 7, mitochondrial |
| NF-κB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| OCMC | O-carboxymethyl-chitosan |
| OCT | Organic cation transporter |
| OS | Overall survival |
| OXPHOS | Oxidative phosphorylation |
| Pck1 | Phosphoenolpyruvate carboxykinase 1 |
| PD-1 | Programmed cell death protein 1 |
| PD-L1 | Programmed death-ligand 1 |
| PEN2 | Presenilin enhancer 2 |
| PET | Positron emission tomography |
| PFK1 | Phosphofructokinase 1 |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PI3K | Phosphoinositide 3-kinase |
| pKa | Acid dissociation constant |
| PKA | Protein kinase A |
| PKNOX2 | PBX/Knotted 1 Homeobox |
| PMAT | Plasma membrane monoamine transporter |
| PPI1 | Proton pump interactor isoform 1 |
| PPP1R3C | Glycogen-targeting regulatory subunit of protein phosphatase 1 |
| ROS | Reactive oxygen species |
| S6K | Ribosomal protein S6 kinase |
| SAH | S-adenosylhomocysteine |
| SCFAs | Short chain fatty acid |
| SESN1 | Sestrin 1 |
| SHBG | Sex hormone-binding globulin |
| SIRT1 | Sirtuin 1 |
| T2DM | Type 2 diabetes mellitus |
| TET3 | tet methylcytosine dioxygenase 3 |
| TNBC | Triple-negative breast cancer |
| TORC2 | Target of rapamycin complex 2 |
| WDTC1 | WD And Tetratricopeptide Repeats 1 |
| WNT | Wingless-type MMTV integration site family |
| WZB117 | 2-fluoro-6-(m-hydroxybenzoyloxy) phenyl m-hydroxybenzoate |
References
- World Health Organization. Model List of Essential Medicines: 21st List 2019. Available online: https://iris.who.int/handle/10665/325771 (accessed on 5 March 2024).
- Ahmad, E.; Sargeant, J.A.; Zaccardi, F.; Khunti, K.; Webb, D.R.; Davies, M.J. Where Does Metformin Stand in Modern Day Management of Type 2 Diabetes? Pharmaceuticals 2020, 13, 427. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.J. Metformin: Historical Overview. Diabetologia 2017, 60, 1566–1576. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Viollet, B. Understanding the Glucoregulatory Mechanisms of Metformin in Type 2 Diabetes Mellitus. Nat. Rev. Endocrinol. 2019, 15, 569–589. [Google Scholar] [CrossRef] [PubMed]
- Inzucchi, S.E.; Lipska, K.J.; Mayo, H.; Bailey, C.J.; McGuire, D.K. Metformin in Patients with Type 2 Diabetes and Kidney Disease: A Systematic Review. JAMA 2014, 312, 2668–2675. [Google Scholar] [CrossRef] [PubMed]
- Raičević, B.; Janković, S. Predictors of Gastrointestinal Complaints in Patients on Metformin Therapy. Open Med. 2023, 18, 20230871. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From Mechanisms of Action to Therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Hundal, R.S.; Krssak, M.; Dufour, S.; Laurent, D.; Lebon, V.; Chandramouli, V.; Inzucchi, S.E.; Schumann, W.C.; Petersen, K.F.; Landau, B.R.; et al. Mechanism by Which Metformin Reduces Glucose Production in Type 2 Diabetes. Diabetes 2000, 49, 2063. [Google Scholar] [CrossRef] [PubMed]
- Cree-Green, M.; Bergman, B.C.; Cengiz, E.; Fox, L.A.; Hannon, T.S.; Miller, K.; Nathan, B.; Pyle, L.; Kahn, D.; Tansey, M.; et al. Metformin Improves Peripheral Insulin Sensitivity in Youth with Type 1 Diabetes. J. Clin. Endocrinol. Metab. 2019, 104, 3265–3278. [Google Scholar] [CrossRef] [PubMed]
- DeFronzo, R.A.; Barzilai, N.; Simonson, D.C. Mechanism of Metformin Action in Obese and Lean Noninsulin-Dependent Diabetic Subjects. J. Clin. Endocrinol. Metab. 1991, 73, 1294–1301. [Google Scholar] [CrossRef]
- Hother-Nielsen, O.; Schmitz, O.; Andersen, P.H.; Beck-Nielsen, H.; Pedersen, O. Metformin Improves Peripheral but Not Hepatic Insulin Action in Obese Patients with Type II Diabetes. Acta Endocrinol. 1989, 120, 257–265. [Google Scholar] [CrossRef]
- Jahn, L.A.; Hartline, L.; Liu, Z.; Barrett, E.J. Metformin Improves Skeletal Muscle Microvascular Insulin Resistance in Metabolic Syndrome. Am. J. Physiol. Endocrinol. Metab. 2022, 322, E173–E180. [Google Scholar] [CrossRef] [PubMed]
- Barroso, E.; Montori-Grau, M.; Wahli, W.; Palomer, X.; Vázquez-Carrera, M. Striking a Gut–Liver Balance for the Antidiabetic Effects of Metformin. Trends Pharmacol. Sci. 2023, 44, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Schäfer, G. Site-Specific Uncoupling and Inhibition of Oxidative Phosphorylation by Biguanides. II. Biochim. Biophys. Acta (BBA) Bioenerg. 1969, 172, 334–337. [Google Scholar] [CrossRef]
- Fontaine, E. Metformin-Induced Mitochondrial Complex I Inhibition: Facts, Uncertainties, and Consequences. Front. Endocrinol. 2018, 9, 753. [Google Scholar] [CrossRef]
- LaMoia, T.E.; Butrico, G.M.; Kalpage, H.A.; Goedeke, L.; Hubbard, B.T.; Vatner, D.F.; Gaspar, R.C.; Zhang, X.M.; Cline, G.W.; Nakahara, K.; et al. Metformin, Phenformin, and Galegine Inhibit Complex IV Activity and Reduce Glycerol-Derived Gluconeogenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2122287119. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Erion, D.M.; Rahimi, Y.; Zhang, X.M.; Braddock, D.T.; Albright, R.A.; Prigaro, B.J.; Wood, J.L.; Bhanot, S.; MacDonald, M.J.; et al. Metformin Suppresses Gluconeogenesis by Inhibiting Mitochondrial Glycerophosphate Dehydrogenase. Nature 2014, 510, 542–546. [Google Scholar] [CrossRef]
- Foretz, M.; Guigas, B.; Viollet, B. Metformin: Update on Mechanisms of Action and Repurposing Potential. Nat. Rev. Endocrinol. 2023, 19, 460–476. [Google Scholar] [CrossRef] [PubMed]
- Lamoia, T.E.; Shulman, G.I. Cellular and Molecular Mechanisms of Metformin Action. Endocr. Rev. 2021, 42, 77–96. [Google Scholar] [CrossRef] [PubMed]
- Cejuela, M.; Martin-Castillo, B.; Menendez, J.A.; Pernas, S. Metformin and Breast Cancer: Where Are We Now? Int. J. Mol. Sci. 2022, 23, 2705. [Google Scholar] [CrossRef]
- Chi Thent, Z.; Hannim Zaidun, N.; Fairuz Azmi, M.; Senin, M.; Haslan, H.; Salehuddin, R.I. Metformin a Therapeutic Paradigm for Colorectal Cancer: Insight into the Molecular Pathway? Curr. Drug Targets 2016, 18, 734–750. [Google Scholar] [CrossRef]
- Ng, C.A.W.; Jiang, A.A.; Toh, E.M.S.; Ng, C.H.; Ong, Z.H.; Peng, S.; Tham, H.Y.; Sundar, R.; Chong, C.S.; Khoo, C.M. Metformin and Colorectal Cancer: A Systematic Review, Meta-Analysis and Meta-Regression. Int. J. Colorectal. Dis. 2020, 35, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Andrzejewski, S.; Siegel, P.M.; St-Pierre, J. Metabolic Profiles Associated with Metformin Efficacy in Cancer. Front. Endocrinol. 2018, 9, 372. [Google Scholar] [CrossRef] [PubMed]
- Col, N.F.; Ochs, L.; Springmann, V.; Aragaki, A.K.; Chlebowski, R.T. Metformin and Breast Cancer Risk: A Meta-Analysis and Critical Literature Review. Breast Cancer Res. Treat. 2012, 135, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.H.; Satkunam, M.; Pond, G.R.; Steinberg, G.R.; Blandino, G.; Schunemann, H.J.; Muti, P. Association of Metformin with Breast Cancer Incidence and Mortality in Patients with Type II Diabetes: A GRADE-Assessed Systematic Review and Meta-Analysis. Cancer Epidemiol. Biomarkers Prev. 2018, 27, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Owen, M.R.; Doran, E.; Halestrap, A.P. Evidence That Metformin Exerts Its Anti-Diabetic Effects through Inhibition of Complex 1 of the Mitochondrial Respiratory Chain. Biochem. J. 2000, 348, 607. [Google Scholar] [CrossRef]
- Jensen, J.B.; Sundelin, E.I.; Jakobsen, S.; Gormsen, L.C.; Munk, O.L.; Frøkiær, J.; Jessen, N. [11C]-Labeled Metformin Distribution in the Liver and Small Intestine Using Dynamic Positron Emission Tomography in Mice Demonstrates Tissue-Specific Transporter Dependency. Diabetes 2016, 65, 1724–1730. [Google Scholar] [CrossRef] [PubMed]
- Liang, X.; Giacomini, K.M. Transporters Involved in Metformin Pharmacokinetics and Treatment Response. J. Pharm. Sci. 2017, 106, 2245–2250. [Google Scholar] [CrossRef]
- Graham, G.G.; Punt, J.; Arora, M.; Day, R.O.; Doogue, M.P.; Duong, J.K.; Furlong, T.J.; Greenfield, J.R.; Greenup, L.C.; Kirkpatrick, C.M.; et al. Clinical Pharmacokinetics of Metformin. Clin. Pharmacokinet. 2011, 50, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Glossmann, H.H.; Lutz, O.M.D. Pharmacology of Metformin—An Update. Eur. J. Pharmacol. 2019, 865, 172782. [Google Scholar] [CrossRef]
- Clarke, J.D.; Dzierlenga, A.L.; Nelson, N.R.; Li, H.; Werts, S.; Goedken, M.J.; Cherrington, N.J. Mechanism of Altered Metformin Distribution in Nonalcoholic Steatohepatitis. Diabetes 2015, 64, 3305–3313. [Google Scholar] [CrossRef]
- Gormsen, L.C.; Sundelin, E.I.; Jensen, J.B.; Vendelbo, M.H.; Jakobsen, S.; Munk, O.L.; Christensen, M.M.H.; Brøsen, K.; Frøkiær, J.; Jessen, N. In Vivo Imaging of Human 11C-Metformin in Peripheral Organs: Dosimetry, Biodistribution, and Kinetic Analyses. J. Nucl. Med. 2016, 57, 1920–1926. [Google Scholar] [CrossRef] [PubMed]
- Alshawi, A.; Agius, L. Low Metformin Causes a More Oxidized Mitochondrial NADH/NAD Redox State in Hepatocytes and Inhibits Gluconeogenesis by a Redox-Independent Mechanism. J. Biol. Chem. 2019, 294, 2839–2853. [Google Scholar] [CrossRef] [PubMed]
- He, L.; Wondisford, F.E. Metformin Action: Concentrations Matter. Cell Metab. 2015, 21, 159–162. [Google Scholar] [CrossRef] [PubMed]
- Wilcock, C.; Bailey, C.J. Accumulation of Metformin by Tissues of the Normal and Diabetic Mouse. Xenobiotica 1994, 24, 49–57. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Goswami, S.; Giacomini, K.M.; Altman, R.B.; Klein, T.E. Metformin Pathways: Pharmacokinetics and Pharmacodynamics. Pharmacogenet Genom. 2012, 22, 820–827. [Google Scholar] [CrossRef] [PubMed]
- Sakellakis, M. Why Metformin Should Not Be Used as an Oxidative Phosphorylation Inhibitor in Cancer Patients. Chemotherapy 2023, 68, 185–189. [Google Scholar] [CrossRef]
- El-Mir, M.Y.; Nogueira, V.; Fontaine, E.; Avéret, N.; Rigoulet, M.; Leverve, X. Dimethylbiguanide Inhibits Cell Respiration via an Indirect Effect Targeted on the Respiratory Chain Complex I. J. Biol. Chem. 2000, 275, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Degli Esposti, M. Inhibitors of NADH-Ubiquinone Reductase: An Overview. Biochim. Biophys. Acta 1998, 1364, 222–235. [Google Scholar] [CrossRef]
- Bridges, H.R.; Jones, A.J.Y.; Pollak, M.N.; Hirst, J. Effects of Metformin and Other Biguanides on Oxidative Phosphorylation in Mitochondria. Biochem. J. 2014, 462, 475–487. [Google Scholar] [CrossRef]
- Bridges, H.R.; Blaza, J.N.; Yin, Z.; Chung, I.; Pollak, M.N.; Hirst, J. Structural Basis of Mammalian Respiratory Complex I Inhibition by Medicinal Biguanides. Science 2023, 379, 351–357. [Google Scholar] [CrossRef]
- Foretz, M.; Hébrard, S.; Leclerc, J.; Zarrinpashneh, E.; Soty, M.; Mithieux, G.; Sakamoto, K.; Andreelli, F.; Viollet, B. Metformin Inhibits Hepatic Gluconeogenesis in Mice Independently of the LKB1/AMPK Pathway via a Decrease in Hepatic Energy State. J. Clin. Investig. 2010, 120, 2355–2369. [Google Scholar] [CrossRef] [PubMed]
- Hunter, R.W.; Hughey, C.C.; Lantier, L.; Sundelin, E.I.; Peggie, M.; Zeqiraj, E.; Sicheri, F.; Jessen, N.; Wasserman, D.H.; Sakamoto, K. Metformin Reduces Liver Glucose Production by Inhibition of Fructose-1-6-Bisphosphatase. Nat. Med. 2018, 24, 1395–1406. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides Suppress Hepatic Glucagon Signalling by Decreasing Production of Cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A Nutrient and Energy Sensor That Maintains Energy Homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Oakhill, J.S.; Scott, J.W.; Kemp, B.E. AMPK Functions as an Adenylate Charge-Regulated Protein Kinase. Trends Endocrinol. Metab. 2012, 23, 125–132. [Google Scholar] [CrossRef]
- Shaw, R.J.; Lamia, K.A.; Vasquez, D.; Koo, S.-H.; Bardeesy, N.; Depinho, R.A.; Montminy, M.; Cantley, L.C. The Kinase LKB1 Mediates Glucose Homeostasis in Liver and Therapeutic Effects of Metformin. Science 2005, 310, 1642–1646. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.-M.; Seo, W.-Y.; Song, K.-H.; Chanda, D.; Kim, Y.D.; Kim, D.-K.; Lee, M.-W.; Ryu, D.; Kim, Y.-H.; Noh, J.-R.; et al. AMPK-Dependent Repression of Hepatic Gluconeogenesis via Disruption of CREB.CRTC2 Complex by Orphan Nuclear Receptor Small Heterodimer Partner. J. Biol. Chem. 2010, 285, 32182–32191. [Google Scholar] [CrossRef] [PubMed]
- Vial, G.; Detaille, D.; Guigas, B. Role of Mitochondria in the Mechanism(s) of Action of Metformin. Front. Endocrinol. 2019, 10, 442419. [Google Scholar] [CrossRef]
- Wilcock, C.; Wyre, N.D.; Bailey, C.J. Subcellular Distribution of Metformin in Rat Liver. J. Pharm. Pharmacol. 1991, 43, 442–444. [Google Scholar] [CrossRef]
- Fullerton, M.D.; Galic, S.; Marcinko, K.; Sikkema, S.; Pulinilkunnil, T.; Chen, Z.P.; O’Neill, H.M.; Ford, R.J.; Palanivel, R.; O’Brien, M.; et al. Single Phosphorylation Sites in Acc1 and Acc2 Regulate Lipid Homeostasis and the Insulin–Sensitizing Effects of Metformin. Nat. Med. 2013, 19, 1649. [Google Scholar] [CrossRef]
- Zhang, C.-S.; Li, M.; Ma, T.; Zong, Y.; Cui, J.; Feng, J.-W.; Wu, Y.-Q.; Lin, S.-Y.; Lin, S.-C. Metformin Activates AMPK through the Lysosomal Pathway. Cell Metab. 2016, 24, 521–522. [Google Scholar] [CrossRef]
- Zhang, C.-S.; Jiang, B.; Li, M.; Zhu, M.; Peng, Y.; Zhang, Y.-L.; Wu, Y.-Q.; Li, T.Y.; Liang, Y.; Lu, Z.; et al. The Lysosomal V-ATPase-Ragulator Complex Is a Common Activator for AMPK and MTORC1, Acting as a Switch between Catabolism and Anabolism. Cell Metab. 2014, 20, 526–540. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Tian, X.; Zhang, B.; Li, M.; Wang, Y.; Yang, C.; Wu, J.; Wei, X.; Qu, Q.; Yu, Y.; et al. Low-Dose Metformin Targets the Lysosomal AMPK Pathway through PEN2. Nature 2022, 603, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Madiraju, A.K.; Qiu, Y.; Perry, R.J.; Rahimi, Y.; Zhang, X.-M.; Zhang, D.; Camporez, J.-P.G.; Cline, G.W.; Butrico, G.M.; Kemp, B.E.; et al. Metformin Inhibits Gluconeogenesis via a Redox-Dependent Mechanism in Vivo. Nat. Med. 2018, 24, 1384–1394. [Google Scholar] [CrossRef] [PubMed]
- Brisson, D.; Vohl, M.C.; St-Pierre, J.; Hudson, T.J.; Gaudet, D. Glycerol: A Neglected Variable in Metabolic Processes? Bioessays 2001, 23, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Lin, E.C. Glycerol Utilization and Its Regulation in Mammals. Annu. Rev. Biochem. 1977, 46, 765–795. [Google Scholar] [CrossRef] [PubMed]
- Saheki, T.; Iijima, M.; Li, M.X.; Kobayashi, K.; Horiuchi, M.; Ushikai, M.; Okumura, F.; Meng, X.J.; Inoue, I.; Tajima, A.; et al. Citrin/Mitochondrial Glycerol-3-Phosphate Dehydrogenase Double Knock-out Mice Recapitulate Features of Human Citrin Deficiency. J. Biol. Chem. 2007, 282, 25041–25052. [Google Scholar] [CrossRef]
- Migoya, E.M.; Bergeron, R.; Miller, J.L.; Snyder, R.N.K.; Tanen, M.; Hilliard, D.; Weiss, B.; Larson, P.; Gutierrez, M.; Jiang, G.; et al. Dipeptidyl Peptidase-4 Inhibitors Administered in Combination with Metformin Result in an Additive Increase in the Plasma Concentration of Active GLP-1. Clin. Pharmacol. Ther. 2010, 88, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Logie, L.; Harthill, J.; Patel, K.; Bacon, S.; Hamilton, D.L.; Macrae, K.; McDougall, G.; Wang, H.-H.; Xue, L.; Jiang, H.; et al. Cellular Responses to the Metal-Binding Properties of Metformin. Diabetes 2012, 61, 1423–1433. [Google Scholar] [CrossRef]
- Galdieri, L.; Gatla, H.; Vancurova, I.; Vancura, A. Activation of AMP-Activated Protein Kinase by Metformin Induces Protein Acetylation in Prostate and Ovarian Cancer Cells. J. Biol. Chem. 2016, 291, 25154–25166. [Google Scholar] [CrossRef]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic Effects of Metformin: From Molecular Mechanisms to Clinical Implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef] [PubMed]
- Ji, X.; Wang, S.; Tang, H.; Zhang, Y.; Zhou, F.; Zhang, L.; Zhu, Q.; Zhu, K.; Liu, Q.; Liu, Y.; et al. PPP1R3C Mediates Metformin-Inhibited Hepatic Gluconeogenesis. Metabolism 2019, 98, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Fernández-Arroyo, S.; Joven, J.; Menendez, J.A. Metformin Targets Histone Acetylation in Cancer-Prone Epithelial Cells. Cell Cycle 2016, 15, 3355–3361. [Google Scholar] [CrossRef] [PubMed]
- Verdone, L. Histone Acetylation in Gene Regulation. Brief. Funct. Genom. Proteomic 2006, 5, 209–221. [Google Scholar] [CrossRef] [PubMed]
- Cuyàs, E.; Verdura, S.; Llorach-Parés, L.; Fernández-Arroyo, S.; Joven, J.; Martin-Castillo, B.; Bosch-Barrera, J.; Brunet, J.; Nonell-Canals, A.; Sanchez-Martinez, M.; et al. Metformin Is a Direct SIRT1-Activating Compound: Computational Modeling and Experimental Validation. Front. Endocrinol. 2018, 9, 657. [Google Scholar] [CrossRef] [PubMed]
- Caton, P.W.; Nayuni, N.K.; Kieswich, J.; Khan, N.Q.; Yaqoob, M.M.; Corder, R. Metformin Suppresses Hepatic Gluconeogenesis through Induction of SIRT1 and GCN5. J. Endocrinol. 2010, 205, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Z.; Bian, Y.; Zhang, Y.; Ren, G.; Li, G. Metformin Activates AMPK/SIRT1/NF-ΚB Pathway and Induces Mitochondrial Dysfunction to Drive Caspase3/GSDME-Mediated Cancer Cell Pyroptosis. Cell Cycle 2020, 19, 1089–1104. [Google Scholar] [CrossRef]
- Zhang, J.; Xiang, H.; Liu, J.; Chen, Y.; He, R.-R.; Liu, B. Mitochondrial Sirtuin 3: New Emerging Biological Function and Therapeutic Target. Theranostics 2020, 10, 8315–8342. [Google Scholar] [CrossRef]
- Li, X. SIRT1 and Energy Metabolism. Acta Biochim. Biophys. Sin. 2013, 45, 51–60. [Google Scholar] [CrossRef]
- Lan, F.; Cacicedo, J.M.; Ruderman, N.; Ido, Y. SIRT1 Modulation of the Acetylation Status, Cytosolic Localization, and Activity of LKB1. J. Biol. Chem. 2008, 283, 27628–27635. [Google Scholar] [CrossRef]
- Stelmaszyk, A.; Mikołajczak, P.; Dworacka, M. Sirtuin 1 as the Mechanism of Action of Agents Used in the Diabetes Mellitus Pharmacotherapy. Eur. J. Pharmacol. 2021, 907, 174289. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Li, J.; Luo, G.; Liu, W.; He, Y.; Wang, F.; Qian, Y.; Fan, C. Pharmacological Activation of SIRT1 by Metformin Prevented Trauma-Induced Heterotopic Ossification through Inhibiting Macrophage Mediated Inflammation. Eur. J. Pharmacol. 2021, 909, 174386. [Google Scholar] [CrossRef] [PubMed]
- Ren, H.; Shao, Y.; Wu, C.; Ma, X.; Lv, C.; Wang, Q. Metformin Alleviates Oxidative Stress and Enhances Autophagy in Diabetic Kidney Disease via AMPK/SIRT1-FoxO1 Pathway. Mol. Cell. Endocrinol. 2020, 500, 110628. [Google Scholar] [CrossRef] [PubMed]
- Felgueiras, R.; Neto, A.C.; Rodrigues, A.R.; Gouveia, A.M.; Almeida, H.; Neves, D. Anti-Oxidant Effect of Metformin through AMPK/SIRT1/PGC-1α/SIRT3– Independent GPx1 Expression in the Heart of Mice with Endometriosis. Horm. Mol. Biol. Clin. Investig. 2022, 43, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Sung, J.Y.; Kim, S.G.; Kang, Y.J.; Choi, H.C. Metformin Mitigates Stress-Induced Premature Senescence by Upregulating AMPKα at Ser485 Phosphorylation Induced SIRT3 Expression and Inactivating Mitochondrial Oxidants. Mech. Ageing Dev. 2022, 206, 111708. [Google Scholar] [CrossRef] [PubMed]
- Zhu, M.; He, J.; Xu, Y.; Zuo, Y.; Zhou, W.; Yue, Z.; Shao, X.; Cheng, J.; Wang, T.; Mou, S. AMPK Activation Coupling SENP1-Sirt3 Axis Protects against Acute Kidney Injury. Mol. Ther. 2023, 31, 3052–3066. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.; Wang, Z.; Zhang, C.; Bian, Y.; Zhang, X.; Liu, X.; Chen, W.; Zhao, Y. Metformin Promotes in vitro Maturation of Oocytes from Aged Mice by Attenuating Mitochondrial Oxidative Stress via SIRT3-Dependent SOD2ac. Front. Cell Dev. Biol. 2022, 10, 1028510. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Gao, W.-N.; Xue, Y.-N.; Zhang, L.-C.; Zhang, J.-J.; Lu, S.-Y.; Yan, X.-Y.; Yu, H.-M.; Su, J.; Sun, L.-K. SIRT3 Aggravates Metformin-Induced Energy Stress and Apoptosis in Ovarian Cancer Cells. Exp. Cell Res. 2018, 367, 137–149. [Google Scholar] [CrossRef]
- Cuyàs, E.; Fernández-Arroyo, S.; Verdura, S.; García, R.Á.-F.; Stursa, J.; Werner, L.; Blanco-González, E.; Montes-Bayón, M.; Joven, J.; Viollet, B.; et al. Metformin Regulates Global DNA Methylation via Mitochondrial One-Carbon Metabolism. Oncogene 2018, 37, 963–970. [Google Scholar] [CrossRef]
- Zhong, T.; Men, Y.; Lu, L.; Geng, T.; Zhou, J.; Mitsuhashi, A.; Shozu, M.; Maihle, N.J.; Carmichael, G.G.; Taylor, H.S.; et al. Metformin Alters DNA Methylation Genome-Wide via the H19/SAHH Axis. Oncogene 2017, 36, 2345–2354. [Google Scholar] [CrossRef]
- García-Calzón, S.; Schrader, S.; Perfilyev, A.; Martinell, M.; Ahlqvist, E.; Ling, C. DNA Methylation Partially Mediates Antidiabetic Effects of Metformin on HbA1c Levels in Individuals with Type 2 Diabetes. Diabetes Res. Clin. Pract. 2023, 202, 110807. [Google Scholar] [CrossRef] [PubMed]
- García-Calzón, S.; Perfilyev, A.; Männistö, V.; de Mello, V.D.; Nilsson, E.; Pihlajamäki, J.; Ling, C. Diabetes Medication Associates with DNA Methylation of Metformin Transporter Genes in the Human Liver. Clin. Epigenetics 2017, 9, 102. [Google Scholar] [CrossRef]
- Hooten, N.N.; Martin-Montalvo, A.; Dluzen, D.F.; Zhang, Y.; Bernier, M.; Zonderman, A.B.; Becker, K.G.; Gorospe, M.; Cabo, R.; Evans, M.K. Metformin-mediated Increase in DICER1 Regulates MicroRNA Expression and Cellular Senescence. Aging Cell 2016, 15, 572–581. [Google Scholar] [CrossRef] [PubMed]
- Demirsoy, İ.H.; Ertural, D.Y.; Balci, Ş.; Çınkır, Ü.; Sezer, K.; Tamer, L.; Aras, N. Profiles of Circulating MiRNAs Following Metformin Treatment in Patients with Type 2 Diabetes. J. Med. Biochem. 2018, 37, 499–506. [Google Scholar] [CrossRef]
- Ortega, F.J.; Mercader, J.M.; Moreno-Navarrete, J.M.; Rovira, O.; Guerra, E.; Esteve, E.; Xifra, G.; Martínez, C.; Ricart, W.; Rieusset, J.; et al. Profiling of Circulating MicroRNAs Reveals Common MicroRNAs Linked to Type 2 Diabetes That Change with Insulin Sensitization. Diabetes Care 2014, 37, 1375–1383. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.C.K.; Than, W.H.; Kwan, B.C.H.; Lai, K.B.; Chan, R.C.K.; Teoh, J.Y.C.; Ng, J.K.C.; Chow, K.M.; Cheng, P.M.S.; Law, M.C.; et al. Adipose and Plasma MicroRNAs MiR-221 and 222 Associate with Obesity, Insulin Resistance, and New Onset Diabetes after Peritoneal Dialysis. Nutrients 2022, 14, 4889. [Google Scholar] [CrossRef] [PubMed]
- Xie, D.; Chen, F.; Zhang, Y.; Shi, B.; Song, J.; Chaudhari, K.; Yang, S.-H.; Zhang, G.J.; Sun, X.; Taylor, H.S.; et al. Let-7 Underlies Metformin-Induced Inhibition of Hepatic Glucose Production. Proc. Natl. Acad. Sci. USA 2022, 119, e2122217119. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Ye, Y.; Wang, B.; Zhao, S. MiR-140-5p Aggravates Insulin Resistance via Directly Targeting GYS1 and PPP1CC in Insulin-Resistant HepG2 Cells. Diabetes Metab. Syndr. Obes. 2021, 14, 2515–2524. [Google Scholar] [CrossRef] [PubMed]
- Vallianou, N.G.; Stratigou, T.; Tsagarakis, S. Metformin and Gut Microbiota: Their Interactions and Their Impact on Diabetes. Hormones 2019, 18, 141–144. [Google Scholar] [CrossRef]
- Petakh, P.; Kamyshna, I.; Kamyshnyi, A. Effects of Metformin on the Gut Microbiota: A Systematic Review. Mol. Metab. 2023, 77, 101805. [Google Scholar] [CrossRef]
- Zhu, X.; Shen, J.; Feng, S.; Huang, C.; Wang, H.; Huo, F.; Liu, H. Akkermansia Muciniphila, Which Is Enriched in the Gut Microbiota by Metformin, Improves Cognitive Function in Aged Mice by Reducing the Proinflammatory Cytokine Interleukin-6. Microbiome 2023, 11, 120. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Lou, X.; Jiang, C.; Ji, X.; Tao, X.; Sun, J.; Bao, Z. Gut Microbiota Is Correlated with Gastrointestinal Adverse Events of Metformin in Patients with Type 2 Diabetes. Front. Endocrinol. 2022, 13, 4030. [Google Scholar] [CrossRef]
- Silamiķele, L.; Silamiķelis, I.; Ustinova, M.; Kalniņa, Z.; Elbere, I.; Petrovska, R.; Kalniņa, I.; Kloviņš, J. Metformin Strongly Affects Gut Microbiome Composition in High-Fat Diet-Induced Type 2 Diabetes Mouse Model of Both Sexes. Front. Endocrinol. 2021, 12, 626359. [Google Scholar] [CrossRef] [PubMed]
- Mueller, N.T.; Differding, M.K.; Zhang, M.; Maruthur, N.M.; Juraschek, S.P.; Miller, E.R.; Appel, L.J.; Yeh, H.-C. Metformin Affects Gut Microbiome Composition and Function and Circulating Short-Chain Fatty Acids: A Randomized Trial. Diabetes Care 2021, 44, 1462–1471. [Google Scholar] [CrossRef]
- Díaz-Perdigones, C.M.; Muñoz-Garach, A.; Álvarez-Bermúdez, M.D.; Moreno-Indias, I.; Tinahones, F.J. Gut Microbiota of Patients with Type 2 Diabetes and Gastrointestinal Intolerance to Metformin Differs in Composition and Functionality from Tolerant Patients. Biomed. Pharmacother. 2022, 145, 112448. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut Microbiota and Intestinal FXR Mediate the Clinical Benefits of Metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Maier, L.; Pruteanu, M.; Kuhn, M.; Zeller, G.; Telzerow, A.; Anderson, E.E.; Brochado, A.R.; Fernandez, K.C.; Dose, H.; Mori, H.; et al. Extensive Impact of Non-Antibiotic Drugs on Human Gut Bacteria. Nature 2018, 555, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Kalender, A.; Selvaraj, A.; Kim, S.Y.; Gulati, P.; Brûlé, S.; Viollet, B.; Kemp, B.E.; Bardeesy, N.; Dennis, P.; Schlager, J.J.; et al. Metformin, Independent of AMPK, Inhibits MTORC1 In a Rag GTPase-Dependent Manner. Cell Metab. 2010, 11, 390. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 Mediates Cellular Energy Response to Control Cell Growth and Survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef]
- Rattan, R.; Giri, S.; Hartmann, L.C.; Shridhar, V. Metformin Attenuates Ovarian Cancer Cell Growth in an AMP-Kinase Dispensable Manner. J. Cell. Mol. Med. 2011, 15, 166. [Google Scholar] [CrossRef]
- Sahra, I.B.; Regazzetti, C.; Robert, G.; Laurent, K.; Le Marchand-Brustel, Y.; Auberger, P.; Tanti, J.F.; Giorgetti-Peraldi, S.; Bost, F. Metformin, Independent of AMPK, Induces MTOR Inhibition and Cell-Cycle Arrest through REDD1. Cancer Res. 2011, 71, 4366–4372. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin, E.; Zhang, Y. Activation of the PTEN/MTOR/STAT3 Pathway in Breast Cancer Stem-like Cells Is Required for Viability and Maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158. [Google Scholar] [CrossRef] [PubMed]
- Korkaya, H.; Paulson, A.; Charafe-Jauffret, E.; Ginestier, C.; Brown, M.; Dutcher, J.; Clouthier, S.G.; Wicha, M.S. Regulation of Mammary Stem/Progenitor Cells by PTEN/Akt/β-Catenin Signaling. PLoS Biol. 2009, 7, e1000121. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. Insulin, Insulin-like Growth Factors and Neoplasia. Best. Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 625–638. [Google Scholar] [CrossRef]
- Dowling, R.J.O.; Niraula, S.; Stambolic, V.; Goodwin, P.J. Metformin in Cancer: Translational Challenges. J. Mol. Endocrinol. 2012, 48, R31–R43. [Google Scholar] [CrossRef] [PubMed]
- Pollak, M. The Insulin and Insulin-like Growth Factor Receptor Family in Neoplasia: An Update. Nat. Rev. Cancer 2012, 12, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Sarfstein, R.; Friedman, Y.; Attias-Geva, Z.; Fishman, A.; Bruchim, I.; Werner, H. Metformin Downregulates the Insulin/IGF-I Signaling Pathway and Inhibits Different Uterine Serous Carcinoma (USC) Cells Proliferation and Migration in P53-Dependent or -Independent Manners. PLoS ONE 2013, 8, 61537. [Google Scholar] [CrossRef] [PubMed]
- Pardoll, D.M. The Blockade of Immune Checkpoints in Cancer Immunotherapy. Nat. Rev. Cancer 2012, 12, 252. [Google Scholar] [CrossRef]
- Chen, L. Co-Inhibitory Molecules of the B7–CD28 Family in the Control of T-Cell Immunity. Nat. Rev. Immunol. 2004, 4, 336–347. [Google Scholar] [CrossRef]
- Chen, L.; Han, X. Anti–PD-1/PD-L1 Therapy of Human Cancer: Past, Present, and Future. J. Clin. Investig. 2015, 125, 3384. [Google Scholar] [CrossRef]
- Cha, J.H.; Yang, W.H.; Xia, W.; Wei, Y.; Chan, L.C.; Lim, S.O.; Li, C.W.; Kim, T.; Chang, S.S.; Lee, H.H.; et al. Metformin Promotes Antitumor Immunity via Endoplasmic-Reticulum-Associated Degradation of PD-L1. Mol. Cell 2018, 71, 606–620.e7. [Google Scholar] [CrossRef]
- Alimoradi, N.; Firouzabadi, N.; Fatehi, R. How Metformin Affects Various Malignancies by Means of MicroRNAs: A Brief Review. Cancer Cell Int. 2021, 21, 207. [Google Scholar] [CrossRef] [PubMed]
- Morales, D.R.; Morris, A.D. Metformin in Cancer Treatment and Prevention. Annu. Rev. Med. 2015, 66, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Arnold, M.; Morgan, E.; Rumgay, H.; Mafra, A.; Singh, D.; Laversanne, M.; Vignat, J.; Gralow, J.R.; Cardoso, F.; Siesling, S.; et al. Current and Future Burden of Breast Cancer: Global Statistics for 2020 and 2040. Breast 2022, 66, 15–23. [Google Scholar] [CrossRef] [PubMed]
- Morgan, E.; Arnold, M.; Gini, A.; Lorenzoni, V.; Cabasag, C.J.; Laversanne, M.; Vignat, J.; Ferlay, J.; Murphy, N.; Bray, F. Global Burden of Colorectal Cancer in 2020 and 2040: Incidence and Mortality Estimates from GLOBOCAN. Gut 2023, 72, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Cancer Today. Available online: https://gco.iarc.fr/today/en (accessed on 4 March 2024).
- Chen, K.; Li, Y.; Guo, Z.; Zeng, Y.; Zhang, W.; Wang, H. Metformin: Current Clinical Applications in Nondiabetic Patients with Cancer. Aging 2020, 12, 3993–4009. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Garczorz, W.; Kosowska, A.; Francuz, T. Antidiabetic Drugs in Breast Cancer Patients. Cancers 2024, 16, 299. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Hajjar, A.; Cryns, V.L.; Trentham-Dietz, A.; Gangnon, R.E.; Heckman-Stoddard, B.M.; Alagoz, O. Breast Cancer Risk for Women with Diabetes and the Impact of Metformin: A Meta-Analysis. Cancer Med. 2023, 12, 11703–11718. [Google Scholar] [CrossRef]
- Bellerba, F.; Chatziioannou, A.C.; Jasbi, P.; Robinot, N.; Keski-Rahkonen, P.; Trolat, A.; Vozar, B.; Hartman, S.J.; Scalbert, A.; Bonanni, B.; et al. Metabolomic Profiles of Metformin in Breast Cancer Survivors: A Pooled Analysis of Plasmas from Two Randomized Placebo-Controlled Trials. J. Transl. Med. 2022, 20, 1–16. [Google Scholar] [CrossRef]
- Chikermane, S.G.; Sharma, M.; Abughosh, S.M.; Aparasu, R.R.; Trivedi, M.V.; Johnson, M.L. Dose-Dependent Relation between Metformin and the Risk of Hormone Receptor-Positive, Her2-Negative Breast Cancer among Postmenopausal Women with Type-2 Diabetes. Breast Cancer Res. Treat. 2022, 195, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Löfling, L.L.; Støer, N.C.; Andreassen, B.K.; Ursin, G.; Botteri, E. Low-Dose Aspirin, Statins, and Metformin and Survival in Patients with Breast Cancers: A Norwegian Population-Based Cohort Study. Breast Cancer Res. 2023, 25, 1–11. [Google Scholar] [CrossRef]
- Yang, J.; Yang, H.; Cao, L.; Yin, Y.; Shen, Y.; Zhu, W. Prognostic Value of Metformin in Cancers: An Updated Meta-Analysis Based on 80 Cohort Studies. Medicine 2022, 101, E31799. [Google Scholar] [CrossRef] [PubMed]
- Najafi, F.; Rajati, F.; Sarokhani, D.; Bavandpour, M.; Moradinazar, M. The Relationship between Metformin Consumption and Cancer Risk: An Updated Umbrella Review of Systematic Reviews and Meta-Analyses. Int. J. Prev. Med. 2023, 14, 90. [Google Scholar] [CrossRef] [PubMed]
- Yu, O.H.Y.; Suissa, S. Metformin and Cancer: Solutions to a Real-World Evidence Failure. Diabetes Care 2023, 46, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Essa, N.M.; Salem, H.F.; Elgendy, M.O.; Gabr, A.; Omran, M.M.; Hassan, N.A.; Tashkandi, H.M.; Harakeh, S.; Boshra, M.S. Efficacy of Metformin as Adjuvant Therapy in Metastatic Breast Cancer Treatment. J. Clin. Med. 2022, 11, 5505. [Google Scholar] [CrossRef] [PubMed]
- Farkhondeh, T.; Amirabadizadeh, A.; Aramjoo, H.; Llorens, S.; Roshanravan, B.; Saeedi, F.; Talebi, M.; Shakibaei, M.; Samarghandian, S. Impact of Metformin on Cancer Biomarkers in Non-Diabetic Cancer Patients: A Systematic Review and Meta-Analysis of Clinical Trials. Curr. Oncol. 2021, 28, 1412. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.; Bellerba, F.; Macis, D.; Bertelsen, B.E.; Guerrieri-Gonzaga, A.; Aristarco, V.; Viste, K.; Mellgren, G.; Di Cola, G.; Costantino, J.; et al. Effect of Metformin and Lifestyle Intervention on Adipokines and Hormones in Breast Cancer Survivors: A Pooled Analysis from Two Randomized Controlled Trials. Breast Cancer Res. Treat. 2024, 2024, 1–11. [Google Scholar] [CrossRef]
- Serageldin, M.A.; Kassem, A.B.; El-Kerm, Y.; Helmy, M.W.; El-Mas, M.M.; El-Bassiouny, N.A. The Effect of Metformin on Chemotherapy-Induced Toxicities in Non-Diabetic Breast Cancer Patients: A Randomised Controlled Study. Drug Saf. 2023, 46, 587. [Google Scholar] [CrossRef]
- Subbiah, V.; Coleman, N.; Piha-Paul, S.A.; Tsimberidou, A.M.; Janku, F.; Rodon, J.; Pant, S.; Dumbrava, E.E.I.; Fu, S.; Hong, D.S.; et al. Phase I Study of MTORC1/2 Inhibitor Sapanisertib (CB-228/TAK-228) in Combination with Metformin in Patients with MTOR/AKT/PI3K Pathway Alterations and Advanced Solid Malignancies. Cancer Res. Commun. 2024, 4, 378. [Google Scholar] [CrossRef]
- Bashraheel, S.S.; Kheraldine, H.; Khalaf, S.; Moustafa, A.E. Al Metformin and HER2-Positive Breast Cancer: Mechanisms and Therapeutic Implications. Biomed. Pharmacother. 2023, 162, 114676. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Tong, Y.; Hong, J.; Huang, O.; Wu, J.; He, J.; Chen, W.; Li, Y.; Chen, X.; Shen, K. Neoadjuvant Docetaxel, Epirubicin, and Cyclophosphamide with or without Metformin in Breast Cancer Patients with Metabolic Abnormality: Results from the Randomized Phase II NeoMET Trial. Breast Cancer Res. Treat. 2023, 197, 525–533. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, K.; Thatcher, A.; Grobe, A.; Hicks, L.; Gu, H.; Sears, D.D.; Ellies, L.G.; Kalachev, L.; Kroll, E. The Combined Treatment with Ketogenic Diet and Metformin Slows Tumor Growth in Two Mouse Models of Triple Negative Breast Cancer. Res. Sq. 2023. [Google Scholar] [CrossRef]
- García-Castillo, V.; López-Urrutia, E.; Villanueva-Sánchez, O.; Ávila-Rodríguez, M.A.; Zentella-Dehesa, A.; Cortés-González, C.; López-Camarillo, C.; Jacobo-Herrera, N.J.; Pérez-Plasencia, C. Targeting Metabolic Remodeling in Triple Negative Breast Cancer in a Murine Model. J. Cancer 2017, 8, 178. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Li, Y.; Zhao, Y.; Shi, F.; Zhou, Q.; Wu, J.; Lyu, S.; Song, Q. Metformin Inducing the Change of Functional and Exhausted Phenotypic Tumor-Infiltrated Lymphocytes and the Correlation with JNK Signal Pathway in Triple-Negative Breast Cancer. Breast Cancer Targets Ther. 2022, 14, 391. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Suo, H.; Gao, L.; Liu, Y.; Chen, B.; Lu, S.; Jin, F.; Cao, Y. Metformin Plays an Antitumor Role by Downregulating Inhibitory Cells and Immune Checkpoint Molecules While Activating Protective Immune Responses in Breast Cancer. Int. Immunopharmacol. 2023, 118, 110038. [Google Scholar] [CrossRef] [PubMed]
- Zilinyi, R.; Czompa, A.; Czegledi, A.; Gajtko, A.; Pituk, D.; Lekli, I.; Tosaki, A. The Cardioprotective Effect of Metformin in Doxorubicin-Induced Cardiotoxicity: The Role of Autophagy. Molecules 2018, 23, 1184. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Alvarez, R.; Martinez-Outschoorn, U.E.; Lamb, R.; Hulit, J.; Howell, A.; Gandara, R.; Sartini, M.; Rubin, E.; Lisanti, M.P.; Sotgia, F. Mitochondrial Dysfunction in Breast Cancer Cells Prevents Tumor Growth: Understanding Chemoprevention with Metformin. Cell Cycle 2013, 12, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Nurzhan, S.; Bekezhankyzy, Z.; Ding, H.; Berdigaliyev, N.; Sergazy, S.; Gulyayev, A.; Shulgau, Z.; Triggle, C.R.; Aljofan, M. The Effect of Different Glucose Concentrations on the Antiproliferative Activity of Metformin in MCF-7 Breast Cancer Cells. Pharmaceutics 2023, 15, 2186. [Google Scholar] [CrossRef]
- Andrzejewski, S.; Klimcakova, E.; Johnson, R.M.; Tabariès, S.; Annis, M.G.; McGuirk, S.; Northey, J.J.; Chénard, V.; Sriram, U.; Papadopoli, D.J.; et al. PGC-1α Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab. 2017, 26, 778–787.e5. [Google Scholar] [CrossRef]
- Janzer, A.; German, N.J.; Gonzalez-Herrera, K.N.; Asara, J.M.; Haigis, M.C.; Struhl, K. Metformin and Phenformin Deplete Tricarboxylic Acid Cycle and Glycolytic Intermediates during Cell Transformation and NTPs in Cancer Stem Cells. Proc. Natl. Acad. Sci. USA 2014, 111, 10574–10579. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.Y.; Liu, Z.; Bi, M.H.; Zhang, J.J.; Han, Z.Q.; Han, X.; Wang, H.Y.; Sun, G.P.; Liu, H. Metformin Induces Apoptosis via a Mitochondria-Mediated Pathway in Human Breast Cancer Cells in Vitro. Exp. Ther. Med. 2016, 11, 1700. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Kumar, S. Metformin Inhibits Human Breast Cancer Cell Growth by Promoting Apoptosis via a ROS-Independent Pathway Involving Mitochondrial Dysfunction: Pivotal Role of Superoxide Dismutase (SOD). Cell. Oncol. 2018, 41, 637–650. [Google Scholar] [CrossRef]
- Min, W.L.; Wang, B.F.; Liang, B.B.; Zhang, L.; Pan, J.Y.; Huang, Y.; Zhao, Y.; Lin, S.; Zhao, Y.H.; Zhang, S.Q.; et al. A ROS/Akt/NF-ΚB Signaling Cascade Mediates Epidermal Growth Factor-Induced Epithelial-Mesenchymal Transition and Invasion in Human Breast Cancer Cells. World J. Oncol. 2022, 13, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Shi, P.; Liu, W.; Tala; Wang, H.; Li, F.; Zhang, H.; Wu, Y.; Kong, Y.; Zhou, Z.; Wang, C.; et al. Metformin Suppresses Triple-Negative Breast Cancer Stem Cells by Targeting KLF5 for Degradation. Cell Discov. 2017, 3, 17010. [Google Scholar] [CrossRef]
- Safari, F.; Momeni, A.; Ramezani, M.; Ansari, Y.; Moghbelinejad, S. Metformin Caused Radiosensitivity of Breast Cancer Cells through the Expression Modulation of MiR-21-5p/SESN1axis. Asian Pac. J. Cancer Prev. 2023, 24, 3715. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Haugrud, A.B.; Schaefer, M.A.; Messerli, S.M.; Miskimins, W.K. Ability of Metformin to Deplete NAD+ Contributes to Cancer Cell Susceptibility to Metformin Cytotoxicity and Is Dependent on NAMPT Expression. Front. Oncol. 2023, 13, 1225220. [Google Scholar] [CrossRef] [PubMed]
- Yu, G.H.; Li, S.F.; Wei, R.; Jiang, Z. Diabetes and Colorectal Cancer Risk: Clinical and Therapeutic Implications. J. Diabetes Res. 2022, 2022, 1747326. [Google Scholar] [CrossRef] [PubMed]
- Lawler, T.; Walts, Z.L.; Steinwandel, M.; Lipworth, L.; Murff, H.J.; Zheng, W.; Warren Andersen, S. Type 2 Diabetes and Colorectal Cancer Risk. JAMA Netw. Open 2023, 6, e2343333. [Google Scholar] [CrossRef]
- Berster, J.M.; Göke, B. Type 2 Diabetes Mellitus as Risk Factor for Colorectal Cancer. Arch. Physiol. Biochem. 2008, 114, 84–98. [Google Scholar] [CrossRef]
- Liu, F.; Yan, L.; Wang, Z.; Lu, Y.; Chu, Y.; Li, X.; Liu, Y.; Rui, D.; Nie, S.; Xiang, H. Metformin Therapy and Risk of Colorectal Adenomas and Colorectal Cancer in Type 2 Diabetes Mellitus Patients: A Systematic Review and Meta-Analysis. Oncotarget 2017, 8, 16017. [Google Scholar] [CrossRef] [PubMed]
- Sehdev, A.; Shih, Y.C.T.; Vekhter, B.; Bissonnette, M.B.; Olopade, O.I.; Polite, B.N. Metformin for Primary Colorectal Cancer Prevention in Patients with Diabetes: A Case-Control Study in a US Population. Cancer 2015, 121, 1071–1078. [Google Scholar] [CrossRef] [PubMed]
- Berkovic, M.C.; Mikulic, D.; Bilic-Curcic, I.; Mrzljak, A. How Far along Are We in Revealing the Connection between Metformin and Colorectal Cancer? World J. Gastroenterol. 2021, 27, 1362. [Google Scholar] [CrossRef] [PubMed]
- Bradley, M.C.; Ferrara, A.; Achacoso, N.; Ehrlich, S.F.; Quesenberry, C.P.; Habel, L.A. A Cohort Study of Metformin and Colorectal Cancer Risk among Patients with Diabetes Mellitus. Cancer Epidemiol. Biomark. Prev. 2018, 27, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Jung, Y.S.; Park, C.H.; Eun, C.S.; Park, D.I.; Han, D.S. Metformin Use and the Risk of Colorectal Adenoma: A Systematic Review and Meta-Analysis. J. Gastroenterol. Hepatol. 2017, 32, 957–965. [Google Scholar] [CrossRef] [PubMed]
- Skinner, H.; Hu, C.; Tsakiridis, T.; Santana-Davila, R.; Lu, B.; Erasmus, J.J.; Doemer, A.J.; Videtic, G.M.M.; Coster, J.; Yang, A.X.; et al. Addition of Metformin to Concurrent Chemoradiation in Patients With Locally Advanced Non-Small Cell Lung Cancer: The NRG-LU001 Phase 2 Randomized Clinical Trial. JAMA Oncol. 2021, 7, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, P.J.; Chen, B.E.; Gelmon, K.A.; Whelan, T.J.; Ennis, M.; Lemieux, J.; Ligibel, J.A.; Hershman, D.L.; Mayer, I.A.; Hobday, T.J.; et al. Effect of Metformin vs Placebo on Invasive Disease-Free Survival in Patients With Breast Cancer: The MA.32 Randomized Clinical Trial. JAMA 2022, 327, 1963–1973. [Google Scholar] [CrossRef] [PubMed]
- Pujalte Martin, M.; Borchiellini, D.; Thamphya, B.; Guillot, A.; Paoli, J.B.; Besson, D.; Hilgers, W.; Priou, F.; El Kouri, C.; Hoch, B.; et al. TAXOMET: A French Prospective Multicentric Randomized Phase II Study of Docetaxel Plus Metformin Versus Docetaxel Plus Placebo in Metastatic Castration-Resistant Prostate Cancer. Clin. Genitourin. Cancer 2021, 19, 501–509. [Google Scholar] [CrossRef] [PubMed]
- Akce, M.; Farran, B.; Switchenko, J.M.; Rupji, M.; Kang, S.; Khalil, L.; Ruggieri-Joyce, A.; Olson, B.; Shaib, W.L.; Wu, C.; et al. Phase II Trial of Nivolumab and Metformin in Patients with Treatment-Refractory Microsatellite Stable Metastatic Colorectal Cancer. J. Immunother. Cancer 2023, 11, e007235. [Google Scholar] [CrossRef]
- Zhang, Y.; Guan, M.; Zheng, Z.; Zhang, Q.; Gao, F.; Xue, Y. Effects of Metformin on CD133+ Colorectal Cancer Cells in Diabetic Patients. PLoS ONE 2013, 8, e81264. [Google Scholar] [CrossRef]
- Bragagnoli, A.C.; Araujo, R.L.C.; Ferraz, M.W.; dos Santos, L.V.; Abdalla, K.C.; Comar, F.; Santos, F.A.; Oliveira, M.A.; Carvalheira, J.B.C.; Cárcano, F.M.; et al. Metformin plus Lrinotecan in Patients with Refractory Colorectal Cancer: A Phase 2 Clinical Trial. Br. J. Cancer 2021, 124, 1072–1078. [Google Scholar] [CrossRef]
- Miranda, V.C.; Braghiroli, M.I.; Faria, L.D.; Bariani, G.; Alex, A.; Bezerra Neto, J.E.; Capareli, F.C.; Sabbaga, J.; Lobo dos Santos, J.F.; Hoff, P.M.; et al. Phase 2 Trial of Metformin Combined With 5-Fluorouracil in Patients with Refractory Metastatic Colorectal Cancer. Clin. Color. Cancer 2016, 15, 321–328.e1. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Lee, K.J.; Seo, Y.; Kwon, J.H.; Yoon, J.P.; Kang, J.Y.; Lee, H.J.; Park, S.J.; Hong, S.P.; Cheon, J.H.; et al. Effects of Metformin on Colorectal Cancer Stem Cells Depend on Alterations in Glutamine Metabolism. Sci. Rep. 2018, 8, 409. [Google Scholar] [CrossRef] [PubMed]
- Durán, R.V.; Mackenzie, E.D.; Boulahbel, H.; Frezza, C.; Heiserich, L.; Tardito, S.; Bussolati, O.; Rocha, S.; Hall, M.N.; Gottlieb, E. HIF-Independent Role of Prolyl Hydroxylases in the Cellular Response to Amino Acids. Oncogene 2013, 32, 4549–4556. [Google Scholar] [CrossRef] [PubMed]
- Bozzi, F.; Mogavero, A.; Varinelli, L.; Belfiore, A.; Manenti, G.; Caccia, C.; Volpi, C.C.; Beznoussenko, G.V.; Milione, M.; Leoni, V.; et al. MIF/CD74 Axis Is a Target for Novel Therapies in Colon Carcinomatosis. J. Exp. Clin. Cancer Res. 2017, 36, 1–15. [Google Scholar] [CrossRef]
- Cunha, A.D.; Costa, F.O.; Carvalheira, J.B.C.; Bragagnoli, A.C. Repurposing Metformin for the Treatment of Gastrointestinal Cancer. World J. Gastroenterol. 2021, 27, 1883–1904. [Google Scholar] [CrossRef]
- Rena, G.; Hardie, D.G.; Pearson, E.R. The Mechanisms of Action of Metformin. Diabetologia 2017, 60, 1577–1585. [Google Scholar] [CrossRef] [PubMed]
- Hosono, K.; Endo, H.; Takahashi, H.; Sugiyama, M.; Uchiyama, T.; Suzuki, K.; Nozaki, Y.; Yoneda, K.; Fujita, K.; Yoneda, M.; et al. Metformin Suppresses Azoxymethane-Induced Colorectal Aberrant Crypt Foci by Activating AMP-Activated Protein Kinase. Mol. Carcinog. 2010, 49, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Tomimoto, A.; Endo, H.; Sugiyama, M.; Fujisawa, T.; Hosono, K.; Takahashi, H.; Nakajima, N.; Nagashima, Y.; Wada, K.; Nakagama, H.; et al. Metformin Suppresses Intestinal Polyp Growth in ApcMin/+ Mice. Cancer Sci. 2008, 99, 2136–2141. [Google Scholar] [CrossRef]
- Zaafar, D.K.; Zaitone, S.A.; Moustafa, Y.M. Role of Metformin in Suppressing 1,2-Dimethylhydrazine-Induced Colon Cancer in Diabetic and Non-Diabetic Mice: Effect on Tumor Angiogenesis and Cell Proliferation. PLoS ONE 2014, 9, e100562. [Google Scholar] [CrossRef]
- Mohamed Suhaimi, N.A.; Phyo, W.M.; Yap, H.Y.; Choy, S.H.Y.; Wei, X.; Choudhury, Y.; Tan, W.J.; Tan, L.A.P.Y.; Foo, R.S.Y.; Tan, S.H.S.; et al. Metformin Inhibits Cellular Proliferation and Bioenergetics in Colorectal Cancer Patient-Derived Xenografts. Mol. Cancer Ther. 2017, 16, 2035–2044. [Google Scholar] [CrossRef] [PubMed]
- Nangia-Makker, P.; Yu, Y.; Vasudevan, A.; Farhana, L.; Rajendra, S.G.; Levi, E.; Majumdar, A.P.N. Metformin: A Potential Therapeutic Agent for Recurrent Colon Cancer. PLoS ONE 2014, 9, e84369. [Google Scholar] [CrossRef] [PubMed]
- Sugiura, K.; Okabayashi, K.; Seishima, R.; Ishida, T.; Shigeta, K.; Tsuruta, M.; Hasegawa, H.; Kitagawa, Y. Metformin Inhibits the Development and Metastasis of Colorectal Cancer. Med. Oncol. 2022, 39, 136. [Google Scholar] [CrossRef] [PubMed]
- Duraiyarasan, S.; Adefuye, M.; Manjunatha, N.; Ganduri, V.; Rajasekaran, K. Colon Cancer and Obesity: A Narrative Review. Cureus 2022, 14, 27589. [Google Scholar] [CrossRef]
- Zakikhani, M.; Dowling, R.J.O.; Sonenberg, N.; Pollak, M.N. The Effects of Adiponectin and Metformin on Prostate and Colon Neoplasia Involve Activation of AMP-Activated Protein Kinase. Cancer Prev. Res. 2008, 1, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Algire, C.; Moiseeva, O.; Deschênes-Simard, X.; Amrein, L.; Petruccelli, L.; Birman, E.; Viollet, B.; Ferbeyre, G.; Pollak, M.N. Metformin Reduces Endogenous Reactive Oxygen Species and Associated DNA Damage. Cancer Prev. Res. 2012, 5, 536–543. [Google Scholar] [CrossRef]
- Scharping, N.E.; Menk, A.V.; Whetstone, R.D.; Zeng, X.; Delgoffe, G.M. Efficacy of PD-1 Blockade Is Potentiated by Metformin-Induced Reduction of Tumor Hypoxia. Cancer Immunol. Res. 2017, 5, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Broadfield, L.A.; Saigal, A.; Szamosi, J.C.; Hammill, J.A.; Bezverbnaya, K.; Wang, D.; Gautam, J.; Tsakiridis, E.E.; Di Pastena, F.; McNicol, J.; et al. Metformin-Induced Reductions in Tumor Growth Involves Modulation of the Gut Microbiome. Mol. Metab. 2022, 61, 101498. [Google Scholar] [CrossRef]
- Bachem, A.; Makhlouf, C.; Binger, K.J.; de Souza, D.P.; Tull, D.; Hochheiser, K.; Whitney, P.G.; Fernandez-Ruiz, D.; Dähling, S.; Kastenmüller, W.; et al. Microbiota-Derived Short-Chain Fatty Acids Promote the Memory Potential of Antigen-Activated CD8+ T Cells. Immunity 2019, 51, 285–297.e5. [Google Scholar] [CrossRef]
- Mogavero, A.; Maiorana, M.V.; Zanutto, S.; Varinelli, L.; Bozzi, F.; Belfiore, A.; Volpi, C.C.; Gloghini, A.; Pierotti, M.A.; Gariboldi, M. Metformin Transiently Inhibits Colorectal Cancer Cell Proliferation as a Result of Either AMPK Activation or Increased ROS Production. Sci. Rep. 2017, 7, 15992. [Google Scholar] [CrossRef]
- Boyle, K.A.; Van Wickle, J.; Hill, R.B.; Marchese, A.; Kalyanaraman, B.; Dwinell, M.B. Mitochondria-Targeted Drugs Stimulate Mitophagy and Abrogate Colon Cancer Cell Proliferation. J. Biol. Chem. 2018, 293, 14891–14904. [Google Scholar] [CrossRef] [PubMed]
- Denise, C.; Paoli, P.; Calvani, M.; Taddei, M.L.; Giannoni, E.; Kopetz, S.; Kazmi, S.M.A.; Pia, M.M.; Pettazzoni, P.; Sacco, E.; et al. 5-Fluorouracil Resistant Colon Cancer Cells Are Addicted to OXPHOS to Survive and Enhance Stem-like Traits. Oncotarget 2015, 6, 41706–41721. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhao, S.; Lu, X.; Shi, Z.; Liu, H.; Zhu, B. Metformin Enhances the Sensitivity of Colorectal Cancer Cells to Cisplatin through ROS-Mediated PI3K/Akt Signaling Pathway. Gene 2020, 745, 144623. [Google Scholar] [CrossRef] [PubMed]
- Wheaton, W.W.; Weinberg, S.E.; Hamanaka, R.B.; Soberanes, S.; Sullivan, L.B.; Anso, E.; Glasauer, A.; Dufour, E.; Mutlu, G.M.; Scott Budigner, G.R.; et al. Metformin Inhibits Mitochondrial Complex I of Cancer Cells to Reduce Tumorigenesis. eLife 2014, 3, e02242. [Google Scholar] [CrossRef] [PubMed]
- Khodaei, F.; Hosseini, S.M.; Omidi, M.; Hosseini, S.F.; Rezaei, M. Cytotoxicity of Metformin against HT29 Colon Cancer Cells Contributes to Mitochondrial Sirt3 Upregulation. J. Biochem. Mol. Toxicol. 2021, 35, e22662. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.T.; Ung, T.T.; Li, S.; Lian, S.; Xia, Y.; Park, S.Y.; Do Jung, Y. Metformin Inhibits Lithocholic Acid-Induced Interleukin 8 Upregulation in Colorectal Cancer Cells by Suppressing ROS Production and NF-KB Activity. Sci. Rep. 2019, 9, 2003. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Lanza-Jacoby, S. Metformin Decreases Growth of Pancreatic Cancer Cells by Decreasing Reactive Oxygen Species: Role of NOX4. Biochem. Biophys. Res. Commun. 2015, 465, 41–46. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.H.; Pu, X.X.; Huang, M.J.; Xiao, J.; Zhou, J.M.; Lin, T.Y.; Lin, E.H. 5-Fluorouracil Upregulates the Activity of Wnt Signaling Pathway in CD133-Positive Colon Cancer Stem-like Cells. Chin. J. Cancer 2010, 29, 810–815. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Kim, S.C.; Ku, J.L. Metformin Increases Chemo-Sensitivity via Gene Downregulation Encoding DNA Replication Proteins in 5-Fu Resistant Colorectal Cancer Cells. Oncotarget 2017, 8, 56546–56557. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y. Metformin Attenuates Cells Stemness and Epithelial-mesenchymal Transition in Colorectal Cancer Cells by Inhibiting the Wnt3a/Β-catenin Pathway. Mol. Med. Rep. 2019, 19, 1203–1209. [Google Scholar] [CrossRef]
- Lord, S.R.; Harris, A.L. Is It Still Worth Pursuing the Repurposing of Metformin as a Cancer Therapeutic? Br. J. Cancer 2023, 128, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, Y.; Miskimins, W.K. Metformin Induces Both Caspase-Dependent and Poly(ADP-Ribose) Polymerase-Dependent Cell Death in Breast Cancer Cells. Mol. Cancer Res. 2011, 9, 603–615. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.S.; Hoyte, C. Review of Biguanide (Metformin) Toxicity. J. Intensive Care Med. 2019, 34, 863–876. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zielonka, J.; Ouari, O.; Lopez, M.; McAllister, D.; Boyle, K.; Barrios, C.S.; Weber, J.J.; Johnson, B.D.; Hardy, M.; et al. Mitochondria-Targeted Analogues of Metformin Exhibit Enhanced Antiproliferative and Radiosensitizing Effects in Pancreatic Cancer Cells. Cancer Res. 2016, 76, 3904–3915. [Google Scholar] [CrossRef]
- De, A.; Wadhwani, A.; Sauraj; Roychowdhury, P.; Kang, J.H.; Ko, Y.T.; Kuppusamy, G. WZB117 Decorated Metformin-Carboxymethyl Chitosan Nanoparticles for Targeting Breast Cancer Metabolism. Polymers 2023, 15, 976. [Google Scholar] [CrossRef] [PubMed]
- Shahbazi, R.; Jafari-Gharabaghlou, D.; Mirjafary, Z.; Saeidian, H.; Zarghami, N. Design and Optimization Various Formulations of PEGylated Niosomal Nanoparticles Loaded with Phytochemical Agents: Potential Anti-Cancer Effects against Human Lung Cancer Cells. Pharmacol. Rep. 2023, 75, 442–455. [Google Scholar] [CrossRef]
- Kumar, V.; Sharma, K.; Sachan, R.; Alhayyani, S.; Al-abbasi, F.A.; Singh, R.; Anwar, F. Co-Drug Development of Gallic Acid and Metformin Targeting the pro-Inflammatory Cytokines for the Treatment of Breast Cancer. J. Biochem. Mol. Toxicol. 2023, 37, e23300. [Google Scholar] [CrossRef] [PubMed]
- Citi, V.; Barresi, E.; Piragine, E.; Spezzini, J.; Testai, L.; Da Settimo, F.; Martelli, A.; Taliani, S.; Calderone, V. Anti-Proliferative Properties of the Novel Hybrid Drug Met-ITC, Composed of the Native Drug Metformin with the Addition of an Isothiocyanate H2S Donor Moiety, in Different Cancer Cell Lines. Int. J. Mol. Sci. 2023, 24, 6131. [Google Scholar] [CrossRef] [PubMed]
- Solier, S.; Müller, S.; Cañeque, T.; Versini, A.; Mansart, A.; Sindikubwabo, F.; Baron, L.; Emam, L.; Gestraud, P.; Pantoș, G.D.; et al. A Druggable Copper-Signalling Pathway That Drives Inflammation. Nature 2023, 617, 386–394. [Google Scholar] [CrossRef]
- Ailuno, G.; Baldassari, S.; Balboni, A.; Pastorino, S.; Zuccari, G.; Cortese, K.; Barbieri, F.; Drava, G.; Florio, T.; Caviglioli, G. Development of Biotinylated Liposomes Encapsulating Metformin for Therapeutic Targeting of Inflammation-Based Diseases. Pharmaceutics 2024, 16, 235. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, Y.; Ding, K.; Li, S.; Liu, T. Biomimetic Nanovesicle Co-Delivery System Impairs Energy Metabolism for Cancer Treatment. J. Nanobiotechnology 2023, 21, 299. [Google Scholar] [CrossRef] [PubMed]
- Torunoglu, S.T.; Zajda, A.; Tampio, J.; Markowicz-Piasecka, M.; Huttunen, K.M. Metformin Derivatives—Researchers’ Friends or Foes? Biochem. Pharmacol. 2023, 215, 115743. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Li, Y.; Liu, Y.; Jia, B.; Liu, X.; Ma, T. Carbonized Polymer Dots Derived from Metformin and L-Arginine for Tumor Cell Membrane- and Mitochondria-Dual Targeting Therapy. Nanoscale 2023, 15, 17922–17935. [Google Scholar] [CrossRef] [PubMed]
- Cheng, G.; Zielonka, J.; Hardy, M.; Ouari, O.; Chitambar, C.R.; Dwinell, M.B.; Kalyanaraman, B. Synergistic Inhibition of Tumor Cell Proliferation by Metformin and Mito-Metformin in the Presence of Iron Chelators. Oncotarget 2019, 10, 3518–3532. [Google Scholar] [CrossRef]
- Ji, P.; Deng, X.C.; Jin, X.K.; Zhang, S.M.; Wang, J.W.; Feng, J.; Chen, W.H.; Zhang, X.Z. Fused Cytomembrane-Camouflaged Nanoparticles for Tumor-Specific Immunotherapy. Adv. Healthc. Mater. 2023, 12, 2370095. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Function/Results | Disease | In Vitro/In Vivo | References |
|---|---|---|---|
| Complex I inhibition | Cancer, Diabetes | In vitro | [26,38,40,41,140,142,143,182,186] |
| Apoptosis induction | Cancer | In vitro | [144,145] |
| Induction of MMP-2, MMP-9, miR21, and miR-155 | Cancer | In vitro | [145] |
| PI3K/Akt/NF-kB pathway inhibition | Cancer | In vitro | [146,191] |
| Targeting KLF5 for degradation | Cancer | In vitro | |
| Modulates miR-21-5p and SESN1 | Cancer | In vitro | [148] |
| Cellular NAD+ depletion | Cancer | In vitro | [149] |
| Reduction in cell proliferation by AMPK activation | Cancer | In vitro | [177,182] |
| Reduction in cell proliferation by G0/G1 cell cycle arrest | Cancer | In vitro | [162,182] |
| Activated ULK1 to stimulate an anti-tumor mitophagy program | Cancer | In vitro | [83] |
| Enhanced cisplatin-induced apoptosis dependent on the generation of ROS | Cancer | In vitro | [185] |
| Mitigation of DNA damage and mutagenesis by reducing ROS production | Cancer | In vitro | [178,188,189] |
| Complex IV inhibition | Diabetes | In vitro | [60] |
| Metformin-induced alterations in aspartate-malate shuttle and allosteric effectors of PFK1 and FBP1 | Diabetes | In vitro | [33] |
| Activation of AMPK-dependent lysosomal pathway (AXIN/LKB1) at low doses: loss of GLP1 secretion upon PEN2 deletion | Diabetes | In vitro, in vivo | [53,54,185] |
| Enhancement of mitochondrial function and anti-hyperglycemic effects by SIRT1 and SIRT3 modulation | Cancer, Diabetes | In vitro, in vivo | [67,76,187] |
| Tumor burden, increased tumor latency, and slower tumor growth | Cancer | In vivo | [135] |
| Local antitumor activity by reduction in M2-like macrophages, M-MDSCs, and Tregs | Cancer | In vivo | [138] |
| Attenuation of doxorubicin-induced cardiotoxicity | Cancer | In vivo | [139] |
| Suppression of colorectal aberrant crypt foci and intestinal polyps development, as well as liver metastasis by activating AMPK | Cancer | In vivo | [170,171,175] |
| Metabolic changes decreasing oxygen consumption, activating AMPK pathway, and causing a reduction in cell growth | Cancer | In vivo | [173] |
| Downregulation of tumor angiogenesis and cell proliferation | Cancer | In vivo | [172] |
| Reduction in stem-like cell subpopulation | Cancer | In vivo | [174,190] |
| Inhibition of the stimulatory effect of a high-energy diet on tumor growth: reduced insulin levels and AKT and FASN expression; changes in the gut microbiome | Cancer | In vivo | [178,180] |
| Reduction in Ras-induced ROS production and associated DNA damage | Cancer | In vivo | [178] |
| Enhanced the effectiveness of anti-PD-1 immunotherapy by mitigation of tumor hypoxia | Cancer | In vivo | [179] |
| Reduced gluconeogenesis and hepatic lipid content by AMPK activation at suprapharmacological doses | Diabetes | In vivo | [47,48,51] |
| Impairment in glucagon-stimulated glucose production by reduction in PKA activity | Diabetes | In vivo | [43,44,59] |
| Suppression of cAMP-stimulated hepatic gluconeogenesis through HAT activity | Diabetes | In vivo | [63] |
| Reduced gluconeogenesis and HbA1c associated with specific miRNAs modulation | Diabetes | In vivo | [86,88] |
| Increase in SCFAs-producing bacteria | Diabetes | In vivo | [92,93,94,95] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amengual-Cladera, E.; Morla-Barcelo, P.M.; Morán-Costoya, A.; Sastre-Serra, J.; Pons, D.G.; Valle, A.; Roca, P.; Nadal-Serrano, M. Metformin: From Diabetes to Cancer—Unveiling Molecular Mechanisms and Therapeutic Strategies. Biology 2024, 13, 302. https://doi.org/10.3390/biology13050302
Amengual-Cladera E, Morla-Barcelo PM, Morán-Costoya A, Sastre-Serra J, Pons DG, Valle A, Roca P, Nadal-Serrano M. Metformin: From Diabetes to Cancer—Unveiling Molecular Mechanisms and Therapeutic Strategies. Biology. 2024; 13(5):302. https://doi.org/10.3390/biology13050302
Chicago/Turabian StyleAmengual-Cladera, Emilia, Pere Miquel Morla-Barcelo, Andrea Morán-Costoya, Jorge Sastre-Serra, Daniel Gabriel Pons, Adamo Valle, Pilar Roca, and Mercedes Nadal-Serrano. 2024. "Metformin: From Diabetes to Cancer—Unveiling Molecular Mechanisms and Therapeutic Strategies" Biology 13, no. 5: 302. https://doi.org/10.3390/biology13050302
APA StyleAmengual-Cladera, E., Morla-Barcelo, P. M., Morán-Costoya, A., Sastre-Serra, J., Pons, D. G., Valle, A., Roca, P., & Nadal-Serrano, M. (2024). Metformin: From Diabetes to Cancer—Unveiling Molecular Mechanisms and Therapeutic Strategies. Biology, 13(5), 302. https://doi.org/10.3390/biology13050302