2.1. Experiment 1: Inverse Relationship between EndoGalC and α-Gal Epitope Expression
As a preliminary test, PEFs were stained with the serially diluted Alexa Fluor 594-labeled IB4 (hereafter referred to as “AF594-IB4”) to know the optimal concentration of AF594-IB4 exhibiting strong binding to the cells. As shown in
Figure 2A, 50–10 μg/mL of AF594-IB4 were found to be highly reactive to the PEFs. Two μg/mL of AF594-IB4 yielded moderate staining for α-Gal epitope expression. Therefore, we hereafter decided to use more than 50 μg/mL of IB4SAP for isolation of α-Gal epitope-negative transfectants.
Figure 2.
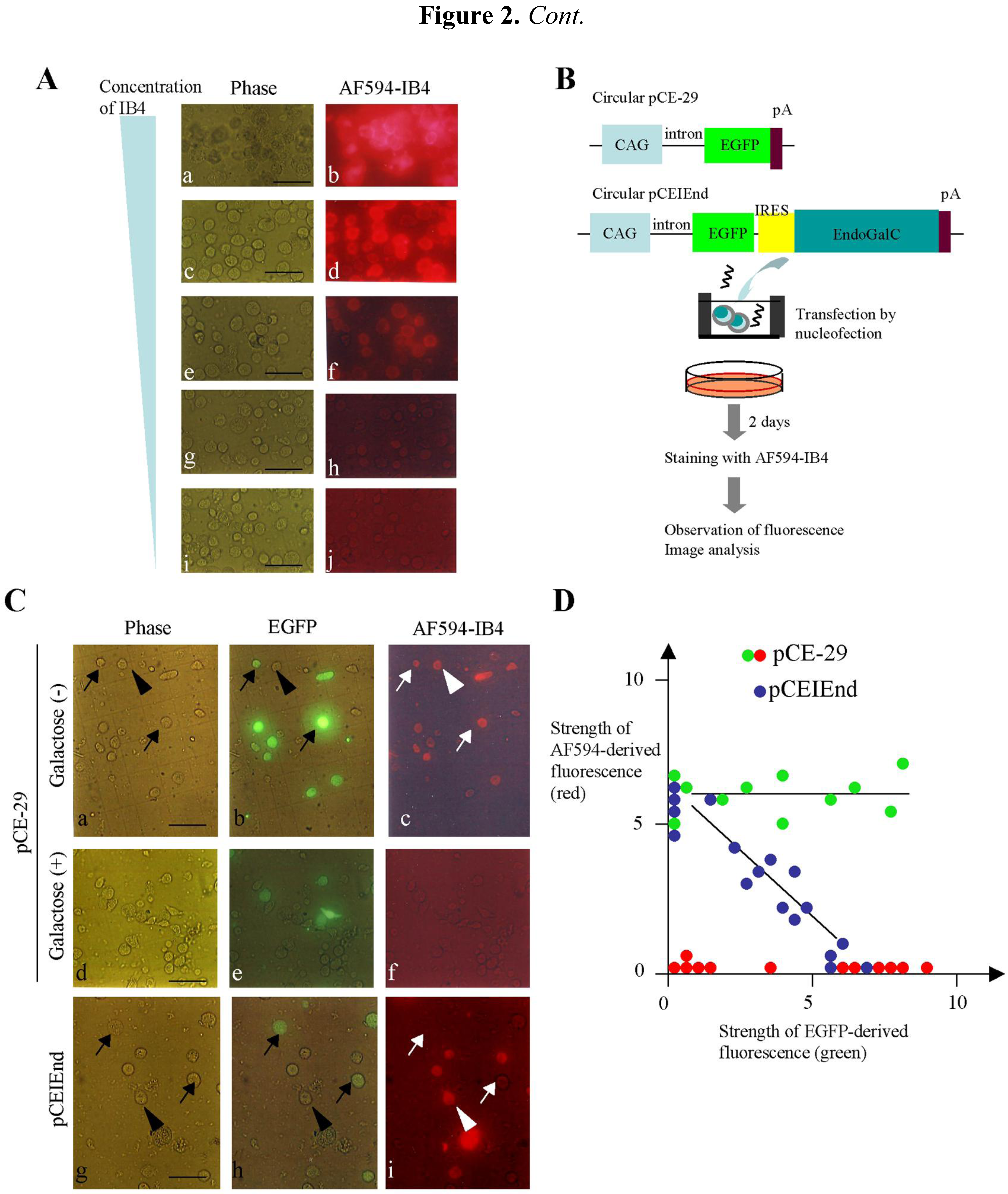
(A) Staining of PEFs with various concentrations [50 (a, b), 10 (c, d), 2 (e, f), 0.4 (g, h) and 0.08 (i, j) μg/mL] of AF594-IB4. Note strong reactivity in the cells stained with 50 to 10 μg/mL of AF594-IB4 (a–d). (B) Expression constructs and experimental flow for examination of the relationship between EndoGalC and α-Gal epitope expression. CAG, approximately 1-kb cytomegalovirus enhancer with chicken β-actin promoter and its 1st intron; EGFP, 0.9-kb enhanced green fluorescent protein; IRES, 0.63-kb internal ribosomal entry site; EndoGalC, 3-kb C. perfringens-derived endo-β-galactosidase C; and pA, 0.56-kb poly(A) sites of rabbit β-globin gene. (C) Cytochemical staining of transfected PEFs with AF594-IB4. Note that the PEFs transfected with pCE-29 were uniformly stained with the lectin, irrespective of the strength of EGFP fluorescence (indicated by arrows and the arrowhead in a–c). In contrast, in the case of transfection with pCEIEnd, PEFs not expressing or weakly expressing EGFP were distinctly stained by the lectin (indicated by the arrowhead in g–i), while PEFs relatively strongly expressing EGFP were almost negative for the staining (indicated by arrows in g–i), suggesting complete loss of the α-Gal epitope from their surface. Staining of the pCE-29-introduced PEFs with AF594-IB4 + galactose abolished the staining completely (d–f). Phase (a, d, g), photographs taken under light; EGFP (b, e, h), photographs taken under light + UV illumination to detect EGFP-derived green fluorescence; and AF594-IB4 (c, f, i), photographs taken under light + UV illumination to detect AF594-derived red fluorescence. Bar = 50 μm. (D) Image analysis of the transfected PEFs shown in (C). The intensity of each cell was measured and plotted, with the arbitrary fluorescence intensity shown in both the abscissa and ordinate axes. The green and blue dots indicate fluorescence measured from the AF594-IB4-stained cells that were transfected with pCE-29 and pCEIEnd, respectively. The red dots indicate fluorescence from the pCE-29-transfected cells that were stained with AF594-IB4 + 50 mM galactose.
Figure 2.
(A) Staining of PEFs with various concentrations [50 (a, b), 10 (c, d), 2 (e, f), 0.4 (g, h) and 0.08 (i, j) μg/mL] of AF594-IB4. Note strong reactivity in the cells stained with 50 to 10 μg/mL of AF594-IB4 (a–d). (B) Expression constructs and experimental flow for examination of the relationship between EndoGalC and α-Gal epitope expression. CAG, approximately 1-kb cytomegalovirus enhancer with chicken β-actin promoter and its 1st intron; EGFP, 0.9-kb enhanced green fluorescent protein; IRES, 0.63-kb internal ribosomal entry site; EndoGalC, 3-kb C. perfringens-derived endo-β-galactosidase C; and pA, 0.56-kb poly(A) sites of rabbit β-globin gene. (C) Cytochemical staining of transfected PEFs with AF594-IB4. Note that the PEFs transfected with pCE-29 were uniformly stained with the lectin, irrespective of the strength of EGFP fluorescence (indicated by arrows and the arrowhead in a–c). In contrast, in the case of transfection with pCEIEnd, PEFs not expressing or weakly expressing EGFP were distinctly stained by the lectin (indicated by the arrowhead in g–i), while PEFs relatively strongly expressing EGFP were almost negative for the staining (indicated by arrows in g–i), suggesting complete loss of the α-Gal epitope from their surface. Staining of the pCE-29-introduced PEFs with AF594-IB4 + galactose abolished the staining completely (d–f). Phase (a, d, g), photographs taken under light; EGFP (b, e, h), photographs taken under light + UV illumination to detect EGFP-derived green fluorescence; and AF594-IB4 (c, f, i), photographs taken under light + UV illumination to detect AF594-derived red fluorescence. Bar = 50 μm. (D) Image analysis of the transfected PEFs shown in (C). The intensity of each cell was measured and plotted, with the arbitrary fluorescence intensity shown in both the abscissa and ordinate axes. The green and blue dots indicate fluorescence measured from the AF594-IB4-stained cells that were transfected with pCE-29 and pCEIEnd, respectively. The red dots indicate fluorescence from the pCE-29-transfected cells that were stained with AF594-IB4 + 50 mM galactose.
![Biology 02 00341 g002]()
To explore the relationship between the EndoGalC and α-Gal epitope expression, we transfected PEFs with the pCEIEnd plasmid (
Figure 2B), which expresses enhanced green fluorescent protein (EGFP) and EndoGalC simultaneously because of the presence of internal ribosomal entry site (IRES) [
11,
12,
13] between the EGFP and EndoGalC genes. PEFs transfected with pCE-29 plasmid were used as the control (
Figure 2B). At two days after transfection, the cells collected by trypsinization were stained with AF594-IB4. In the case of pCEIEnd transfection, the cells strongly expressing EGFP were almost completely negative for IB4 staining (
Figure 2C, arrows in g–i), whereas those not expressing or weakly expressing EGFP showed distinct staining (
Figure 2C, arrowheads in g–i). In the case of pCE-29 transfection, all the cells were stained with lectin, irrespective of EGFP expression (
Figure 2C, arrows and arrowheads in a–c). However, incubation of the pCE-29-transfected cells with AF594-IB4 + 50 mM galactose abolished the IB4-specific staining (d–f in
Figure 2C). The image analysis confirmed these observations (green dots
vs. red dots in
Figure 2D). Also, there was an inverse relationship between EndoGalC and α-Gal epitope expression in the pCEIEnd-transfected cells (blue dots in
Figure 2D). Thus, transgene high-expressors exhibited greatly reduced expression levels of the α-Gal epitope.
2.2. Experiment 2: Enrichment of Transgene High-Expressors with Toxin-Conjugated IB4 Treatment
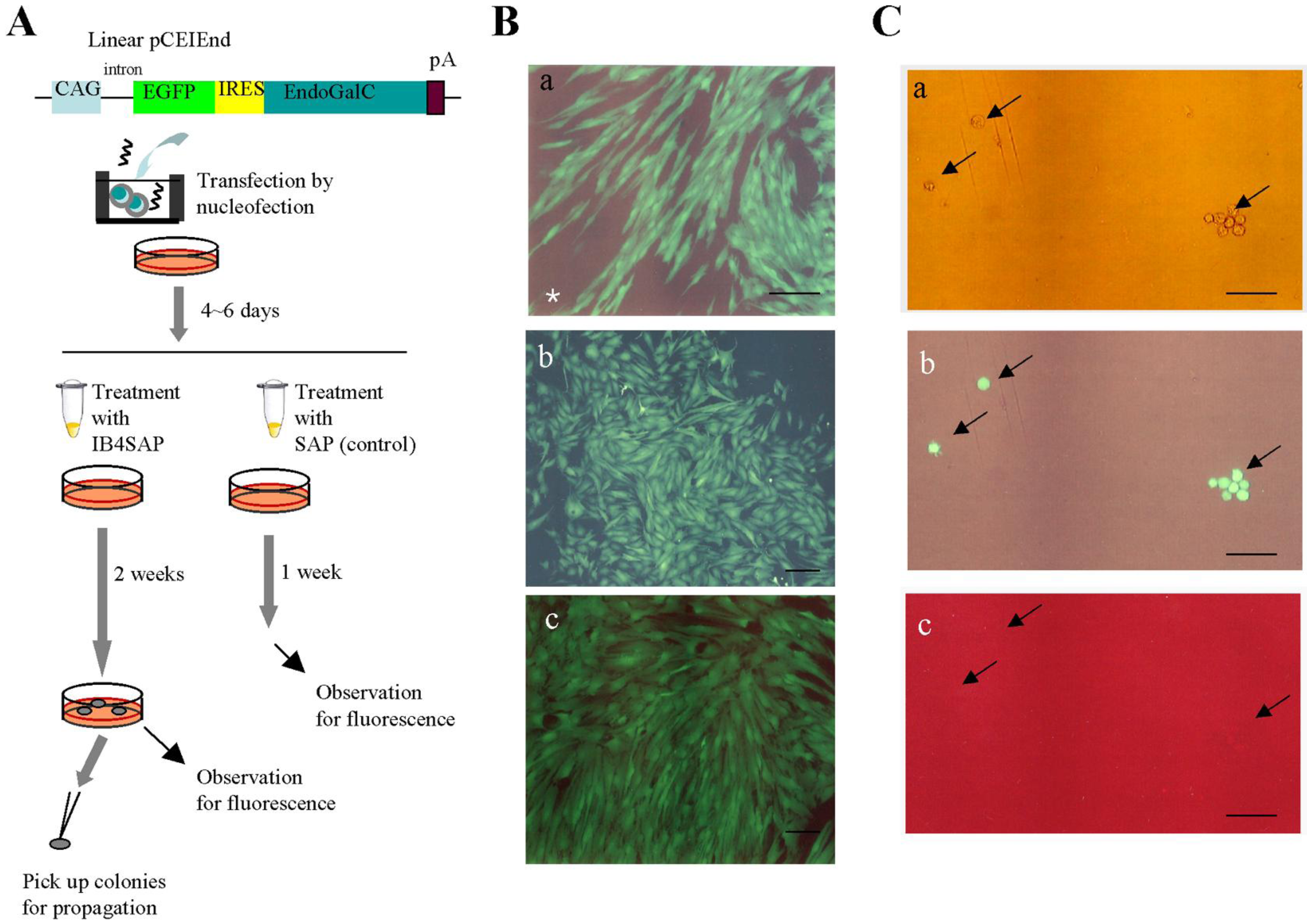
Next, we performed IB4SAP treatment to enrich transgene high-expressors exhibiting distinct EGFP fluorescence, but reduced expression levels of the α-Gal epitope. As depicted in
Figure 3A, PEFs transfected with pCEIEnd were cultured for four to six days in normal medium (without drug selection). The cells were subsequently split into two groups: one treated with IB4SAP and the other with SAP (control). After treatment, the cells were reseeded in a 60-mm tissue culture dish containing normal medium. Within two days, extensive cell death was observed in the IB4SAP-treated group, in contrast to the SAP-treated cells (data not shown). One week after reseeding, the cells in the control group reached confluency and were found to contain a mixture of fluorescent and non-fluorescent cells determined by a fluorescence microscope [
Figure 3B(a)]. In contrast, almost all the emerging colonies obtained two weeks after IB4SAP treatment exhibited bright and strong EGFP-derived green fluorescence [
Figure 3B(b,c)], even though there were some nonfluorescent colonies (
Table 1). Subsequently, some of these fluorescent colonies were subjected to staining with AF594-IB4. All cells exhibited green fluorescence [
Figure 3C(b), arrows] but lost AF594-derived red fluorescence [
Figure 3C(c), arrows], indicating greatly reduced levels of the α-Gal epitope on their surface. Long-term (more than six months; over 40 passages) cultivation did not alter their phenotype (high levels of EGFP expression and greatly reduced levels of the α-Gal epitope expression) (data not shown). Moreover, staining of nonfluorescent colonies with lectin resulted in the appearance of red fluorescence (data not shown), suggesting that they were likely to be non-transfectants that survived after IB4SAP treatment. We also found that these nonfluorescent cells could be excluded by repeated treatment with IB4SAP (data not shown).
2.3. Experiment 3: Targeted Toxin-Based Enrichment of Transgene High-Expressors Is Applicable to Multidrug-Resistant Cells
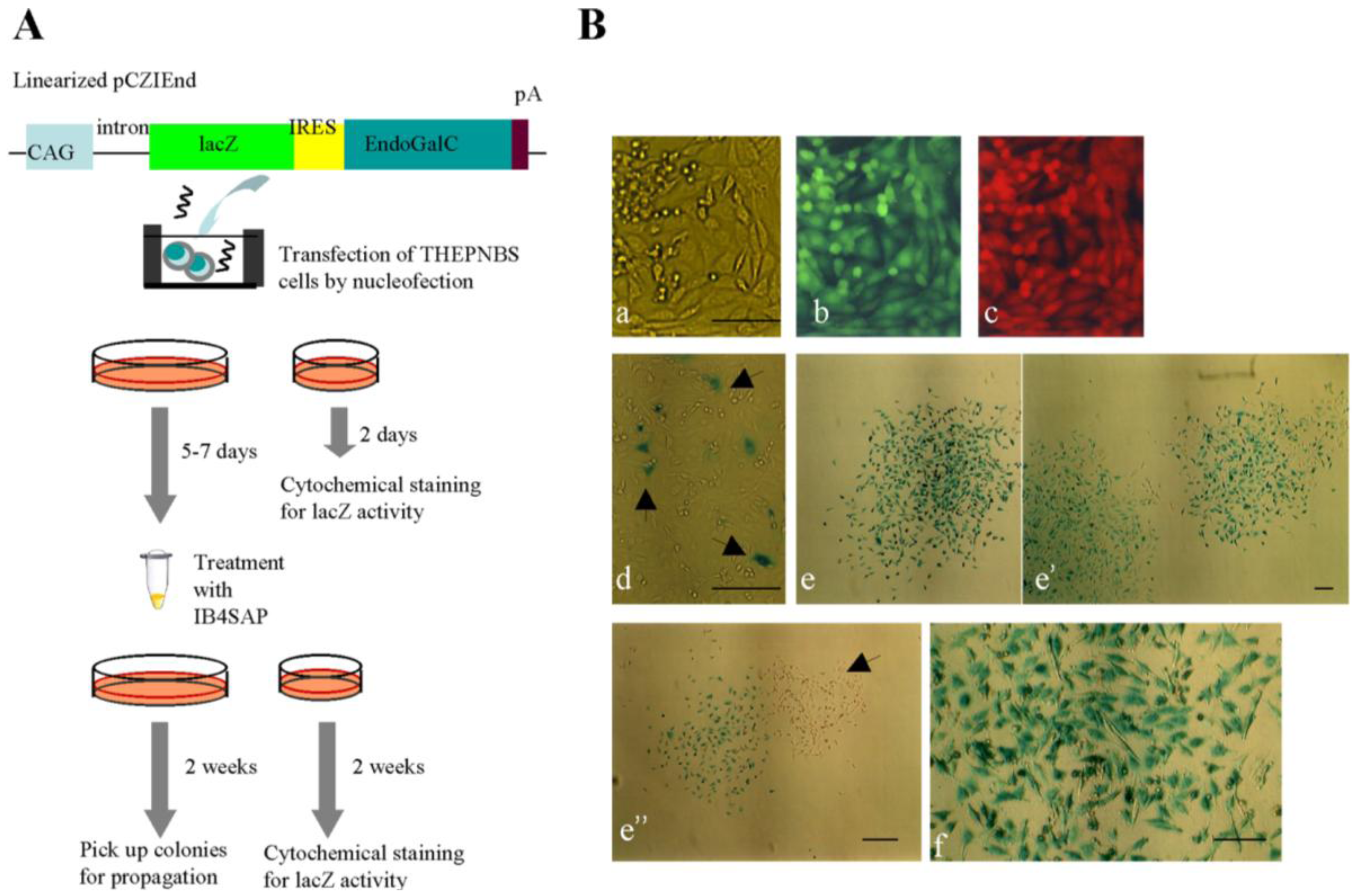
To extend the usefulness of this novel system for multidrug-resistant cells, we introduced a transgene (
Figure 4A) into a multidrug-resistant porcine cell line THEPNBS, which carries two fluorescent marker genes (EGFP and tdTomato) as well as five drug resistance genes (
puro,
neo,
hyg,
Sh ble, and
zeo) [
14]. As expected, simultaneous expression of EGFP and tdTomato was observed in the parental THEPNBS cells [
Figure 4B(b,c)]. As depicted in
Figure 4A, the THEPNBS cells were transfected with pCZIEnd carrying the lacZ gene that codes for
E. coli-derived β-galactosidase, which can be easily detected by cytochemical staining with its substrate, X-Gal. At two days after transfection, only a few cells exhibited lacZ activity [
Figure 4B(d), arrowheads], whereas the majority of cells had negative results for staining [
Figure 4B(d)]. At five to seven days after transfection, the cells harvested by trypsinization were subjected to IB4SAP treatment and then cultured in normal medium. Two weeks after reseeding, the emerging colonies were fixed and examined by cytochemical staining for lacZ activity. As expected, almost all the colonies (26/30) obtained were distinctly stained [
Figure 4B(e,e’)], and higher magnification also showed uniform distribution of lacZ activity throughout the colonies [
Figure 4B(f)]. However, a few colonies (4/30) were negative for X-Gal staining [
Figure 4B(e’’), arrowhead]. As mentioned previously, these colonies may be derived from non-transfectants that survived after IB4SAP treatment. Nevertheless, our results demonstrate that this IB4SAP-based drug-free selection system is applicable to multidrug-resistant cells.
Figure 3.
(
A) Experimental flow for examining whether enrichment of cells with high transgene expression can be achieved with IB4SAP treatment. Abbreviations are the same in
Figure 2A. (
B) Micrographs taken under light + UV illumination. (
a), The cells obtained one week after SAP treatment; and (
b) and (
c), the emerging colonies obtained two weeks after IB4SAP treatment. In (
a), a mixture of fluorescent cells (successfully transfected with pCEIEnd) and nonfluorescent untransfected cells is indicated by an asterisk. The cells in (
b) and (
c) are from two independent colonies. Bar = 100 μm. (
C) Dissociated cells isolated from a colony described in (
c) of
B. These cells were then stained with AF594-IB4. The arrows indicate the isolated cells in (
a) showing bright green fluorescence (
b) but negative for lectin staining (
c). (
a), photograph taken under light; (
b) and (
c), photographs taken under light + UV illumination. Bar = 100 μm.
Figure 3.
(
A) Experimental flow for examining whether enrichment of cells with high transgene expression can be achieved with IB4SAP treatment. Abbreviations are the same in
Figure 2A. (
B) Micrographs taken under light + UV illumination. (
a), The cells obtained one week after SAP treatment; and (
b) and (
c), the emerging colonies obtained two weeks after IB4SAP treatment. In (
a), a mixture of fluorescent cells (successfully transfected with pCEIEnd) and nonfluorescent untransfected cells is indicated by an asterisk. The cells in (
b) and (
c) are from two independent colonies. Bar = 100 μm. (
C) Dissociated cells isolated from a colony described in (
c) of
B. These cells were then stained with AF594-IB4. The arrows indicate the isolated cells in (
a) showing bright green fluorescence (
b) but negative for lectin staining (
c). (
a), photograph taken under light; (
b) and (
c), photographs taken under light + UV illumination. Bar = 100 μm.
Table 1.
Summary of Experiment 2.
Table 1.
Summary of Experiment 2.
| Experiment 1 | No. of colonies generated after IB4SAP treatment | No. of colonies inspected for EGFP fluorescence | No. colonies with various degrees of fluorescence 2 |
| ++ | + | +/− | − |
| 1 | 20 | 15 | 11 | 2 | 0 | 2 |
| 2 | 33 | 24 | 20 | 3 | 0 | 1 |
| 3 | 14 | 10 | 10 | 0 | 0 | 0 |
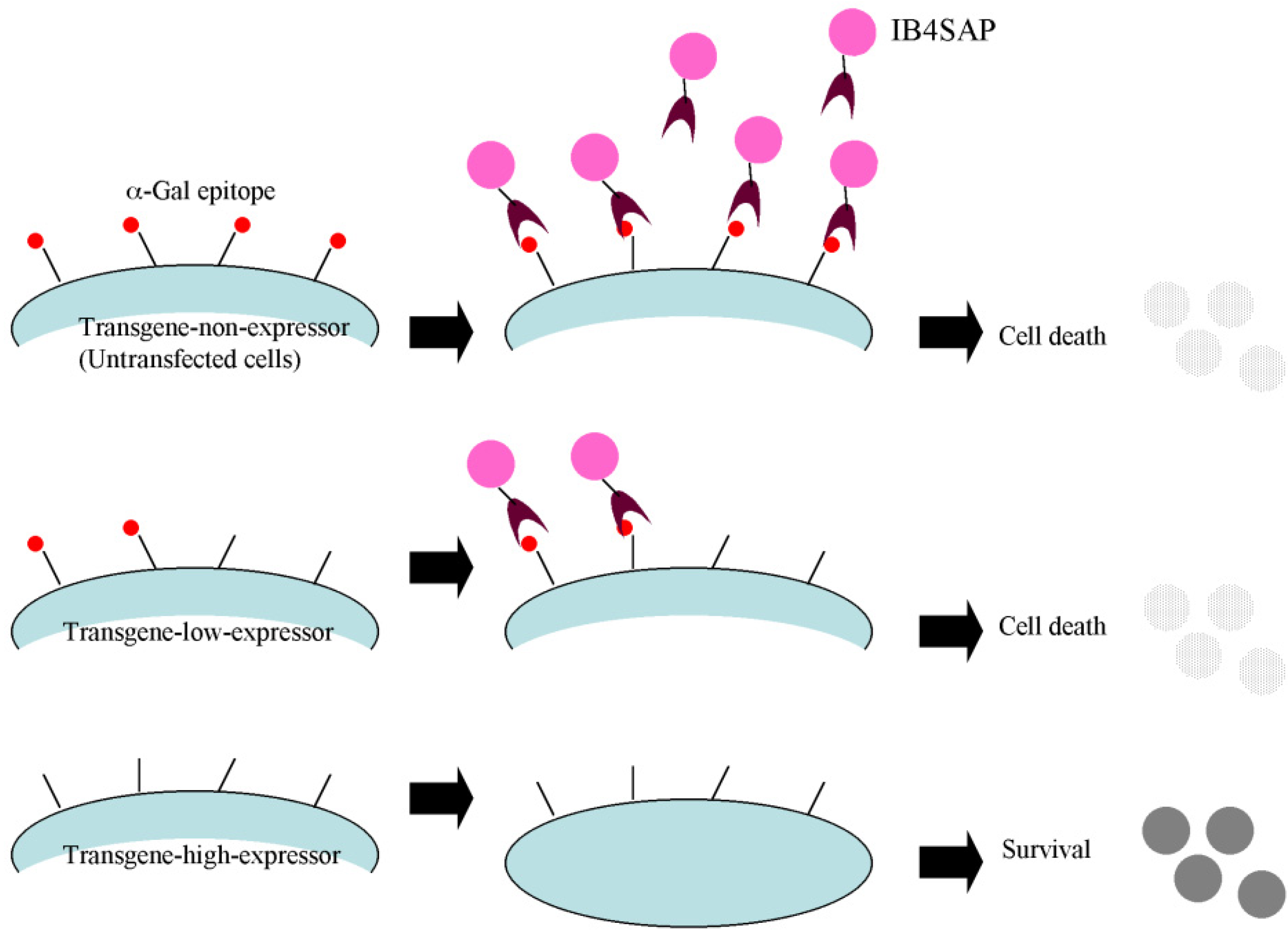
Targeted toxin technology using IB4SAP was first applied for specific elimination of porcine cells that were untransfected, or weakly expressed the EndoGalC gene, to create genetically modified cells suitable for pig-to-human xenotransplantation [
8]. On the basis of this previous study, we decided to utilize this novel system for selecting transgene high-expressors, as described in
Figure 1. Here, we confirm that this system works well. Unfortunately, this system is only applicable to mammalian cells that express the α-Gal epitope. Human and Old World monkey cells that do not express such an epitope due to mutations in the α-GalT gene [
15] cannot be used. Theoretically, if these α-Gal epitope-negative cells are genetically engineered to express α-GalT, then the system would become applicable.
Figure 4.
(A) Experimental flow for examining whether enrichment of cells with high transgene expression in the multidrug-resistant cells (THEPNBS). (B) The THEPNBS cells (a–c) express both EGFP-derived green fluorescence (b) and tdTomato-derived red fluorescence (c) under light + UV illumination. (d) Cytochemical staining of THEPNBS cells for lacZ activity at two day after transfection. The arrowheads indicate the cells exhibiting blue deposits in their cytoplasm, while the other cells were negative for such staining, probably reflecting unsuccessful transfection in those cells. (e–e'') Two weeks after IB4SAP treatment, the colonies were stained for lacZ activity. An arrowhead in (e'') indicates some colonies with no staining for lacZ activity (f) Magnified view of a stained colony demonstrates uniform distribution of the lacZ protein throughout the cells. Bar = 100 μm.
Figure 4.
(A) Experimental flow for examining whether enrichment of cells with high transgene expression in the multidrug-resistant cells (THEPNBS). (B) The THEPNBS cells (a–c) express both EGFP-derived green fluorescence (b) and tdTomato-derived red fluorescence (c) under light + UV illumination. (d) Cytochemical staining of THEPNBS cells for lacZ activity at two day after transfection. The arrowheads indicate the cells exhibiting blue deposits in their cytoplasm, while the other cells were negative for such staining, probably reflecting unsuccessful transfection in those cells. (e–e'') Two weeks after IB4SAP treatment, the colonies were stained for lacZ activity. An arrowhead in (e'') indicates some colonies with no staining for lacZ activity (f) Magnified view of a stained colony demonstrates uniform distribution of the lacZ protein throughout the cells. Bar = 100 μm.
The present system is based on the coexpression of the EndoGalC gene and a GOI in an α-Gal epitope-expressing cell. To achieve this, we used a 0.63-kb IRES sequence that enables simultaneous expression of at least two proteins from a single mRNA [
11,
12,
13]. Subsequently, increased expression of EndoGalC accelerated the digestion of the α-Gal epitope on the cell surface. This reverse correlation between EndoGalC and α-Gal epitope expression was confirmed in the current study (see
Figure 2D). Notably, because EndoGalC is produced by bacteria, such as
C. perfringens, rather than mammalian cells, there is a possibility that EndoGalC expression in mammalian cells would affect cellular properties such as proliferation rate, cell behavior (including cell migration and differentiation), and cellular metabolism. Watanabe
et al. [
16] attempted to address this problem by producing transgenic mice that exhibited systematic expression of EndoGalC. Their results showed that although mice at the newborn stage transiently exhibited growth retardation with abnormal keratinogenesis, those at adult stages gained weight normally and showed normal skin formation. Their internal organs were also normal, and the reproductive ability was not impaired. The same research group later demonstrated that mouse NIH3T3 cells, transfected with an EndoGalC-expression vector, exhibited greater proliferative activity than the untransfected parental cells [
17]. Therefore, it is likely that the EndoGalC-expressing cells proliferate faster than intact cells. Therefore, careful attention should be paid to examine whether cellular behavior (including cell proliferation) is altered before and after introducing an EndoGalC-expression vector.
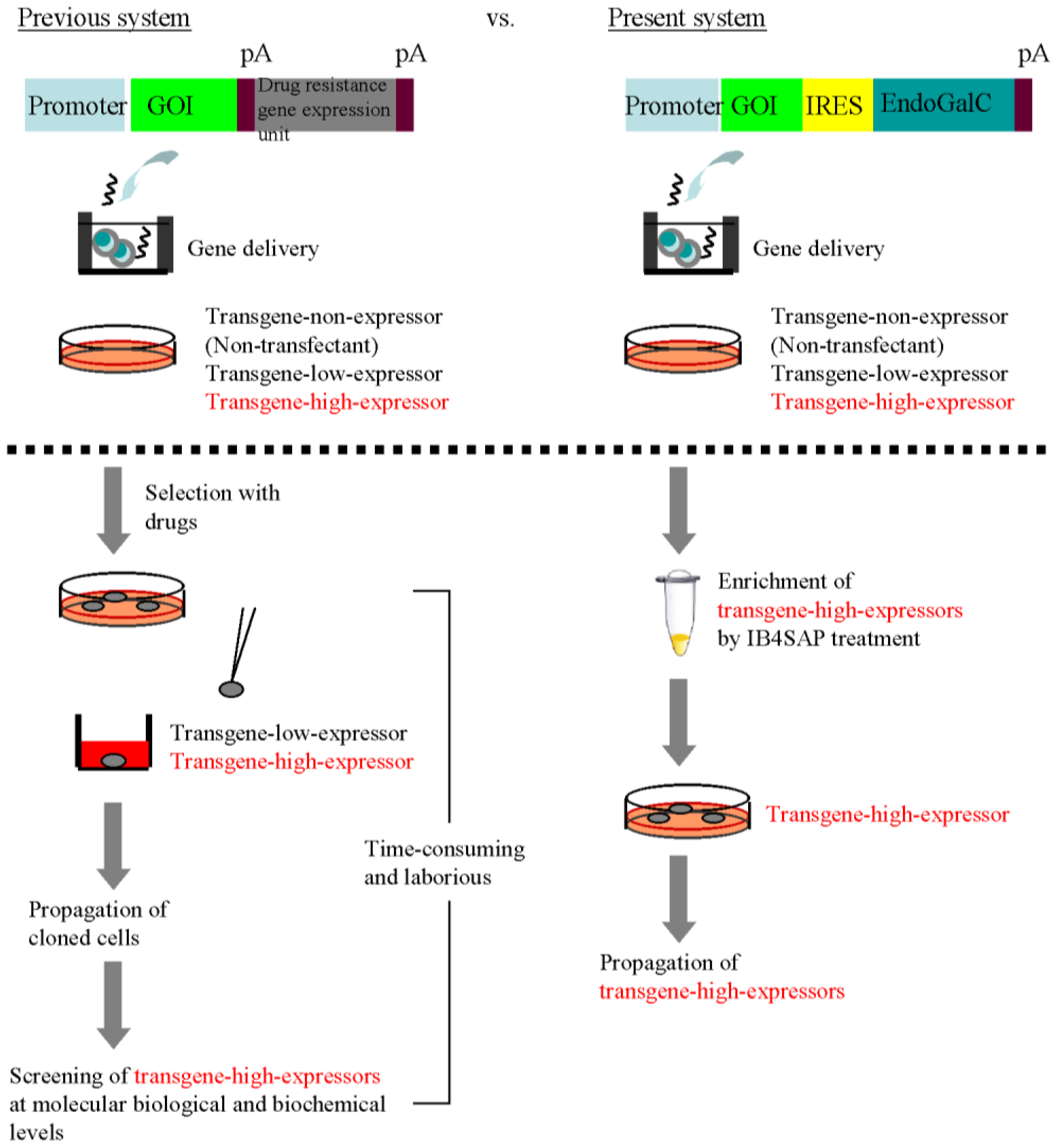
The most excellent property of this system appears to be simple acquisition of transgene high-expressors without drug selection and subsequent molecular biological and biochemical screening of isolated clones. In the traditional cloning approach for isolating transgene high-expressors, the isolation of drug-resistant cells and subsequent characterization of individual clones that have been clonally propagated are essential steps (
Figure 5, previous system). Characterizing the isolated clones at the molecular biological and biochemical levels is time-consuming and laborious. In contrast, our EndoGalC/IB4SAP-based system does not require either drug selection or subsequent characterization of the isolated clones (
Figure 5, current system). The colonies that survive after transfection of an EndoGalC-expressing vector and subsequent IB4SAP treatment should strongly express the GOI. The results presented in this study prove our proposed hypothesis (
Figure 1). Our concern regarding this new system is the procedure to be used to eliminate unwanted cells, which are likely to be untransfected cells escaping from IB4SAP-mediated cell death. However, we experienced a very low incidence of this issue [
Table 1;
Figure 4B(
e’’), arrowhead]. Because these cells still express the α-Gal epitope on their surface, they can be eliminated by repeated IB4SAP treatment.
Figure 5.
Comparison between the previous system for the cloning of recombinant cells and our present EndoGalC/targeted toxin-based system.
Figure 5.
Comparison between the previous system for the cloning of recombinant cells and our present EndoGalC/targeted toxin-based system.
Moreover, this EndoGalC/IB4SAP-based system for the acquisition of transgene high-expressors would be particularly valuable for researchers who wish to perform large-scale production of therapeutically important recombinant proteins (e.g., immunoglobulins) by using mammalian cells. In this case, the drug-free selectable cultivation of cells with high transgene expression is in great demand. To test whether our system can address this demand, we are currently attempting to produce recombinant proteins that can be secreted into the medium by introducing a gene encoding for secreted alkaline phosphatase (SEAP). This system is also applicable to other α-Gal epitope-expressing cells. For example, we successfully obtained transgene high-expressors in mouse NIH3T3 cells using this technology (data not shown). Furthermore, this system appears to be useful in the xenotransplantation field. We have recently succeeded in isolating porcine cells with greatly reduced expression of the α-Gal epitope after transfection with a vector expressing small interfering RNA (siRNA) for α-GalT [
18]. We showed that when these isolated siRNA-expressing cells were used as donor cells for somatic cell nuclear transfer (SCNT) experiments in pigs, the resulting cloned blastocysts exhibited a significant reduction in α-Gal epitope expression. This result has encouraged us to plan studies for producing α-Gal epitope-negative cloned piglets by using SCNT.

 ,
, 



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

