Analysis of T-DNA/Host-Plant DNA Junction Sequences in Single-Copy Transgenic Barley Lines

Abstract
:
1. Introduction
2. Experimental
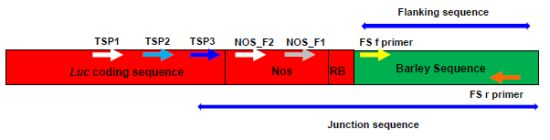
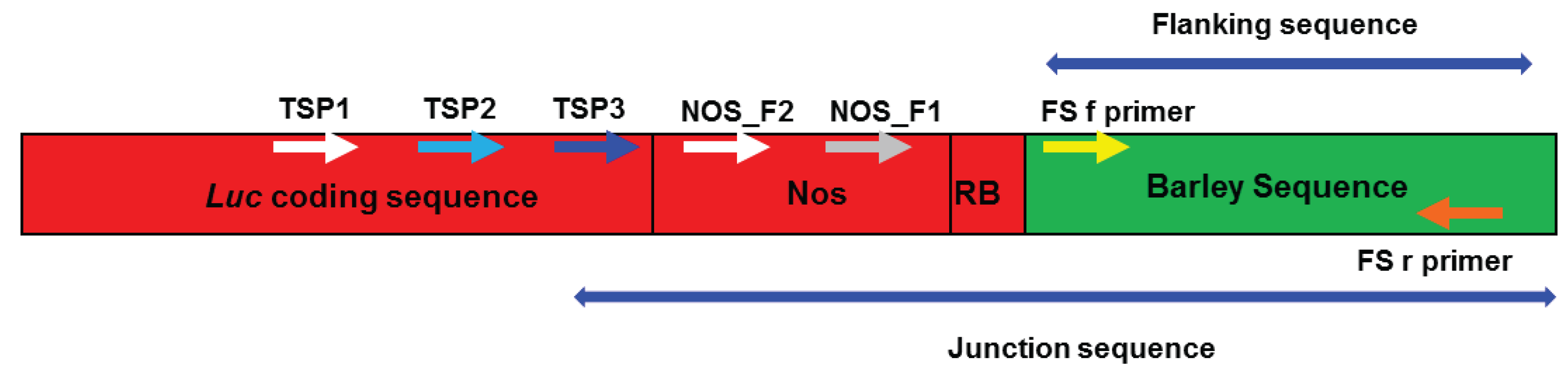
2.1. Amplifying Junction Sequences
2.2. Direct Sequencing

2.3. Verification of Junction Sequences

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Line | FS_f primer (5' to 3') | FS_r primer (5' to 3') | TSP3 and FS_r amplicon size (bp) | FS_f and FS_r amplicon size (bp) |
|---|---|---|---|---|
| 92-05-01 | TGCATTTGCGGACTAATCAT | GAACAAGGGTGCGAAAGAAA | 711 | 221 |
| 90-06-01 | AACCCTTTCATCCGAACATC | GCCTGTTTACCGTCCGTCTA | 656 | 270 |
| 84-13-01 | CGTGTATGGTGTATACTAGCGTAAGA | GGGACGGGGTCTTTAGTTTC | 536 | 170 |
| 84-15-01 | GGCACGGTACAGTCCGTTTA | CACTCAAGCAGACCTGGACA | 681 | 315 |
| 87-10-01 | TCCCGTCAGTCAGTGAGATG | AGCAGGAGCCGATGAACG | 811 | 252 |
| 88-06-01 | TTGCCTACTTGCCTTGCTTT | TTGTTTCCCAATCACCACCT | 810 | 241 |
| 88-13-01 | CCTGCCAAACTGATCCAAAT | ACGGATTCACTGTCGCTGTC | 778 | 220 |
| 89-02-01 | GGGTGGATCTAGCGTACGAG | AGATCTGCACCGCATGAAG | N/A a | 247 |
| 89-07-01 | TGTGCAAAGCAGTGTGTGAA | GATCGCATGCATGTACTCGT | 770 | 281 |
| 85-01-01 | GCATGCATTTCAGTGCTGTT | AGCTGCATGCTCCTGTTCTT | 847 | 285 |
| 85-06-01 | TGCCAGACCAGCTTTAATCA | CCTCTCAACAATGCCATGAA | 957 | 354 |
| 86-03-01 | ATCTCCCACTGATGCTCGAC | TCATGGATATGTCGCCTGTC | 857 | 321 |
| 86-05-01 | GCCTGTTGTTGGGAGTCG | GCCACTCCTTTCAGGAACTAAA | 764 | 259 |
| 85-03-01 | AGCCTGCGAGAATCTCTGGT | ACGATGCTCCATCATCATCA | 807 | 296 |
| 91-02-01 | CCACAAACCCTTACGCTATCA | GATGCTTCTGCGTGCAAGT | 787 | 289 |
| 92-02-01 | ATGAGCACCAATCATCACCA | CGCATGATTACGACAATCCA | 818 | 294 |
| 90-04-01 | GGTCATAATTAAACCCGCACT | CTAACGTGCATCGACTCCAA | 598 | 238 |
| 71-09-01 | AGCGAAGACGACAAGAGCAT | AAACAAAGGCGGTCAATGTG | 784 | 294 |
| 83-06-01 | AGGAAGTCGGAGCATAATTGA | AGGTAGGTATGGTGGCTGTTT | 797 | 292 |
2.4. Analysis of T-DNA Flanking Sequences
3. Results
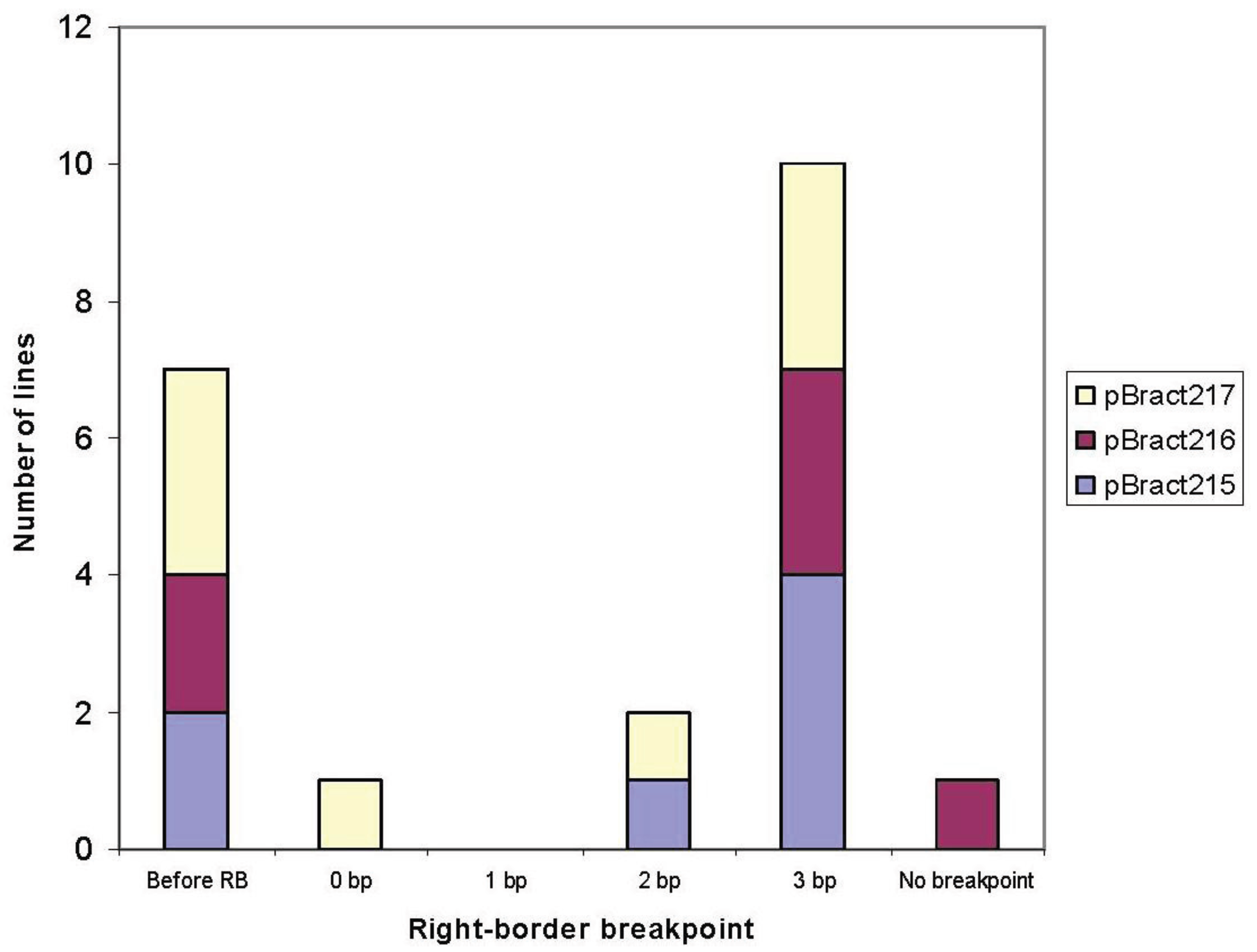
3.1. Right-Border Breakpoints
| Construct | Line | Right-border breakpoint * | Barley flanking sequence obtained (bp) |
|---|---|---|---|
| pBract215 | 88-06-01 | 3 | 559 |
| 88-13-01 | 3 | 877 (with 1 gap) | |
| 85-01-01 | 3 | 1,050 | |
| 85-03-01 | −7 | 632 | |
| 85-06-01 | 3 | 918 | |
| 91-02-01 | 2 | 417 | |
| 71-09-01 | −20 | 1,007 | |
| pBract216 | 90-06-01 | 3 | 1,170 |
| 84-13-01 | 3 | 178 | |
| 84-15-01 | −36 | 627 | |
| 87-10-01 | 3 | 622 | |
| 90-04-01 | −10 | 263 | |
| 84-14-01 | None | 0 | |
| pBract217 | 92-05-01 | −15 | 1,551 |
| 89-02-01 | 0 | 688 | |
| 89-07-01 | −81 | 1,145 (with 1 gap) | |
| 86-03-01 | 3 | 901 | |
| 86-05-01 | 3 | 1,092 (with 2 gaps) | |
| 92-02-01 | 2 | 631 | |
| 83-06-01 | 3 | 448 | |
| 86-09-01 | −1 | 0 |

| Line | Construct | Right-border breakpoint | Barley sequence obtained (bp) | Additional DNA present after the right-border breakpoint |
|---|---|---|---|---|
| 87-10-01 | pBract216 | 3 | 622 | 167 bp of reverse-complement sequence from the left-border region. Sequence begins in the CaMv 35 s promoter (in the T-DNA) and ends 2 bp into the left-border repeat. |
| 86-09-01 | pBract217 | −1 | 0 | At least 315 bp of reverse-complement sequence from the left-border region, starting at the left-border repeat and going into backbone DNA. |
| 89-02-01 | pBract217 | 0 | 688 | 1,055 bp of reverse-complement backbone sequence from the right-border region. Construct sequence ends 2 bp into the right-border repeat from the 3' end |
3.2. Analysis of Barley Flanking DNA

3.3. Relationship between Insertion Site and Transgene Expression
4. Discussion
4.1. T-DNA Integration at the Right-Border
| Construct | Line | Luciferase activity (RLU/µg protein) | Flanking sequence features | Right-border breakpoint |
|---|---|---|---|---|
| pBract215 | 88-13-01 | 27028 | Transposon directly adjacent to T-DNA | 3 |
| 85-01-01 | 28751 | Barley EST 780 bp upstream of T-DNA | 3 | |
| 88-06-01 | 39222 | Homology to H. Vulgare cultivar Bowman contig | 3 | |
| 85-06-01 | 64696 | Retrotransposon directly adjacent to T-DNA | 3 | |
| 85-03-01 | 65924 | Homology to H. vulgare chromosome 5 | −7 | |
| 71-09-01 | 93115 | Alignment to transposon 310 bp upstream of T-DNA | −20 | |
| 91-02-01 | 96837 | Homology to T. aestivum genomic sequence | 2 | |
| pBract216 | 84-15-01 | 69628 | Alignment to repeat element 20 bp upstream from T-DNA | −36 |
| 90-04-01 | 80286 | Transposon directly adjacent to T-DNA | −10 | |
| 90-06-01 | 98937 | Barley EST directly adjacent to T-DNA | 3 | |
| 84-13-01 | 107106 | Transposon directly adjacent to T-DNA | 3 | |
| 84-14-01 | 109189 | No flanking sequence obtained | N/A | |
| 87-10-01 | 115756 | Region of homology to B. distachyon | 3 | |
| pBract217 | 89-07-01 | 140921 | Retrotransposon directly adjacent to T-DNA | −81 |
| 89-02-01 | 278776 | T. aestivum EST 384 bp upstream of T-DNA | 0 | |
| 86-03-01 | 286903 | Retrotransposon 16 bp upstream of T-DNA | 3 | |
| 92-05-01 | 308998 | Retrotransposon directly adjacent to T-DNA | −15 | |
| 86-09-01 | 312466 | No flanking sequence obtained | −1 | |
| 86-05-01 | 364120 | Retrotransposon about 500 bp upstream of T-DNA | 3 | |
| 83-06-01 | 486226 | Retrotransposon directly adjacent to T-DNA | 3 | |
| 92-02-01 | 590787 | T. aestivum EST 526 bp upstream of T-DNA | 2 |
4.2. Analysis of Barley Flanking DNA
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Harwood, W.A.; Bilham, L.; Smedley, M.; Snape, J.W.; Charlton, A.; Oehlschlager, S.; Ibrahim, A.; Waugh, R. Methods for the Analysis of GM Wheat and Barley Seed for Unexpected Consequences of the Transgene Insertion; Final Report for Food Standards Agency Project G02002: London, UK, 2004. [Google Scholar]
- Salvo-Garrido, H.; Travella, S.; Bilham, L.J.; Harwood, W.A.; Snape, J.W. The distribution of transgene insertion sites in barley determined by physical and genetic mapping. Genetics 2004, 167, 1371–1379. [Google Scholar] [CrossRef]
- Francis, K.E.; Spiker, S. Identification of Arabidopsis thaliana transformants without selection reveals a high occurrence of silenced T-DNA integrations. Plant J. 2005, 41, 464–477. [Google Scholar] [CrossRef]
- Kim, S.I.; Veena; Gelvin, S.B. Genome-wide analysis of Agrobacterium T-DNA integration sites in the Arabidopsis genome generated under non-selective conditions. Plant J. 2007, 51, 779–791. [Google Scholar] [CrossRef]
- Sessions, A.; Burke, E.; Presting, G.; Aux, G.; McElver, J.; Patton, D.; Dietrich, B.; Ho, P.; Bacwaden, J.; Ko, C.; et al. A high-throughput Arabidopsis reverse genetics system. Plant Cell 2002, 14, 2985–2994. [Google Scholar] [CrossRef]
- Alonso, J.M.; Stepanova, A.N.; Leisse, T.J.; Kim, C.J.; Chen, H.; Shinn, P.; Stevenson, D.K.; Zimmerman, J.; Barajas, P.; Cheuk, R.; et al. Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 2003, 301, 653–657. [Google Scholar] [CrossRef]
- Tzafrir, I.; Dickerman, A.; Brazhnik, O.; Nguyen, Q.; McElver, J.; Frye, C.; Patton, D.; Meinke, D. The Arabidopsis SeedGenes Project. Nucleic Acids Res. 2003, 31, 90–93. [Google Scholar] [CrossRef]
- Sallaud, C.; Gay, C.; Larmande, P.; Bès, M.; Piffanelli, P.; Piégu, B.; Droc, G.; Regad, F.; Bourgeois, E.; Meynard, D.; et al. High throughput T-DNA insertion mutagenesis in rice: A first step towards in silico reverse genetics. Plant J. 2004, 39, 450–464. [Google Scholar] [CrossRef]
- Wang, N.; Long, T.; Yao, W.; Xiong, L.; Zhang, Q.; Wu, C. Mutant resources for the functional analysis of the rice genome. Mol. Plant 2013, 6, 596–604. [Google Scholar] [CrossRef]
- Bartlett, J.G.; Alves, S.C.; Smedley, M.; Snape, J.W.; Harwood, W.A. High-throughput Agrobacterium-mediated barley transformation. Plant Methods 2008, 4, e22. [Google Scholar] [CrossRef]
- Basic Local Alignment Search Tool (BLAST). Available online: http://www.ncbi.nlm.nih.gov/blast/Blast.cgi/ (accessed on 9 December 2013).
- ClustalW2 Multiple Sequence Alignment. Available online: http://www.ebi.ac.uk/Tools/clustalw2/index.html/ (accessed on 9 December 2013).
- ITMI Triticeae Repeat Sequence Database (TREP). Available online: http://wheat.pw.usda.gov/ITMI/Repeats/ (accessed on 9 December 2013).
- Bartlett, J.G.; Snape, J.W.; Harwood, W.A. Intron-mediated enhancement as a method for increasing transgene expression levels in barley. Plant Biotechnol. J. 2009, 7, 856–866. [Google Scholar] [CrossRef]
- The Barley Genome Sequencing Consortium. A physical, genetic and functional sequence assembly of the barley genome. Nature 2012, 491, 711–716. [Google Scholar]
- Ramanathan, V.; Veluthambi, K. Transfer of non-T-DNA portions of the Agrobacterium tumefaciens Ti plasmid pTiA6 from the left terminus of TL-DNA. Plant Mol. Biol. 1995, 28, 1149–1154. [Google Scholar] [CrossRef]
- Van der Graaff, E.; Den Dulk-Ras, A.; Hooykaas, P.J.J. Deviating T-DNA transfer from Agrobacterium tumefaciens to plants. Plant Mol. Biol. 1996, 31, 677–681. [Google Scholar] [CrossRef]
- Tinland, B.; Schoumacher, F.; Gloeckler, V.; Bravo-Angel, A.M.; Hohn, B. The Agrobacterium tumefaciens virulence D2 protein is responsible for precise integration of T-DNA into the plant genome. EMBO J. 1995, 14, 3585–3595. [Google Scholar]
- Van Lijsebettens, M.; Inzé, D.; Schell, J.; van Montagu, M. Transformed cell clones as a tool to study T-DNA integration mediated by Agrobacterium tumefaciens. J. Mol. Biol. 1986, 188, 129–145. [Google Scholar] [CrossRef]
- Herman, L.; Jacobs, A.; van Montagu, M.; Depicker, A. Plant chromosome/marker gene fusion assay for study of normal and truncated T-DNA integration events. Mol. Gen. Genet. 1990, 224, 248–256. [Google Scholar] [CrossRef]
- Wu, H.; Sparks, C.A.; Jones, H.D. Characterisation of T-DNA loci and vector backbone sequences in transgenic wheat produced by Agrobacterium-mediated transformation. Mol. Breed. 2006, 18, 195–208. [Google Scholar] [CrossRef]
- Gambino, G.; Chitarra, W.; Maghuly, F.; Laimer, M.; Boccacci, P.; Marinoni, D.T.; Gribaudo, I. Characterization of T-DNA insertions in transgenic grapevines obtained by Agrobacterium-mediated transformation. Mol. Breed. 2009, 24, 305–320. [Google Scholar] [CrossRef]
- Zhang, J.; Cai, L.; Cheng, J.; Mao, H.; Fan, X.; Meng, Z.; Chan, K.M.; Zhang, H.; Qi, J.; Ji, L.; et al. Transgene integration and organization in Cotton (Gossypium hirsutum L.) genome. Transgenic Res. 2008, 17, 293–306. [Google Scholar] [CrossRef]
- Gheysen, G.; Angenon, G.; van Montagu, M. Agrobacterium-Mediated Plant Transformation: A Scientifically Intriguing Story with Significant Applications. In Transgenic Plant Research; Lindsey, K., Ed.; Harwood Academic: Amsterdam, The Netherlands, 1998; pp. 1–33. [Google Scholar]
- Wenck, A.; Czakó, M.; Kanevski, I.; Márton, L. Frequent collinear long transfer of DNA inclusive of the whole binary vector during Agrobacterium-mediated transformation. Plant Mol. Biol. 1997, 34, 913–922. [Google Scholar] [CrossRef]
- Yin, Z.; Wang, G.L. Evidence of multiple complex patterns of T-DNA integration into the rice genome. Theor. Appl. Genet. 2000, 100, 461–470. [Google Scholar] [CrossRef]
- Sha, Y.; Li, S.; Pei, Z.; Luo, L.; Tian, Y.; He, C. Generation and flanking sequence analysis of a rice T-DNA tagged population. Theor. Appl. Genet. 2004, 108, 306–314. [Google Scholar] [CrossRef]
- Vicient, C.M.; Jääskeläinen, M.J.; Kalendar, R.; Schulman, A.H. Active retrotransposons are a common feature of grass genomes. Plant Physiol. 2001, 125, 1283–1292. [Google Scholar] [CrossRef]
- Zhao, T.; Palotta, M.; Langridge, P.; Prasad, M.; Graner, A.; Schulze-Lefert, P.; Koprek, T. Mapped Ds/T-DNA launch pads for functional genomics in barley. Plant J. 2006, 47, 811–826. [Google Scholar] [CrossRef]
- Lindsey, K.; Wei, W.; Clarke, M.C.; McArdle, H.F.; Rooke, L.M.; Topping, J.F. Tagging genomic sequences that direct transgene expression by activation of a promoter trap in plants. Transgenic Res. 1993, 2, 33–47. [Google Scholar] [CrossRef]
- Szabados, L.; Kovács, I.; Oberschall, A.; Ábrahám, E.; Kerekes, I.; Zsigmond, L.; Nagy, R.; Alvarado, M.; Krasovskaja, I.; Gál, M.; et al. Distribution of 1,000 sequenced T-DNA tags in the Arabidopsis genome. Plant J. 2002, 32, 233–242. [Google Scholar] [CrossRef]
- Feschotte, C.; Jiang, N.; Wessler, S.R. Plant transposable elements: Where genetics meets genomics. Nat. Rev. Genet. 2002, 3, 329–341. [Google Scholar] [CrossRef]
- Travella, S.; Ross, S.M.; Harden, J.; Everett, C.; Snape, J.W.; Harwood, W.A. A comparison of transgenic barley lines produced by particle bombardment and Agrobacterium-mediated techniques. Plant Cell Rep. 2005, 23, 780–789. [Google Scholar] [CrossRef]
- Zhang, J.; Guo, D.; Chang, Y.; You, C.; Li, X.; Dai, X.; Weng, Q.; Zhang, J.; Chen, G.; Li, X.; et al. Non-random distribution of T-DNA insertions at various levels of the genome hierarchy as revealed by analysing 13 804 T-DNA flanking sequences from an enhancer-trap mutant library. Plant J. 2007, 49, 947–959. [Google Scholar] [CrossRef]
- Ainley, W.M.; Sastry-Dent, L.; Welter, M.E.; Murray, M.G.; Zeitler, B.; Amora, R.; Corbin, D.R.; Miles, R.R.; Arnold, N.L.; Strange, T.L.; et al. Trait stacking via targeted genome editing. Plant Biotechnol. J. 2013, 11, 1126–1134. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bartlett, J.G.; Smedley, M.A.; Harwood, W.A. Analysis of T-DNA/Host-Plant DNA Junction Sequences in Single-Copy Transgenic Barley Lines. Biology 2014, 3, 39-55. https://doi.org/10.3390/biology3010039
Bartlett JG, Smedley MA, Harwood WA. Analysis of T-DNA/Host-Plant DNA Junction Sequences in Single-Copy Transgenic Barley Lines. Biology. 2014; 3(1):39-55. https://doi.org/10.3390/biology3010039
Chicago/Turabian StyleBartlett, Joanne G., Mark A. Smedley, and Wendy A. Harwood. 2014. "Analysis of T-DNA/Host-Plant DNA Junction Sequences in Single-Copy Transgenic Barley Lines" Biology 3, no. 1: 39-55. https://doi.org/10.3390/biology3010039
APA StyleBartlett, J. G., Smedley, M. A., & Harwood, W. A. (2014). Analysis of T-DNA/Host-Plant DNA Junction Sequences in Single-Copy Transgenic Barley Lines. Biology, 3(1), 39-55. https://doi.org/10.3390/biology3010039