High Throughput Screening in Duchenne Muscular Dystrophy: From Drug Discovery to Functional Genomics

Abstract
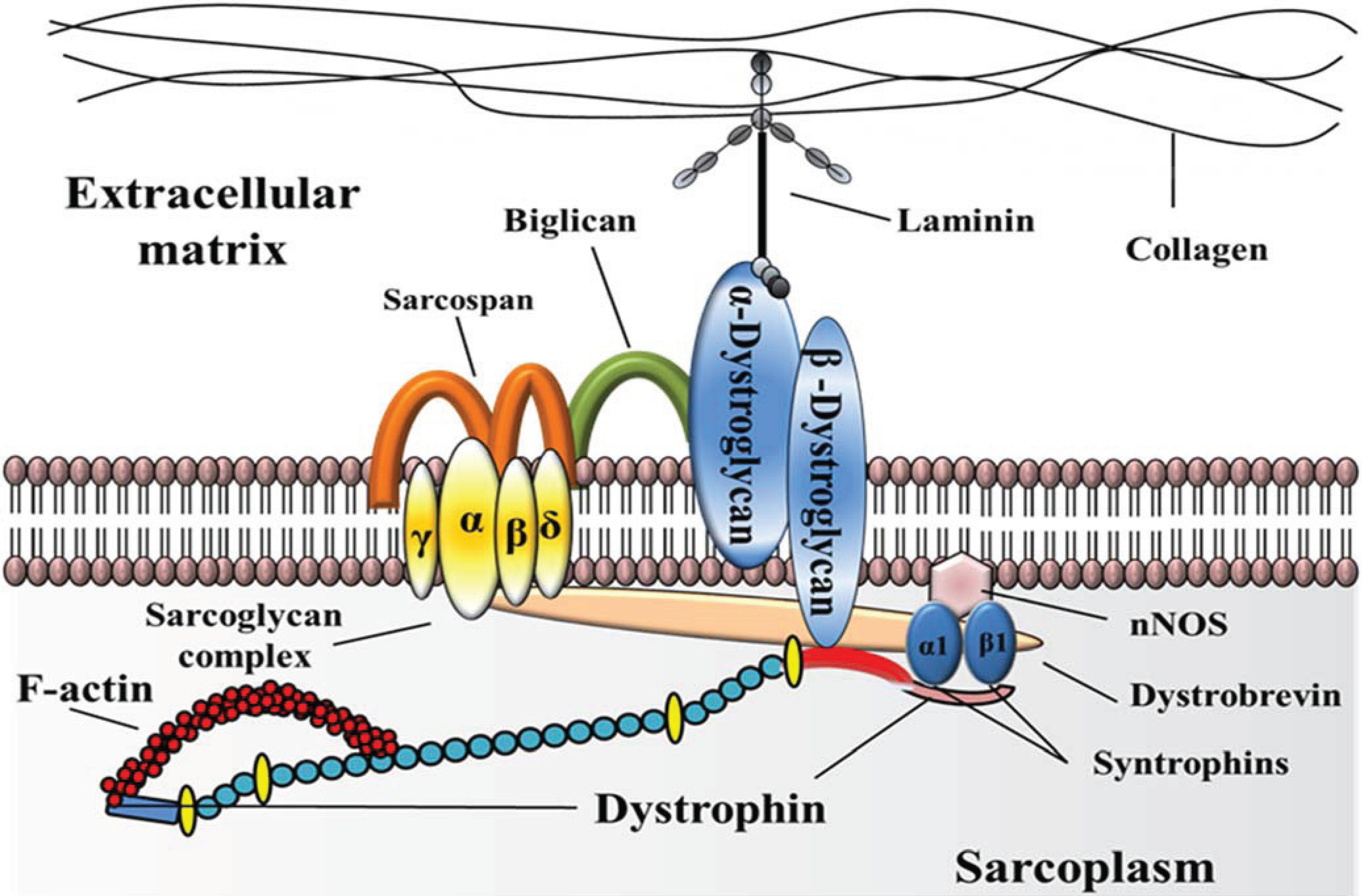
:1. Introduction

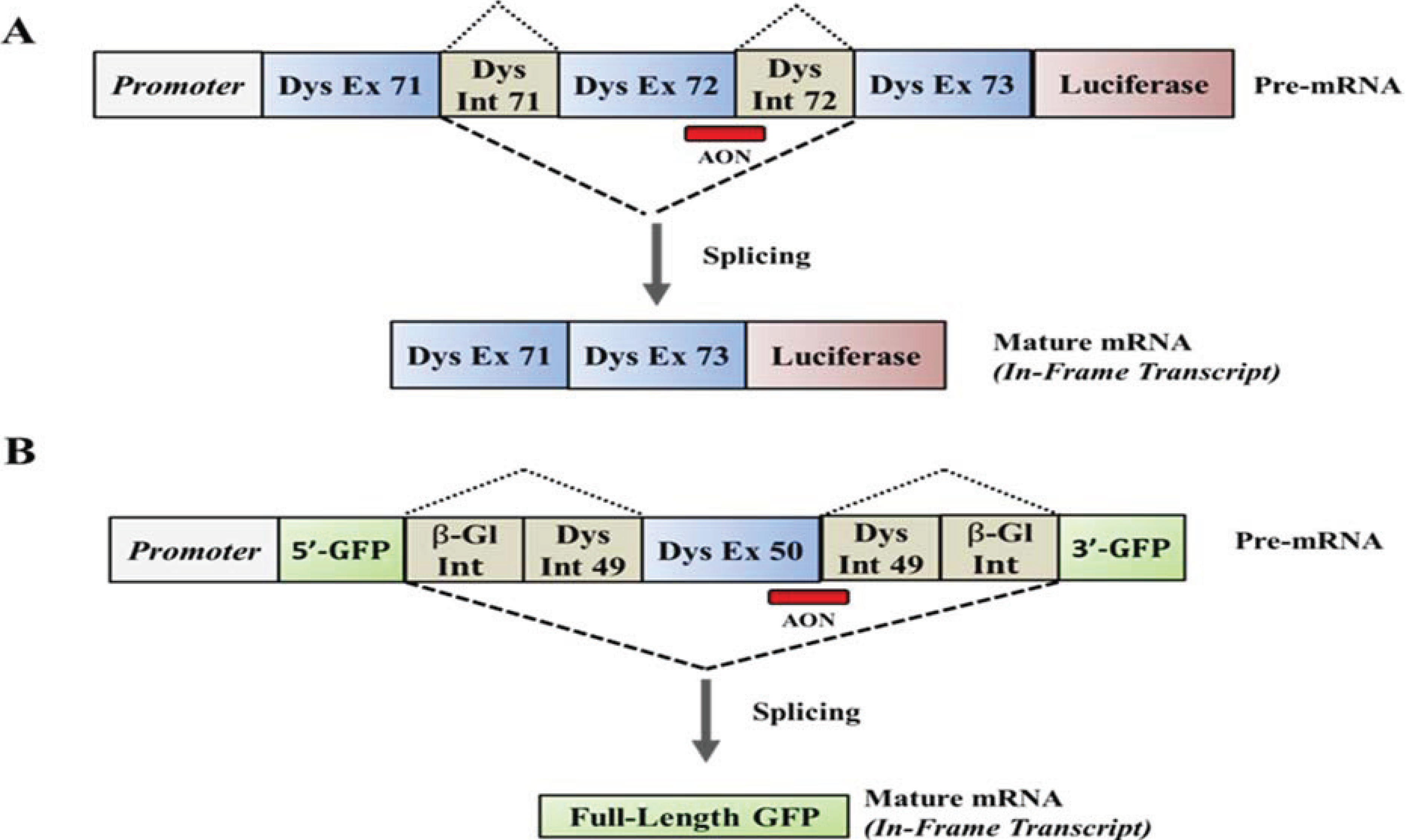
2. HTS of Compounds Capable of Suppressing Nonsense Mutations

{kind=link}
{kind=link}
{kind=link}
| Approach and Lead Candidate/s | Size of Library | Reporter or Assay | Screening System | Animal Model Used to Validate Hits In-Vivo | References |
|---|---|---|---|---|---|
| Read-Through of Premature Stop Codons | |||||
| PTC124 (Translarna) | 800,000 Compounds | Luciferase Reporter | Human HEK 293 Cells | Mdx Mice | [28] |
| Compounds that Enhance AON Activity | |||||
| Podophyllotoxin and HDAC inhibitors | 10,000 Compounds | Luciferase Reporter | Human HEK 293 Cells | None | [78] |
| Utrophin Upregulation | |||||
| SMTC1100 | Not Specified | Luciferase Reporter | Murine H2K Cells | Mdx Mice | [41,81,82] |
| Compounds that Alter Glycosylation | |||||
| Lobeline | 1,124 Compounds | Chemiluminescent Assay | Murine C2C12 Mbs | None | [84] |
| Inhibitors of TGF-β Signaling | |||||
| Several hundred hits | 2,500 Compounds | Luciferase Reporter | Human HEK 293 Cells | None | [85] |
| PDE5 Inhibitors | |||||
| Aminophylline and Sinedafil | 2,640 Compounds | Survival Assay | Zebrafish sapjie and sapjie-like | Mdx5cv Mice | [86,87,88] |
| Regulators of Cell Differentiation and Lineage Commitment | |||||
| Geraldol and Bromopride | 1,600 Compounds | Morphological Assays | Human Muscle Cells | None | [89] |
3. HTS of Small Molecules that Enhance Skipping of the Dystrophin Gene

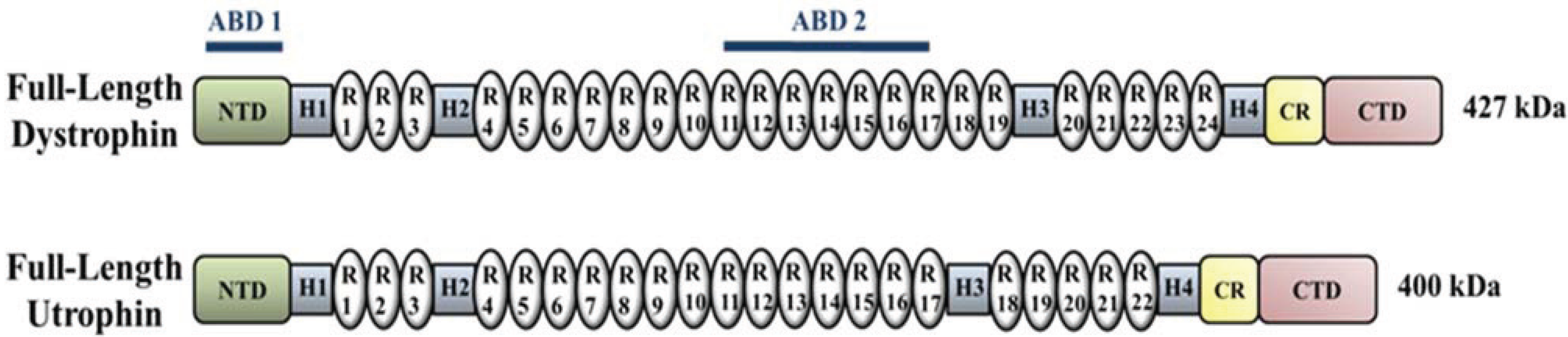
4. Upregulation of Utrophin for the Treatment of DMD

5. Screening of Compounds that Alter Glycosylation of Members of the Dgc
6. HTS in Regenerative Approaches for DMD
7. HTS of Compounds for DMD Using Zebrafish Models
8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hoffman, E.P.; Brown, R.H., Jr.; Kunkel, L.M. Dystrophin: The protein product of the Duchenne muscular dystrophy locus. Cell 1987, 51, 919–928. [Google Scholar] [CrossRef] [PubMed]
- Monaco, A.P.; Neve, R.L.; Colletti-Feener, C.; Bertelson, C.J.; Kurnit, D.M.; Kunkel, L.M. Isolation of candidate cDNAs for portions of the Duchenne muscular dystrophy gene. Nature 1986, 323, 646–650. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Lloyd-Puryear, M. Report of MDA muscle disease symposium on newborn screening for Duchenne muscular dystrophy. Muscle Nerve 2013, 48, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Durbeej, M.; Campbell, K.P. Muscular dystrophies involving the dystrophin-glycoprotein complex: An overview of current mouse models. Curr. Opin. Genet. Dev. 2002, 12, 349–361. [Google Scholar] [CrossRef] [PubMed]
- Hopf, F.W.; Turner, P.R.; Steinhardt, R.A. Calcium misregulation and the pathogenesis of muscular dystrophy. Subcell. Biochem. 2007, 45, 429–464. [Google Scholar] [PubMed]
- Angelini, C. The role of corticosteroids in muscular dystrophy: A critical appraisal. Muscle Nerve 2007, 36, 424–435. [Google Scholar] [CrossRef] [PubMed]
- Moxley, R.T., III; Ashwal, S.; Pandya, S.; Connolly, A.; Florence, J.; Mathews, K.; Baumbach, L.; McDonald, C.; Sussman, M.; Wade, C. Practice parameter: Corticosteroid treatment of Duchenne dystrophy. Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Practice Committee of the Child Neurology Society. Neurology 2005, 64, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Griggs, R.C.; Herr, B.E.; Reha, A.; Elfring, G.; Atkinson, L.; Cwik, V.; McColl, E.; Tawil, R.; Pandya, S.; McDermott, M.P.; Bushby, K. Corticosteroids in Duchenne muscular dystrophy: Major variations in practice. Muscle Nerve 2013, 48, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Dunckley, M.G.; Love, D.R.; Davies, K.E.; Walsh, F.S.; Morris, G.E.; Dickson, G. Retroviral-mediated transfer of a dystrophin minigene into mdx mouse myoblasts in vitro. FEBS Lett. 1992, 296, 128–134. [Google Scholar] [CrossRef] [PubMed]
- Haecker, S.E.; Stedman, H.H.; Balice-Gordon, R.J.; Smith, D.B.; Greelish, J.P.; Mitchell, M.A.; Wells, A.; Sweeney, H.L.; Wilson, J.M. In vivo expression of full-length human dystrophin from adenoviral vectors deleted of all viral genes. Hum. Gene Ther. 1996, 7, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.; Nalbantoglu, J.; Howell, J.M.; Davies, L.; Fletcher, S.; Amalfitano, A.; Petrof, B.J.; Kamen, A.; Massie, B.; Karpati, G. Dystrophin expression in muscle following gene transfer with a fully deleted (“gutted”) adenovirus is markedly improved by trans-acting adenoviral gene products. Hum. Gene Ther. 2001, 12, 1741–1755. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.M.; Li, S.; Harper, S.Q.; Welikson, R.; Bourque, D.; DelloRusso, C.; Hauschka, S.D.; Chamberlain, J.S. Viral vectors for gene transfer of micro-, mini-, or full-length dystrophin. Neuromuscul. Disord. 2002, 12, S23–S29. [Google Scholar] [CrossRef] [PubMed]
- Gregorevic, P.; Blankinship, M.J.; Allen, J.M.; Crawford, R.W.; Meuse, L.; Miller, D.G.; Russell, D.W.; Chamberlain, J.S. Systemic delivery of genes to striated muscles using adeno-associated viral vectors. Nat. Med. 2004, 10, 828–834. [Google Scholar] [CrossRef] [PubMed]
- Lai, Y.; Yue, Y.; Liu, M.; Ghosh, A.; Engelhardt, J.F.; Chamberlain, J.S.; Duan, D. Efficient in vivo gene expression by trans-splicing adeno-associated viral vectors. Nat. Biotechnol. 2005, 23, 1435–1439. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.H.; Mack, L.M.; Choi, S.Y.; Ontell, M.; Kochanek, S.; Clemens, P.R. DNA from both high-capacity and first-generation adenoviral vectors remains intact in skeletal muscle. Hum. Gene Ther. 1999, 10, 365–373. [Google Scholar] [CrossRef] [PubMed]
- Kimura, E.; Li, S.; Gregorevic, P.; Fall, B.M.; Chamberlain, J.S. Dystrophin delivery to muscles of mdx mice using lentiviral vectors leads to myogenic progenitor targeting and stable gene expression. Mol. Ther. 2010, 18, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Odom, G.L.; Gregorevic, P.; Allen, J.M.; Chamberlain, J.S. Gene therapy of mdx mice with large truncated dystrophins generated by recombination using rAAV6. Mol. Ther. 2011, 19, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Mann, C.J.; Honeyman, K.; Cheng, A.J.; Ly, T.; Lloyd, F.; Fletcher, S.; Morgan, J.E.; Partridge, T.A.; Wilton, S.D. Antisense-induced exon skipping and synthesis of dystrophin in the mdx mouse. Proc. Natl. Acad. Sci. USA 2001, 98, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Van Deutekom, J.C.; Bremmer-Bout, M.; Janson, A.A.; Ginjaar, I.B.; Baas, F.; den Dunnen, J.T.; van Ommen, G.J. Antisense-induced exon skipping restores dystrophin expression in DMD patient derived muscle cells. Hum. Mol. Genet. 2001, 10, 1547–1554. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Janson, A.A.; Kaman, W.E.; Bremmer-Bout, M.; den Dunnen, J.T.; Baas, F.; van Ommen, G.J.; van Deutekom, J.C. Therapeutic antisense-induced exon skipping in cultured muscle cells from six different DMD patients. Hum. Mol. Genet. 2003, 12, 907–914. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Lu, Q.; Wood, M. Effective exon skipping and restoration of dystrophin expression by peptide nucleic acid antisense oligonucleotides in mdx mice. Mol. Ther. 2008, 16, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Goyenvalle, A.; Babbs, A.; Wright, J.; Wilkins, V.; Powell, D.; Garcia, L.; Davies, K.E. Rescue of severely affected dystrophin/utrophin-deficient mice through scAAV-U7snRNA-mediated exon skipping. Hum. Mol. Genet. 2012, 21, 2559–2571. [Google Scholar] [CrossRef] [PubMed]
- Cirak, S.; Feng, L.; Anthony, K.; Arechavala-Gomeza, V.; Torelli, S.; Sewry, C.; Morgan, J.E.; Muntoni, F. Restoration of the dystrophin-associated glycoprotein complex after exon skipping therapy in Duchenne muscular dystrophy. Mol. Ther. 2012, 20, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Barton-Davis, E.R.; Cordier, L.; Shoturma, D.I.; Leland, S.E.; Sweeney, H.L. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J. Clin. Invest. 1999, 104, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Hamed, S.; Hadley, D.W.; Gropman, A.L.; Burstein, A.H.; Escolar, D.M.; Hoffman, E.P.; Fischbeck, K.H. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann. Neurol. 2001, 49, 706–711. [Google Scholar] [CrossRef] [PubMed]
- Politano, L.; Nigro, G.; Nigro, V.; Piluso, G.; Papparella, S.; Paciello, O.; Comi, L.I. Gentamicin administration in Duchenne patients with premature stop codon. Preliminary results. Acta Myol. 2003, 22, 15–21. [Google Scholar] [PubMed]
- Malik, V.; Rodino-Klapac, L.R.; Viollet, L.; Wall, C.; King, W.; Al-Dahhak, R.; Lewis, S.; Shilling, C.J.; Kota, J.; Serrano-Munuera, C.; et al. Gentamicin-induced readthrough of stop codons in Duchenne muscular dystrophy. Ann. Neurol. 2010, 67, 771–780. [Google Scholar] [PubMed]
- Welch, E.M.; Barton, E.R.; Zhuo, J.; Tomizawa, Y.; Friesen, W.J.; Trifillis, P.; Paushkin, S.; Patel, M.; Trotta, C.R.; Hwang, S.; et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature 2007, 447, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Kayali, R.; Ku, J.M.; Khitrov, G.; Jung, M.E.; Prikhodko, O.; Bertoni, C. Read-through compound 13 restores dystrophin expression and improves muscle function in the mdx mouse model for Duchenne muscular dystrophy. Hum. Mol. Genet. 2012, 21, 4007–4020. [Google Scholar] [CrossRef] [PubMed]
- Bertoni, C.; Morris, G.E.; Rando, T.A. Strand bias in oligonucleotide-mediated dystrophin gene editing. Hum. Mol. Genet. 2005, 14, 221–233. [Google Scholar] [CrossRef] [PubMed]
- Kayali, R.; Bury, F.; Ballard, M.; Bertoni, C. Site directed gene repair of the dystrophin gene mediated by PNA-ssODNs. Hum. Mol. Genet. 2010, 19, 3266–3281. [Google Scholar] [CrossRef] [PubMed]
- Nik-Ahd, F.; Bertoni, C. Ex vivo gene editing of the dystrophin gene in muscle stem cells using peptide nucleic acid single stranded oligodeoxynucleotides (PNA-SsODNs) induces stable expression of dystrophin in a mouse model for Duchenne muscular dystrophy. Stem Cells 2014, 32, 1817–1830. [Google Scholar] [CrossRef] [PubMed]
- Chapdelaine, P.; Pichavant, C.; Rousseau, J.; Paques, F.; Tremblay, J.P. Meganucleases can restore the reading frame of a mutated dystrophin. Gene Ther. 2010, 17, 846–858. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, J.; Chapdelaine, P.; Boisvert, S.; Almeida, L.P.; Corbeil, J.; Montpetit, A.; Tremblay, J.P. Endonucleases: Tools to correct the dystrophin gene. J. Gene Med. 2011, 13, 522–537. [Google Scholar] [CrossRef] [PubMed]
- Ousterout, D.G.; Perez-Pinera, P.; Thakore, P.I.; Kabadi, A.M.; Brown, M.T.; Qin, X.; Fedrigo, O.; Mouly, V.; Tremblay, J.P.; Gersbach, C.A. Reading frame correction by targeted genome editing restores dystrophin expression in cells from Duchenne muscular dystrophy patients. Mol. Ther. 2013, 21, 1718–1726. [Google Scholar] [CrossRef]
- Tinsley, J.M.; Potter, A.C.; Phelps, S.R.; Fisher, R.; Trickett, J.I.; Davies, K.E. Amelioration of the dystrophic phenotype of mdx mice using a truncated utrophin transgene. Nature 1996, 384, 349–353. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.; Deconinck, N.; Fisher, R.; Kahn, D.; Phelps, S.; Gillis, J.M.; Davies, K. Expression of full-length utrophin prevents muscular dystrophy in mdx mice. Nat. Med. 1998, 4, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, R.; Nalbanoglu, J.; Tinsley, J.M.; Massie, B.; Davies, K.E.; Karpati, G. Efficient utrophin expression following adenovirus gene transfer in dystrophic muscle. Biochem. Biophys. Res. Commun. 1998, 242, 244–247. [Google Scholar] [CrossRef] [PubMed]
- Wakefield, P.M.; Tinsley, J.M.; Wood, M.J.; Gilbert, R.; Karpati, G.; Davies, K.E. Prevention of the dystrophic phenotype in dystrophin/utrophin-deficient muscle following adenovirus-mediated transfer of a utrophin minigene. Gene Ther. 2000, 7, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Krag, T.O.; Bogdanovich, S.; Jensen, C.J.; Fischer, M.D.; Hansen-Schwartz, J.; Javazon, E.H.; Flake, A.W.; Edvinsson, L.; Khurana, T.S. Heregulin ameliorates the dystrophic phenotype in mdx mice. Proc. Natl. Acad. Sci. USA 2004, 101, 13856–13860. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.M.; Fairclough, R.J.; Storer, R.; Wilkes, F.J.; Potter, A.C.; Squire, S.E.; Powell, D.S.; Cozzoli, A.; Capogrosso, R.F.; Lambert, A.; et al. Daily treatment with SMTC1100, a novel small molecule utrophin upregulator, dramatically reduces the dystrophic symptoms in the mdx mouse. PLoS One 2011, 6, e19189. [Google Scholar] [CrossRef] [PubMed]
- Bogdanovich, S.; Krag, T.O.; Barton, E.R.; Morris, L.D.; Whittemore, L.A.; Ahima, R.S.; Khurana, T.S. Functional improvement of dystrophic muscle by myostatin blockade. Nature 2002, 420, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; McPherron, A.C.; Winik, N.; Lee, S.J. Loss of myostatin attenuates severity of muscular dystrophy in mdx mice. Ann. Neurol. 2002, 52, 832–836. [Google Scholar] [CrossRef] [PubMed]
- Haidet, A.M.; Rizo, L.; Handy, C.; Umapathi, P.; Eagle, A.; Shilling, C.; Boue, D.; Martin, P.T.; Sahenk, Z.; Mendell, J.R.; Kaspar, B.K. Long-term enhancement of skeletal muscle mass and strength by single gene administration of myostatin inhibitors. Proc. Natl. Acad. Sci. USA 2008, 105, 4318–4322. [Google Scholar] [CrossRef] [PubMed]
- Cohn, R.D.; van Erp, C.; Habashi, J.P.; Soleimani, A.A.; Klein, E.C.; Lisi, M.T.; Gamradt, M.; ap Rhys, C.M.; Holm, T.M.; Loeys, B.L.; et al. Angiotensin II type 1 receptor blockade attenuates TGF-beta-induced failure of muscle regeneration in multiple myopathic states. Nat. Med. 2007, 13, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Bish, L.T.; Yarchoan, M.; Sleeper, M.M.; Gazzara, J.A.; Morine, K.J.; Acosta, P.; Barton, E.R.; Sweeney, H.L. Chronic losartan administration reduces mortality and preserves cardiac but not skeletal muscle function in dystrophic mice. PLoS One 2011, 6, e20856. [Google Scholar] [PubMed]
- Spurney, C.F.; Sali, A.; Guerron, A.D.; Iantorno, M.; Yu, Q.; Gordish-Dressman, H.; Rayavarapu, S.; van der Meulen, J.; Hoffman, E.P.; Nagaraju, K. Losartan decreases cardiac muscle fibrosis and improves cardiac function in dystrophin-deficient mdx mice. J. Cardiovasc. Pharmacol. Ther. 2011, 16, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Messina, S.; Bitto, A.; Aguennouz, M.; Minutoli, L.; Monici, M.C.; Altavilla, D.; Squadrito, F.; Vita, G. Nuclear factor kappa-B blockade reduces skeletal muscle degeneration and enhances muscle function in Mdx mice. Exp. Neurol. 2006, 198, 234–241. [Google Scholar] [CrossRef] [PubMed]
- Grounds, M.D.; Torrisi, J. Anti-TNFalpha (remicade) therapy protects dystrophic skeletal muscle from necrosis. FASEB J. 2004, 18, 676–682. [Google Scholar] [CrossRef]
- Pierno, S.; Nico, B.; Burdi, R.; Liantonio, A.; Didonna, M.P.; Cippone, V.; Fraysse, B.; Rolland, J.F.; Mangieri, D.; Andreetta, F.; et al. Role of tumour necrosis factor alpha, but not of cyclo-oxygenase-2-derived eicosanoids, on functional and morphological indices of dystrophic progression in mdx mice: A pharmacological approach. Neuropathol. Appl. Neurobiol. 2007, 33, 344–359. [Google Scholar] [CrossRef] [PubMed]
- Hodgetts, S.; Radley, H.; Davies, M.; Grounds, M.D. Reduced necrosis of dystrophic muscle by depletion of host neutrophils, or blocking TNFalpha function with Etanercept in mdx mice. Neuromuscul. Disord. 2006, 16, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Piers, A.T.; Lavin, T.; Radley-Crabb, H.G.; Bakker, A.J.; Grounds, M.D.; Pinniger, G.J. Blockade of TNF in vivo using cV1q antibody reduces contractile dysfunction of skeletal muscle in response to eccentric exercise in dystrophic mdx and normal mice. Neuromuscul. Disord. 2011, 21, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Peterson, J.M.; Kline, W.; Canan, B.D.; Ricca, D.J.; Kaspar, B.; Delfin, D.A.; DiRienzo, K.; Clemens, P.R.; Robbins, P.D.; Baldwin, A.S.; et al. Peptide-based inhibition of NF-kappaB rescues diaphragm muscle contractile dysfunction in a murine model of Duchenne muscular dystrophy. Mol. Med. 2011, 17, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Malik, V.; Rodino-Klapac, L.R.; Mendell, J.R. Emerging drugs for Duchenne muscular dystrophy. Expert. Opin. Emerg. Drugs 2012, 17, 261–277. [Google Scholar] [CrossRef] [PubMed]
- Ruegg, U.T. Pharmacological prospects in the treatment of Duchenne muscular dystrophy. Curr. Opin. Neurol. 2013, 26, 577–584. [Google Scholar] [CrossRef] [PubMed]
- Fairclough, R.J.; Wood, M.J.; Davies, K.E. Therapy for Duchenne muscular dystrophy: Renewed optimism from genetic approaches. Nat. Rev. Genet. 2013, 14, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Rahimov, F.; Kunkel, L.M. The cell biology of disease: Cellular and molecular mechanisms underlying muscular dystrophy. J. Cell Biol. 2013, 201, 499–510. [Google Scholar] [CrossRef] [PubMed]
- Prior, T.W.; Bartolo, C.; Pearl, D.K.; Papp, A.C.; Snyder, P.J.; Sedra, M.S.; Burghes, A.H.; Mendell, J.R. Spectrum of small mutations in the dystrophin coding region. Am. J. Hum. Genet. 1995, 57, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.; Gilbert, W.; Gorini, L. Streptomycin, suppression, and the code. Proc. Natl. Acad. Sci. USA 1964, 51, 883–890. [Google Scholar] [CrossRef] [PubMed]
- Anderson, W.F.; Gorini, L.; Breckenridge, L. Role of ribosomes in streptomycin-activated suppression. Proc. Natl. Acad. Sci. USA 1965, 54, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Howard, M.; Frizzell, R.A.; Bedwell, D.M. Aminoglycoside antibiotics restore CFTR function by overcoming premature stop mutations. Nat. Med. 1996, 2, 467–469. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.J. Correction of genetic disease by making sense from nonsense. J. Clin. Invest. 1999, 104, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Du, M.; Jones, J.R.; Lanier, J.; Keeling, K.M.; Lindsey, J.R.; Tousson, A.; Bebok, Z.; Whitsett, J.A.; Dey, C.R.; Colledge, W.H.; et al. Aminoglycoside suppression of a premature stop mutation in a Cftr-/- mouse carrying a human CFTR-G542X transgene. J. Mol. Med. 2002, 80, 595–604. [Google Scholar] [CrossRef] [PubMed]
- Manuvakhova, M.; Keeling, K.; Bedwell, D.M. Aminoglycoside antibiotics mediate context-dependent suppression of termination codons in a mammalian translation system. RNA 2000, 6, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Damoiseaux, R.; Nahas, S.; Gao, K.; Hu, H.; Pollard, J.M.; Goldstine, J.; Jung, M.E.; Henning, S.M.; Bertoni, C.; et al. Nonaminoglycoside compounds induce readthrough of nonsense mutations. J. Exp. Med. 2009, 206, 2285–2297. [Google Scholar] [CrossRef] [PubMed]
- Auld, D.S.; Thorne, N.; Maguire, W.F.; Inglese, J. Mechanism of PTC124 activity in cell-based luciferase assays of nonsense codon suppression. Proc. Natl. Acad. Sci. USA 2009, 106, 3585–3590. [Google Scholar] [CrossRef] [PubMed]
- Auld, D.S.; Lovell, S.; Thorne, N.; Lea, W.A.; Maloney, D.J.; Shen, M.; Rai, G.; Battaile, K.P.; Thomas, C.J.; Simeonov, A.; et al. Molecular basis for the high-affinity binding and stabilization of firefly luciferase by PTC124. Proc. Natl. Acad. Sci. USA 2010, 107, 4878–4883. [Google Scholar] [CrossRef] [PubMed]
- Peltz, S.W.; Welch, E.M.; Jacobson, A.; Trotta, C.R.; Naryshkin, N.; Sweeney, H.L.; Bedwell, D.M. Nonsense suppression activity of PTC124 (ataluren). Proc. Natl. Acad. Sci. USA 2009, 106, E64. [Google Scholar] [CrossRef] [PubMed]
- McElroy, S.P.; Nomura, T.; Torrie, L.S.; Warbrick, E.; Gartner, U.; Wood, G.; McLean, W.H.I. A lack of premature termination codon read-through efficacy of PTC124 (Ataluren) in a diverse array of reporter assays. PLoS Biol. 2013, 11, e1001593. [Google Scholar] [CrossRef] [PubMed]
- Kerem, E.; Hirawat, S.; Armoni, S.; Yaakov, Y.; Shoseyov, D.; Cohen, M.; Nissim-Rafinia, M.; Blau, H.; Rivlin, J.; Aviram, M.; et al. Effectiveness of PTC124 treatment of cystic fibrosis caused by nonsense mutations: A prospective phase II trial. Lancet 2008, 372, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Sermet-Gaudelus, I.; Boeck, K.D.; Casimir, G.J.; Vermeulen, F.; Leal, T.; Mogenet, A.; Roussel, D.; Fritsch, J.; Hanssens, L.; Hirawat, S.; et al. Ataluren (PTC124) induces cystic fibrosis transmembrane conductance regulator protein expression and activity in children with nonsense mutation cystic fibrosis. Am. J. Respir. Crit. Care Med. 2010, 182, 1262–1272. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.; Narayan, S.B.; Chen, J.; Meyers, G.D.; Bennett, M.J. PTC124 improves readthrough and increases enzymatic activity of the CPT1A R160X nonsense mutation. J. Inherit. Metab. Dis. 2011, 34, 443–447. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Overlack, N.; Wolfrum, U.; Nagel-Wolfrum, K. PTC124-mediated translational readthrough of a nonsense mutation causing Usher syndrome type 1C. Hum. Gene Ther. 2011, 22, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Goldmann, T.; Overlack, N.; Moller, F.; Belakhov, V.; van Wyk, M.; Baasov, T.; Wolfrum, U.; Nagel-Wolfrum, K. A comparative evaluation of NB30, NB54 and PTC124 in translational read-through efficacy for treatment of an USH1C nonsense mutation. EMBO Mol. Med. 2012, 4, 1186–1199. [Google Scholar] [CrossRef] [PubMed]
- Guglieri, M.; Bushby, K. Molecular treatments in Duchenne muscular dystrophy. Curr. Opin. Pharmacol. 2010, 10, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.E.; Ku, J.M.; Du, L.; Hu, H.; Gatti, R.A. Synthesis and evaluation of compounds that induce readthrough of premature termination codons. Bioorg. Med. Chem. Lett. 2011, 21, 5842–5848. [Google Scholar] [CrossRef] [PubMed]
- Du, L.; Jung, M.E.; Damoiseaux, R.; Completo, G.; Fike, F.; Ku, J.M.; Nahas, S.; Piao, C.; Hu, H.; Gatti, R.A. A new series of small molecular weight compounds induce read through of all three types of nonsense mutations in the ATM gene. Mol. Ther. 2013, 21, 1653–1660. [Google Scholar] [CrossRef]
- O’Leary, D.A.; Sharif, O.; Anderson, P.; Tu, B.; Welch, G.; Zhou, Y.; Caldwell, J.S.; Engels, I.H.; Brinker, A. Identification of small molecule and genetic modulators of AON-induced dystrophin exon skipping by high-throughput screening. PLoS One 2009, 4, e8348. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wu, B.; Zillmer, A.; Lu, P.; Benrashid, E.; Wang, M.; Doran, T.; Shaban, M.; Wu, X.; Lu, Q.L. Guanine analogues enhance antisense oligonucleotide-induced exon skipping in dystrophin gene in vitro and in vivo. Mol. Ther. 2010, 18, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Kendall, G.C.; Mokhonova, E.I.; Moran, M.; Sejbuk, N.E.; Wang, D.W.; Silva, O.; Wang, R.T.; Martinez, L.; Lu, Q.L.; Damoiseaux, R.; et al. Dantrolene enhances antisense-mediated exon skipping in human and mouse models of Duchenne muscular dystrophy. Sci. Transl. Med. 2012, 4, 164ra160. [Google Scholar] [CrossRef] [PubMed]
- Chancellor, D.R.; Davies, K.E.; de Moor, O.; Dorgan, C.R.; Johnson, P.D.; Lambert, A.G.; Lawrence, D.; Lecci, C.; Maillol, C.; Middleton, P.J.; et al. Discovery of 2-arylbenzoxazoles as upregulators of utrophin production for the treatment of Duchenne muscular dystrophy. J. Med. Chem. 2011, 54, 3241–3250. [Google Scholar] [CrossRef] [PubMed]
- De Moor, O.; Dorgan, C.R.; Johnson, P.D.; Lambert, A.G.; Lecci, C.; Maillol, C.; Nugent, G.; Poignant, S.D.; Price, P.D.; Pye, R.J.; et al. Discovery and SAR of 2-arylbenzotriazoles and 2-arylindazoles as potential treatments for Duchenne muscular dystrophy. Bioorg. Med. Chem. Lett. 2011, 21, 4828–4831. [Google Scholar] [CrossRef] [PubMed]
- Moorwood, C.; Lozynska, O.; Suri, N.; Napper, A.D.; Diamond, S.L.; Khurana, T.S. Drug discovery for Duchenne muscular dystrophy via utrophin promoter activation screening. PLoS One 2011, 6, e26169. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, P.V.; Pang, M.; Marshall, J.L.; Kung, R.; Nelson, S.F.; Stalnaker, S.H.; Wells, L.; Crosbie-Watson, R.H.; Baum, L.G. High throughput screening for compounds that alter muscle cell glycosylation identifies new role for N-glycans in regulating sarcolemmal protein abundance and laminin binding. J. Biol. Chem. 2012, 287, 22759–22770. [Google Scholar] [CrossRef] [PubMed]
- Cash, J.N.; Angerman, E.B.; Kirby, R.J.; Merck, L.; Seibel, W.L.; Wortman, M.D.; Papoian, R.; Nelson, S.; Thompson, T.B. Development of a small-molecule screening method for inhibitors of cellular response to myostatin and activin A. J. Biomol. Screen. 2013, 18, 837–844. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, G.; Kunkel, L.M. Zebrafish based small molecule screens for novel DMD drugs. Drug Discov. Today Technol. 2013, 10, e91–e96. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, G.; Karpf, J.A.; Myers, J.A.; Alexander, M.S.; Guyon, J.R.; Kunkel, L.M. Drug screening in a zebrafish model of Duchenne muscular dystrophy. Proc. Natl. Acad. Sci. USA 2011, 108, 5331–5336. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, G.; Gasperini, M.J.; Myers, J.A.; Widrick, J.J.; Eran, A.; Serafini, P.R.; Alexander, M.S.; Pletcher, M.T.; Morris, C.A.; Kunkel, L.M. Dystrophic muscle improvement in zebrafish via increased heme oxygenase signaling. Hum. Mol. Genet. 2013. [Google Scholar] [CrossRef]
- Nierobisz, L.S.; Cheatham, B.; Buehrer, B.M.; Sexton, J.Z. High-content screening of human primary muscle satellite cells for new therapies for muscular atrophy/dystrophy. Curr. Chem. Genomics Transl. Med. 2013, 7, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Saccone, V.; Consalvi, S.; Giordani, L.; Mozzetta, C.; Barozzi, I.; Sandona, M.; Ryan, T.; Rojas-Munoz, A.; Madaro, L.; Fasanaro, P.; et al. HDAC-regulated myomiRs control BAF60 variant exchange and direct the functional phenotype of fibro-adipogenic progenitors in dystrophic muscles. Genes Dev. 2014, 28, 841–857. [Google Scholar] [CrossRef] [PubMed]
- Xu, C.; Tabebordbar, M.; Iovino, S.; Ciarlo, C.; Liu, J.; Castiglioni, A.; Price, E.; Liu, M.; Barton, E.R.; Kahn, C.R.; et al. A zebrafish embryo culture system defines factors that promote vertebrate myogenesis across species. Cell 2013, 155, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Dunckley, M.G.; Manoharan, M.; Villiet, P.; Eperon, I.C.; Dickson, G. Modification of splicing in the dystrophin gene in cultured Mdx muscle cells by antisense oligoribonucleotides. Hum. Mol. Genet. 1998, 7, 1083–1090. [Google Scholar] [CrossRef] [PubMed]
- Wilton, S.D.; Lloyd, F.; Carville, K.; Fletcher, S.; Honeyman, K.; Agrawal, S.; Kole, R. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul. Disord. 1999, 9, 330–338. [Google Scholar] [CrossRef] [PubMed]
- Lu, Q.L.; Mann, C.J.; Lou, F.; Bou-Gharios, G.; Morris, G.E.; Xue, S.A.; Fletcher, S.; Partridge, T.A.; Wilton, S.D. Functional amounts of dystrophin produced by skipping the mutated exon in the mdx dystrophic mouse. Nat. Med. 2003, 9, 1009–1014. [Google Scholar] [CrossRef] [PubMed]
- Kinali, M.; Arechavala-Gomeza, V.; Feng, L.; Cirak, S.; Hunt, D.; Adkin, C.; Guglieri, M.; Ashton, E.; Abbs, S.; Nihoyannopoulos, P.; et al. Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: A single-blind, placebo-controlled, dose-escalation, proof-of-concept study. Lancet Neurol. 2009, 8, 918–928. [Google Scholar] [CrossRef] [PubMed]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of PRO051 in Duchenne’s muscular dystrophy. N. Engl. J. Med. 2011, 364, 1513–1522. [Google Scholar] [CrossRef] [PubMed]
- Mendell, J.R.; Rodino-Klapac, L.R.; Sahenk, Z.; Roush, K.; Bird, L.; Lowes, L.P.; Alfano, L.; Gomez, A.M.; Lewis, S.; Kota, J.; et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann. Neurol. 2013, 74, 637–647. [Google Scholar] [CrossRef] [PubMed]
- Voit, T.; Topaloglu, H.; Straub, V.; Muntoni, F.; Deconinck, N.; Campion, G.; de Kimpe, S.J.; Eagle, M.; Guglieri, M.; Hood, S.; et al. Safety and efficacy of drisapersen for the treatment of Duchenne muscular dystrophy (DEMAND II): An exploratory, randomised, placebo-controlled phase 2 study. Lancet Neurol. 2014, 13, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Nissim-Rafinia, M.; Aviram, M.; Randell, S.H.; Shushi, L.; Ozeri, E.; Chiba-Falek, O.; Eidelman, O.; Pollard, H.B.; Yankaskas, J.R.; Kerem, B. Restoration of the cystic fibrosis transmembrane conductance regulator function by splicing modulation. EMBO Rep. 2004, 5, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Riessland, M.; Brichta, L.; Hahnen, E.; Wirth, B. The benzamide M344, a novel histone deacetylase inhibitor, significantly increases SMN2 RNA/protein levels in spinal muscular atrophy cells. Hum. Genet. 2006, 120, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Sazani, P.; Kang, S.H.; Maier, M.A.; Wei, C.; Dillman, J.; Summerton, J.; Manoharan, M.; Kole, R. Nuclear antisense effects of neutral, anionic and cationic oligonucleotide analogs. Nucleic Acids Res. 2001, 29, 3965–3974. [Google Scholar] [PubMed]
- Compound Information. Available online: http://www.msdiscovery.com/spectrum.html (accessed on 10 November 2014).
- Chemical Libraries. Available online: http://www.mssr.ucla.edu/lib.html (accessed on 10 November 2014).
- Verhaart, I.E.; Aartsma-Rus, A. The effect of 6-thioguanine on alternative splicing and antisense-mediated exon skipping treatment for duchenne muscular dystrophy. PLoS Curr. 2012, 4. [Google Scholar] [CrossRef]
- Bertorini, T.E.; Palmieri, G.M.; Griffin, J.; Igarashi, M.; Hinton, A.; Karas, J.G. Effect of dantrolene in Duchenne muscular dystrophy. Muscle Nerve 1991, 14, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, J.G.; Johnson, S.R.; Samaha, F.J. Dantrolene normalizes serum creatine kinase in MDX mice. Muscle Nerve 1990, 13, 268–269. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.L.; Huynh, T.; Uaesoontrachoon, K.; Tatem, K.; Phadke, A.; van der Meulen, J.H.; Yu, Q.; Nagaraju, K. Effects of dantrolene therapy on disease phenotype in dystrophin deficient mdx Mice. PLoS Curr. 2013, 5. [Google Scholar] [CrossRef]
- Bertorini, T.E.; Bhattacharya, S.K.; Palmieri, G.M.; Chesney, C.M.; Pifer, D.; Baker, B. Muscle calcium and magnesium content in Duchenne muscular dystrophy. Neurology 1982, 32, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Gailly, P.; Boland, B.; Himpens, B.; Casteels, R.; Gillis, J.M. Critical evaluation of cytosolic calcium determination in resting muscle fibres from normal and dystrophic (mdx) mice. Cell Calcium 1993, 14, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Morine, K.J.; Sleeper, M.M.; Barton, E.R.; Sweeney, H.L. Overexpression of SERCA1a in the mdx diaphragm reduces susceptibility to contraction-induced damage. Hum. Gene Ther. 2010, 21, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Gehrig, S.M.; van der Poel, C.; Sayer, T.A.; Schertzer, J.D.; Henstridge, D.C.; Church, J.E.; Lamon, S.; Russell, A.P.; Davies, K.E.; Febbraio, M.A.; et al. Hsp72 preserves muscle function and slows progression of severe muscular dystrophy. Nature 2012, 484, 394–398. [Google Scholar] [CrossRef] [PubMed]
- Bellinger, A.M.; Reiken, S.; Carlson, C.; Mongillo, M.; Liu, X.; Rothman, L.; Matecki, S.; Lacampagne, A.; Marks, A.R. Hypernitrosylated ryanodine receptor calcium release channels are leaky in dystrophic muscle. Nat. Med. 2009, 15, 325–330. [Google Scholar] [CrossRef] [PubMed]
- Love, D.R.; Hill, D.F.; Dickson, G.; Spurr, N.K.; Byth, B.C.; Marsden, R.F.; Walsh, F.S.; Edwards, Y.H.; Davies, K.E. An autosomal transcript in skeletal muscle with homology to dystrophin. Nature 1989, 339, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Khurana, T.S.; Hoffman, E.P.; Kunkel, L.M. Identification of a chromosome 6-encoded dystrophin-related protein. J. Biol. Chem. 1990, 265, 16717–16720. [Google Scholar] [PubMed]
- Tinsley, J.M.; Blake, D.J.; Roche, A.; Fairbrother, U.; Riss, J.; Byth, B.C.; Knight, A.E.; Kendrick-Jones, J.; Suthers, G.K.; Love, D.R. Primary structure of dystrophin-related protein. Nature 1992, 360, 591–593. [Google Scholar] [CrossRef] [PubMed]
- Gramolini, A.O.; Angus, L.M.; Schaeffer, L.; Burton, E.A.; Tinsley, J.M.; Davies, K.E.; Changeux, J.P.; Jasmin, B.J. Induction of utrophin gene expression by heregulin in skeletal muscle cells: Role of the N-box motif and GA binding protein. Proc. Natl. Acad. Sci. USA 1999, 96, 3223–3227. [Google Scholar] [CrossRef] [PubMed]
- Khurana, T.S.; Rosmarin, A.G.; Shang, J.; Krag, T.O.; Das, S.; Gammeltoft, S. Activation of utrophin promoter by heregulin via the ets-related transcription factor complex GA-binding protein alpha/beta. Mol. Biol. Cell 1999, 10, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Desantis, A.; Onori, A.; di Certo, M.G.; Mattei, E.; Fanciulli, M.; Passananti, C.; Corbi, N. Novel activation domain derived from Che-1 cofactor coupled with the artificial protein Jazz drives utrophin upregulation. Neuromuscul. Disord. 2009, 19, 158–162. [Google Scholar] [CrossRef] [PubMed]
- Mattei, E.; Corbi, N.; di Certo, M.G.; Strimpakos, G.; Severini, C.; Onori, A.; Desantis, A.; Libri, V.; Buontempo, S.; Floridi, A.; et al. Utrophin up-regulation by an artificial transcription factor in transgenic mice. PLoS One 2007, 2, e774. [Google Scholar] [CrossRef] [PubMed]
- Onori, A.; Desantis, A.; Buontempo, S.; di Certo, M.G.; Fanciulli, M.; Salvatori, L.; Passananti, C.; Corbi, N. The artificial 4-zinc-finger protein Bagly binds human utrophin promoter A at the endogenous chromosomal site and activates transcription. Biochem. Cell Biol. 2007, 85, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Corbi, N.; Libri, V.; Fanciulli, M.; Tinsley, J.M.; Davies, K.E.; Passananti, C. The artificial zinc finger coding gene “Jazz” binds the utrophin promoter and activates transcription. Gene Ther. 2000, 7, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Tian, C.; Danialou, G.; Gilbert, R.; Petrof, B.J.; Karpati, G.; Nalbantoglu, J. Targeting artificial transcription factors to the utrophin A promoter: Effects on dystrophic pathology and muscle function. J. Biol. Chem. 2008, 283, 34720–34727. [Google Scholar] [CrossRef] [PubMed]
- Moorwood, C.; Khurana, T.S. Duchenne muscular dystrophy drug discovery-the application of utrophin promoter activation screening. Expert. Opin. Drug Discov. 2013, 8, 569–581. [Google Scholar] [CrossRef] [PubMed]
- Tinsley, J.; Robinson, N.; Wilson, F.X.; Horne, G.; Fairclough, R.J.; Davies, K. Future clinical and biomarker development for SMTC1100, the first utrophin modulator to enter clinical trials for Duchenne Muscular Dystrophy (DMD). Neuromuscul. Disorders 2013, 23, 813. [Google Scholar] [CrossRef]
- Bertoni, C. Therapy development for neuromuscular diseases: Translating hope into promise. Future. Neurol. 2013, 8. [Google Scholar] [CrossRef]
- Moorwood, C.; Soni, N.; Patel, G.; Wilton, S.D.; Khurana, T.S. A cell-based high-throughput screening assay for posttranscriptional utrophin upregulation. J. Biomol. Screen. 2013, 18, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.H.; Jayasinha, V.; Xia, B.; Hoyte, K.; Martin, P.T. Overexpression of the cytotoxic T cell GalNAc transferase in skeletal muscle inhibits muscular dystrophy in mdx mice. Proc. Natl. Acad. Sci. USA 2002, 99, 5616–5621. [Google Scholar] [CrossRef] [PubMed]
- Charge, S.B.; Rudnicki, M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev. 2004, 84, 209–238. [Google Scholar] [CrossRef] [PubMed]
- Cheung, T.H.; Rando, T.A. Molecular regulation of stem cell quiescence. Nat. Rev. Mol. Cell Biol. 2013, 14, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Brack, A.S.; Rando, T.A. Tissue-specific stem cells: Lessons from the skeletal muscle satellite cell. Cell Stem Cell 2012, 10, 504–514. [Google Scholar] [CrossRef] [PubMed]
- Khanjyan, M.V.; Yang, J.; Kayali, R.; Caldwell, T.; Bertoni, C. A high-content, high-throughput siRNA screen identifies cyclin D2 as a potent regulator of muscle progenitor cell fusion and a target to enhance muscle regeneration. Hum. Mol. Genet. 2013, 22, 3283–3295. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Cohen, J.S. Myostatin-related muscle hypertrophy. Available online: http://www.ncbi.nlm.nih.gov/gtr/conditions/C2931112/ (accessed online: 10 October 2014).
- McPherron, A.C.; Lee, S.J. Double muscling in cattle due to mutations in the myostatin gene. Proc. Natl. Acad. Sci. USA 1997, 94, 12457–12461. [Google Scholar] [CrossRef] [PubMed]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997, 387, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J.; McPherron, A.C. Myostatin and the control of skeletal muscle mass. Curr. Opin. Genet. Dev. 1999, 9, 604–607. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Massague, J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell 2003, 113, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Wagner, K.R.; Fleckenstein, J.L.; Amato, A.A.; Barohn, R.J.; Bushby, K.; Escolar, D.M.; Flanigan, K.M.; Pestronk, A.; Tawil, R.; Wolfe, G.I.; et al. A phase I/IItrial of MYO-029 in adult subjects with muscular dystrophy. Ann. Neurol. 2008, 63, 561–571. [Google Scholar] [CrossRef]
- Wahrmann, J.P.; Winand, R.; Luzzati, D. Effect of cyclic AMP on growth and morphological differentiation of an established myogenic cell line. Nat. New Biol. 1973, 245, 112–113. [Google Scholar] [CrossRef] [PubMed]
- Berdeaux, R.; Stewart, R. cAMP signaling in skeletal muscle adaptation: Hypertrophy, metabolism, and regeneration. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1–E17. [Google Scholar] [CrossRef] [PubMed]
- Cash, J.N.; Angerman, E.B.; Kattamuri, C.; Nolan, K.; Zhao, H.; Sidis, Y.; Keutmann, H.T.; Thompson, T.B. Structure of myostatin.follistatin-like 3: N-terminal domains of follistatin-type molecules exhibit alternate modes of binding. J. Biol. Chem. 2012, 287, 1043–1053. [Google Scholar] [CrossRef] [PubMed]
- Thies, R.S.; Chen, T.; Davies, M.V.; Tomkinson, K.N.; Pearson, A.A.; Shakey, Q.A.; Wolfman, N.M. GDF-8 propeptide binds to GDF-8 and antagonizes biological activity by inhibiting GDF-8 receptor binding. Growth Factors 2001, 18, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.; Flechner, L.; Montminy, M.; Berdeaux, R. CREB is activated by muscle injury and promotes muscle regeneration. PLoS One 2011, 6, e24714. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.E.; Ginty, D.D.; Fan, C.M. Protein kinase A signalling via CREB controls myogenesis induced by Wnt proteins. Nature 2005, 433, 317–322. [Google Scholar] [CrossRef] [PubMed]
- Keren, A.; Keren-Politansky, A.; Bengal, E. A p38 MAPK-CREB pathway functions to pattern mesoderm in Xenopus. Dev. Biol. 2008, 322, 86–94. [Google Scholar] [CrossRef] [PubMed]
- Zalin, R.J.; Montague, W. Changes in cyclic AMP, adrenylate cyclase and protein kinase levels during the development of embryonic chick skeletal muscle. Exp. Cell Res. 1975, 93, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Lynch, G.S.; Ryall, J.G. Role of beta-adrenoceptor signaling in skeletal muscle: Implications for muscle wasting and disease. Physiol. Rev. 2008, 88, 729–767. [Google Scholar] [CrossRef] [PubMed]
- Dahm, R.; Geisler, R. Learning from small fry: The zebrafish as a genetic model organism for aquaculture fish species. Mar. Biotechnol. (NY) 2006, 8, 329–345. [Google Scholar] [CrossRef]
- Kari, G.; Rodeck, U.; Dicker, A.P. Zebrafish: An emerging model system for human disease and drug discovery. Clin. Pharmacol. Ther. 2007, 82, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Lieschke, G.J.; Currie, P.D. Animal models of human disease: Zebrafish swim into view. Nat. Rev. Genet. 2007, 8, 353–367. [Google Scholar] [PubMed]
- Santoriello, C.; Zon, L.I. Hooked! Modeling human disease in zebrafish. J. Clin. Invest. 2012, 122, 2337–2343. [Google Scholar] [CrossRef] [PubMed]
- Stern, H.M.; Zon, L.I. Cancer genetics and drug discovery in the zebrafish. Nat. Rev. Cancer 2003, 3, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Berghmans, S.; Jette, C.; Langenau, D.; Hsu, K.; Stewart, R.; Look, T.; Kanki, J.P. Making waves in cancer research: New models in the zebrafish. Biotechniques 2005, 39, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Rubinstein, A.L. Zebrafish: From disease modeling to drug discovery. Curr. Opin. Drug Discov. Devel. 2003, 6, 218–223. [Google Scholar] [PubMed]
- Zon, L.I.; Peterson, R.T. In vivo drug discovery in the zebrafish. Nat. Rev. Drug Discov. 2005, 4, 35–44. [Google Scholar] [CrossRef] [PubMed]
- Seeley, M.; Huang, W.; Chen, Z.; Wolff, W.O.; Lin, X.; Xu, X. Depletion of zebrafish titin reduces cardiac contractility by disrupting the assembly of Z-discs and A-bands. Circ. Res. 2007, 100, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Granato, M.; van Eeden, F.J.; Schach, U.; Trowe, T.; Brand, M.; Furutani-Seiki, M.; Haffter, P.; Hammerschmidt, M.; Heisenberg, C.P.; Jiang, Y.J.; et al. Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 1996, 123, 399–413. [Google Scholar] [PubMed]
- Bassett, D.I.; Bryson-Richardson, R.J.; Daggett, D.F.; Gautier, P.; Keenan, D.G.; Currie, P.D. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development 2003, 130, 5851–5860. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.E.; Bryson-Richardson, R.J.; Berger, S.; Jacoby, A.S.; Cole, N.J.; Hollway, G.E.; Berger, J.; Currie, P.D. The zebrafish candyfloss mutant implicates extracellular matrix adhesion failure in laminin alpha2-deficient congenital muscular dystrophy. Proc. Natl. Acad. Sci. USA 2007, 104, 7092–7097. [Google Scholar] [CrossRef] [PubMed]
- Steffen, L.S.; Guyon, J.R.; Vogel, E.D.; Howell, M.H.; Zhou, Y.; Weber, G.J.; Zon, L.I.; Kunkel, L.M. The zebrafish runzel muscular dystrophy is linked to the titin gene. Dev. Biol. 2007, 309, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.; Kawahara, G.; Gundry, S.R.; Chen, A.T.; Lencer, W.I.; Zhou, Y.; Zon, L.I.; Kunkel, L.M.; Beggs, A.H. The zebrafish dag1 mutant: A novel genetic model for dystroglycanopathies. Hum. Mol. Genet. 2011, 20, 1712–1725. [Google Scholar] [CrossRef] [PubMed]
- Parsons, M.J.; Campos, I.; Hirst, E.M.; Stemple, D.L. Removal of dystroglycan causes severe muscular dystrophy in zebrafish embryos. Development 2002, 129, 3505–3512. [Google Scholar] [PubMed]
- Gibbs, E.M.; Horstick, E.J.; Dowling, J.J. Swimming into prominence: The zebrafish as a valuable tool for studying human myopathies and muscular dystrophies. FEBS J. 2013, 280, 4187–4197. [Google Scholar] [CrossRef] [PubMed]
- Guyon, J.R.; Goswami, J.; Jun, S.J.; Thorne, M.; Howell, M.; Pusack, T.; Kawahara, G.; Steffen, L.S.; Galdzicki, M.; Kunkel, L.M. Genetic isolation and characterization of a splicing mutant of zebrafish dystrophin. Hum. Mol. Genet. 2009, 18, 202–211. [Google Scholar] [CrossRef] [PubMed]
- Collins, F. An audience with Francis Collins. Interviewed by Asher Mullard. Nat. Rev. Drug Discov. 2011, 10, 14. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gintjee, T.J.J.; Magh, A.S.H.; Bertoni, C. High Throughput Screening in Duchenne Muscular Dystrophy: From Drug Discovery to Functional Genomics. Biology 2014, 3, 752-780. https://doi.org/10.3390/biology3040752
Gintjee TJJ, Magh ASH, Bertoni C. High Throughput Screening in Duchenne Muscular Dystrophy: From Drug Discovery to Functional Genomics. Biology. 2014; 3(4):752-780. https://doi.org/10.3390/biology3040752
Chicago/Turabian StyleGintjee, Thomas J.J., Alvin S.H. Magh, and Carmen Bertoni. 2014. "High Throughput Screening in Duchenne Muscular Dystrophy: From Drug Discovery to Functional Genomics" Biology 3, no. 4: 752-780. https://doi.org/10.3390/biology3040752
APA StyleGintjee, T. J. J., Magh, A. S. H., & Bertoni, C. (2014). High Throughput Screening in Duchenne Muscular Dystrophy: From Drug Discovery to Functional Genomics. Biology, 3(4), 752-780. https://doi.org/10.3390/biology3040752