
Will Lipidation of ApoA1 through Interaction with ABCA1 at the Intestinal Level Affect the Protective Functions of HDL?

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Difficulties in Translational Studies Related to High-Density Lipoprotein (HDL) Metabolism
2.1. Absence of Reliable Method to Biochemically Characterize HDL Particles
2.2. Lack of Clinically Relevant Pre-Clinical Animal Models for Drug Development
2.3. Early Indications that High-Density Lipoprotein Cholesterol HDL-C Is Not Always a Reliable Biomarker of Cardiovascular (CV) Risk
2.4. Role of Cholesteryl Ester Transfer Protein (CETP) in HDL Recycling and ApoA1 Exchange
3. HDL Function: Roles of ABCA1, ABCG1 and SRB1 in Cholesterol Efflux from Macrophages
3.1. Macrophage as Preferred Target Cell for the Antiatherosclerotic/Atheroprotective Role of HDL
3.1.1. ABCA1
3.1.2. Macrophage Cholesterol Efflux as a Potential Marker of HDL Function and CV Risk
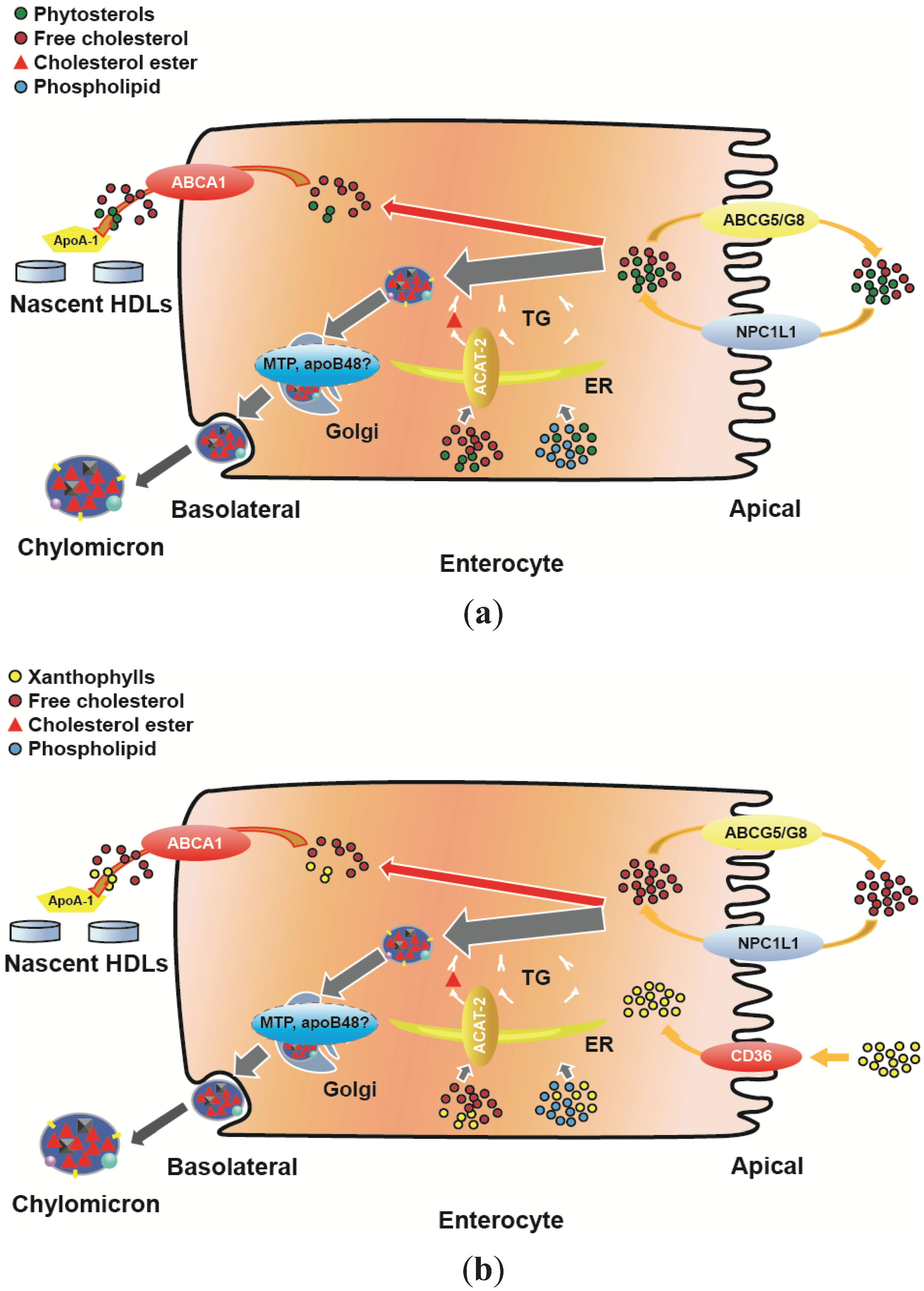
4. HDL Function at the Intestinal Level
4.1. Uptake of Cholesterol
4.2. Uptake of Phytosterols
4.3. Uptake of Xanthophyll


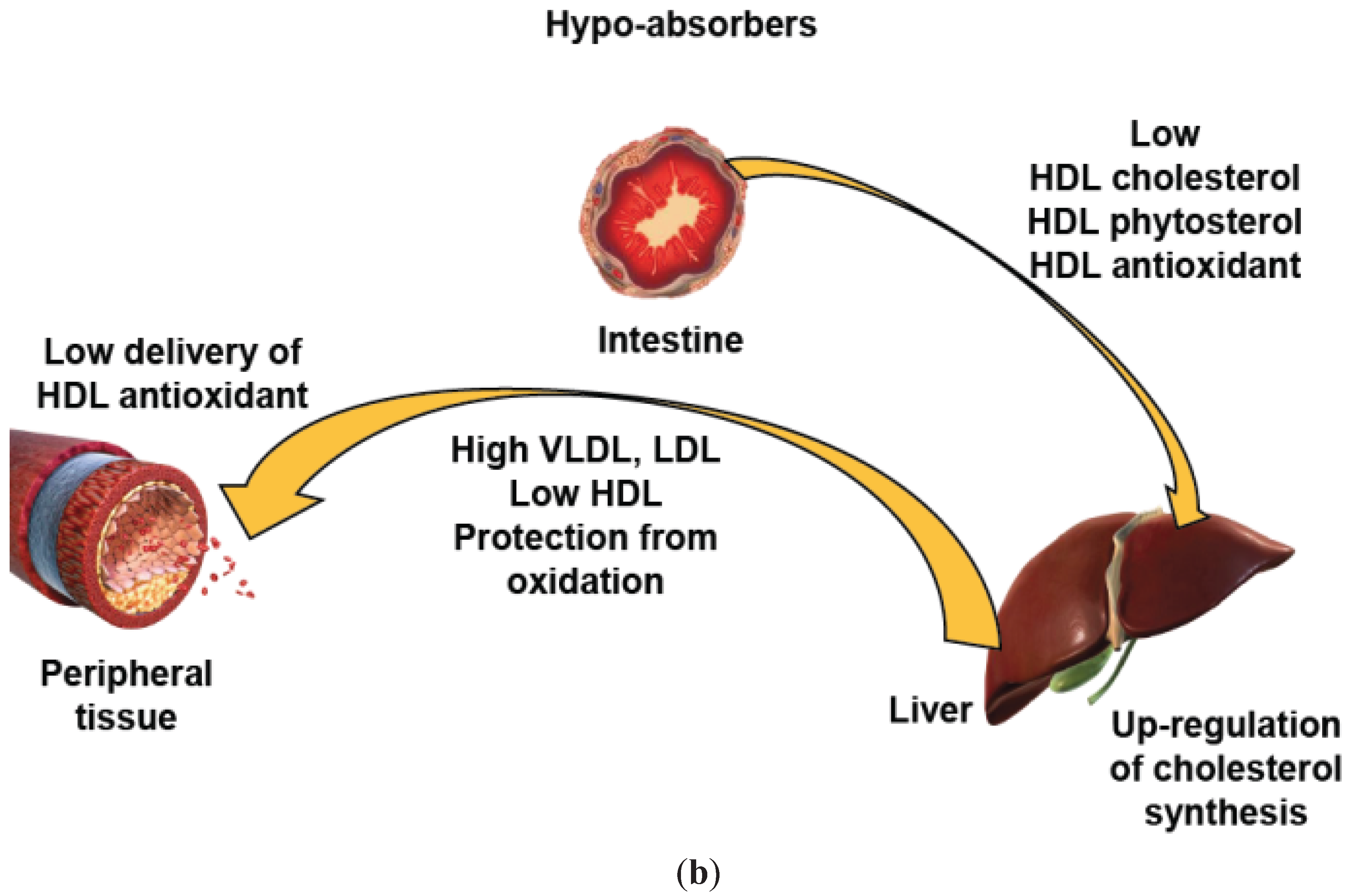
5. HDL Functionality at the Intestinal Level
6. A Key Role of HDL of Intestinal Origin in HDL Protection?


7. Summary
Acknowledgments
Conflicts of Interest
References
- Gordon, T.; Castelli, W.P.; Hjortland, M.C.; Kannel, W.B.; Dawber, T.R. High density lipoprotein as a protective factor against coronary heart disease. The Framingham Study. Am. J. Med. 1977, 62, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Alexander, E.T.; Weibel, G.L.; Billheimer, J.; Rothblat, G.H. The role of reverse cholesterol transport in animals and humans and relationship to atherosclerosis. J. Lipid Res. 2009, 50, S189–S194. [Google Scholar] [CrossRef] [PubMed]
- Linsel-Nitschke, P.; Tall, A.R. HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat. Rev. Drug Discov. 2005, 4, 193–205. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J.; Hovingh, G.K. HDL and cardiovascular disease. Lancet 2014, 384, 618–625. [Google Scholar] [CrossRef] [PubMed]
- Tuteja, S.; Rader, D.J. High-density lipoproteins in the prevention of cardiovascular disease: Changing the paradigm. Clin. Pharmacol. Ther. 2014, 96, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Barter, P.J.; Caulfield, M.; Eriksson, M.; Grundy, S.M.; Kastelein, J.J.; Komajda, M.; Lopez-Sendon, J.; Mosca, L.; Tardif, J.C.; Waters, D.D.; et al. Effects of torcetrapib in patients at high risk for coronary events. N. Engl. J. Med. 2007, 357, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef] [PubMed]
- HPS2-THRIVE Collaborative Group. HPS2-THRIVE randomized placebo-controlled trial in 25,673 high-risk patients of ER niacin/laropiprant: Trial design, pre-specified muscle and liver outcomes, and reasons for stopping study treatment. Eur. Heart J. 2013, 34, 1279–1291. [Google Scholar]
- Lincoff, A.M.; Tardif, J.C.; Schwartz, G.G.; Nicholls, S.J.; Ryden, L.; Neal, B.; Malmberg, K.; Wedel, H.; Buse, J.B.; Henry, R.R.; et al. Effect of aleglitazar on cardiovascular outcomes after acute coronary syndrome in patients with type 2 diabetes mellitus: The AleCardio randomized clinical trial. JAMA 2014, 311, 1515–1525. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Olsson, A.G.; Abt, M.; Ballantyne, C.M.; Barter, P.J.; Brumm, J.; Chaitman, B.R.; Holme, I.M.; Kallend, D.; Leiter, L.A.; et al. Effects of dalcetrapib in patients with a recent acute coronary syndrome. N. Engl. J. Med. 2012, 367, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Tardif, J.C.; Ballantyne, C.M.; Barter, P.; Dasseux, J.L.; Fayad, Z.A.; Guertin, M.C.; Kastelein, J.J.; Keyserling, C.; Klepp, H.; Koenig, W.; et al. Effects of the high-density lipoprotein mimetic agent CER-001 on coronary atherosclerosis in patients with acute coronary syndromes: A randomized trial. Eur. Heart J. 2014, 35, 3277–3286. [Google Scholar] [CrossRef] [PubMed]
- Rader, D.J. Spotlight on HDL biology: New insights in metabolism, function, and translation. Cardiovasc. Res. 2014, 103, 337–340. [Google Scholar] [CrossRef] [PubMed]
- Keene, D.; Price, C.; Shun-Shin, M.J.; Francis, D.P. Effect on cardiovascular risk of high density lipoprotein targeted drug treatments niacin, fibrates, and CETP inhibitors: Meta-analysis of randomised controlled trials including 117,411 patients. BMJ 2014, 349. [Google Scholar] [CrossRef] [PubMed]
- Millar, J.S. The Effect of CETP Inhibition with Anacetrapib on the Metabolism of PCSK9. Arterioscler. Thromb. Vasc. Biol. 2013, Abstract 118. [Google Scholar]
- Dong, B.; Singh, A.B.; Fung, C.; Kan, K.; Liu, J. CETP inhibitors downregulate hepatic LDL receptor and PCSK9 expression in vitro and in vivo through a SREBP2 dependent mechanism. Atherosclerosis 2014, 235, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Niesor, E.J.; Magg, C.; Ogawa, N.; Okamoto, H.; von der Mark, E.; Matile, H.; Schmid, G.; Clerc, R.G.; Chaput, E.; Blum-Kaelin, D.; et al. Modulating cholesteryl ester transfer protein activity maintains efficient pre-beta-HDL formation and increases reverse cholesterol transport. J. Lipid Res. 2010, 51, 3443–3454. [Google Scholar] [CrossRef] [PubMed]
- Rosenson, R.S.; Brewer, H.B., Jr.; Chapman, M.J.; Fazio, S.; Hussain, M.M.; Kontush, A.; Krauss, R.M.; Otvos, J.D.; Remaley, A.T.; Schaefer, E.J.; et al. HDL measures, particle heterogeneity, proposed nomenclature, and relation to atherosclerotic cardiovascular events. Clin. Chem. 2011, 57, 392–410. [Google Scholar] [CrossRef] [PubMed]
- Vaisar, T.; Pennathur, S.; Green, P.S.; Gharib, S.A.; Hoofnagle, A.N.; Cheung, M.C.; Byun, J.; Vuletic, S.; Kassim, S.; Singh, P.; et al. Shotgun proteomics implicates protease inhibition and complement activation in the antiinflammatory properties of HDL. J. Clin. Investig. 2007, 117, 746–756. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; Lhomme, M.; Chapman, M.J. Unraveling the complexities of the HDL lipidome. J. Lipid Res. 2013, 54, 2950–2963. [Google Scholar] [CrossRef] [PubMed]
- Stahlman, M.; Fagerberg, B.; Adiels, M.; Ekroos, K.; Chapman, J.M.; Kontush, A.; Boren, J. Dyslipidemia, but not hyperglycemia and insulin resistance, is associated with marked alterations in the HDL lipidome in type 2 diabetic subjects in the DIWA cohort: Impact on small HDL particles. Biochim. Biophys. Acta 2013, 1831, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Tunaru, S.; Kero, J.; Schaub, A.; Wufka, C.; Blaukat, A.; Pfeffer, K.; Offermanns, S. PUMA-G and HM74 are receptors for nicotinic acid and mediate its anti-lipolytic effect. Nat. Med. 2003, 9, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Kamanna, V.S.; Zhang, M.C.; Kashyap, M.L. Niacin inhibits surface expression of ATP synthase beta chain in HepG2 cells: Implications for raising HDL. J. Lipid Res. 2008, 49, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.H.; Kamanna, V.S.; Ganji, S.H.; Xiong, X.M.; Kashyap, M.L. Niacin increases HDL biogenesis by enhancing DR4-dependent transcription of ABCA1 and lipidation of apolipoprotein A-I in HepG2 cells. J. Lipid Res. 2012, 53, 941–950. [Google Scholar] [CrossRef] [PubMed]
- Staels, B.; Auwerx, J. Regulation of apo A-I gene expression by fibrates. Atherosclerosis 1998, 137, S19–S23. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, C.A.; Roy, A.; Ebert, D.L. Genomic evidence for the absence of a functional cholesteryl ester transfer protein gene in mice and rats. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 2003, 135, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Ha, Y.C.; Barter, P.J. Differences in plasma cholesteryl ester transfer activity in sixteen vertebrate species. Comp. Biochem. Physiol. B 1982, 71, 265–269. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Iwamoto, Y.; Maki, M.; Sotani, T.; Yonemori, F.; Wakitani, K. Effect of JTT-705 on cholesteryl ester transfer protein and plasma lipid levels in normolipidemic animals. Eur. J. Pharmacol. 2003, 466, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Niesor, E.J.; Chaput, E.; Staempfli, A.; Blum, D.; Derks, M.; Kallend, D. Effect of dalcetrapib, a CETP modulator, on non-cholesterol sterol markers of cholesterol homeostasis in healthy subjects. Atherosclerosis 2011, 219, 761–767. [Google Scholar] [CrossRef] [PubMed]
- Niesor, E.J.; Chaput, E.; Mary, J.L.; Staempfli, A.; Topp, A.; Stauffer, A.; Wang, H.; Durrwell, A. Effect of compounds affecting ABCA1 expression and CETP activity on the HDL pathway involved in intestinal absorption of Lutein and Zeaxanthin. Lipids 2014, 49, 1233–1243. [Google Scholar] [CrossRef] [PubMed]
- Morton, R.E.; Izem, L. Cholesteryl ester transfer proteins from different species do not have equivalent activities. J. Lipid Res. 2014, 55, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, M.; Carlson, L.A.; Miettinen, T.A.; Angelin, B. Stimulation of fecal steroid excretion after infusion of recombinant proapolipoprotein A-I. Potential reverse cholesterol transport in humans. Circulation 1999, 100, 594–598. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, G.; Sirtori, M.; Gianfranceschi, G.; Sirtori, C.R. Relation between the HDL apoproteins and AI isoproteins in subjects with the AIMilano abnormality. Metab. Clin. Exp. 1981, 30, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Favari, E.; Gomaraschi, M.; Zanotti, I.; Bernini, F.; Lee-Rueckert, M.; Kovanen, P.T.; Sirtori, C.R.; Franceschini, G.; Calabresi, L. A unique protease-sensitive high density lipoprotein particle containing the apolipoprotein A-I(Milano) dimer effectively promotes ATP-binding Cassette A1-mediated cell cholesterol efflux. J. Biol. Chem. 2007, 282, 5125–5132. [Google Scholar] [CrossRef] [PubMed]
- Franceschini, G.; Vecchio, G.; Gianfranceschi, G.; Magani, D.; Sirtori, C.R. Apolipoprotein AIMilano. Accelerated binding and dissociation from lipids of a human apolipoprotein variant. J. Biol. Chem. 1985, 260, 16321–16325. [Google Scholar] [PubMed]
- Perez-Mendez, O.; Bruckert, E.; Franceschini, G.; Duhal, N.; Lacroix, B.; Bonte, J.P.; Sirtori, C.; Fruchart, J.C.; Turpin, G.; Luc, G.; et al. Metabolism of apolipoproteins AI and AII in subjects carrying similar apoAI mutations, apoAI Milano and apoAI Paris. Atherosclerosis 2000, 148, 317–325. [Google Scholar] [CrossRef] [PubMed]
- Gursky, O.; Jones, M.K.; Mei, X.; Segrest, J.P.; Atkinson, D. Structural basis for distinct functions of the naturally occurring Cys mutants of human apolipoprotein A-I. J. Lipid Res. 2013, 54, 3244–3257. [Google Scholar] [CrossRef] [PubMed]
- Acton, S.; Rigotti, A.; Landschulz, K.T.; Xu, S.; Hobbs, H.H.; Krieger, M. Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 1996, 271, 518–520. [Google Scholar] [CrossRef] [PubMed]
- El Bouhassani, M.; Gilibert, S.; Moreau, M.; Saint-Charles, F.; Treguier, M.; Poti, F.; Chapman, M.J.; Le, G.W.; Lesnik, P.; Huby, T.; et al. Cholesteryl ester transfer protein expression partially attenuates the adverse effects of SR-BI receptor deficiency on cholesterol metabolism and atherosclerosis. J. Biol. Chem. 2011, 286, 17227–17238. [Google Scholar] [CrossRef] [PubMed]
- Niesor, E.J.; von der Mark, E.; Calabresi, L.; Averna, M.; Cefalu, A.B.; Tarugi, P.; Nilsson, P.; Dernick, G. Lipid and apoprotein composition of HDL in partial or complete CETP deficiency. Curr. Vasc. Pharmacol. 2012, 10, 422–431. [Google Scholar] [CrossRef] [PubMed]
- Rohatgi, A.; Khera, A.; Berry, J.D.; Givens, E.G.; Ayers, C.R.; Wedin, K.E.; Neeland, I.J.; Yuhanna, I.S.; Rader, D.R.; de Lemos, J.A.; et al. HDL cholesterol efflux capacity and incident cardiovascular events. N. Engl. J. Med. 2014, 371, 2383–2393. [Google Scholar] [CrossRef] [PubMed]
- Khera, A.V.; Cuchel, M.; de la Llera-Moya, M.; Rodrigues, A.; Burke, M.F.; Jafri, K.; French, B.C.; Phillips, J.A.; Mucksavage, M.L.; Wilensky, R.L.; et al. Cholesterol efflux capacity, high-density lipoprotein function, and atherosclerosis. N. Engl. J. Med. 2011, 364, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Kunitake, S.T.; Mendel, C.M.; Hennessy, L.K. Interconversion between apolipoprotein A-I-containing lipoproteins of pre-beta and alpha electrophoretic mobilities. J. Lipid Res. 1992, 33, 1807–1816. [Google Scholar] [PubMed]
- Lagrost, L.; Gambert, P.; Dangremont, V.; Athias, A.; Lallemant, C. Role of cholesteryl ester transfer protein (CETP) in the HDL conversion process as evidenced by using anti-CETP monoclonal antibodies. J. Lipid Res. 1990, 31, 1569–1575. [Google Scholar] [PubMed]
- Rye, K.A.; Barter, P.J. Formation and metabolism of prebeta-migrating, lipid-poor apolipoprotein A-I. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Maugeais, C.; Perez, A.; von der Mark, E.; Magg, C.; Pflieger, P.; Niesor, E.J. Evidence for a role of CETP in HDL remodeling and cholesterol efflux: Role of cysteine 13 of CETP. Biochim. Biophys. Acta 2013, 1831, 1644–1650. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.; Sniderman, A.; Melone, M.; Brown, P.E.; Otvos, J.D.; Mente, A.; Schulze, K.; McQueen, M.J.; Anand, S.S.; Yusuf, S.; et al. Elevated cholesteryl ester transfer protein (CETP) activity, a major determinant of the atherogenic dyslipidemia, and atherosclerotic cardiovascular disease in South Asians. Eur. J. Prev. Cardiol. 2014. [Google Scholar] [PubMed]
- Chapman, M.J. Therapeutic elevation of HDL-cholesterol to prevent atherosclerosis and coronary heart disease. Pharmacol. Ther. 2006, 111, 893–908. [Google Scholar] [CrossRef] [PubMed]
- Adorni, M.P.; Zimetti, F.; Billheimer, J.T.; Wang, N.; Rader, D.J.; Phillips, M.C.; Rothblat, G.H. The roles of different pathways in the release of cholesterol from macrophages. J. Lipid Res. 2007, 48, 2453–2462. [Google Scholar] [CrossRef] [PubMed]
- Knight, B.L. ATP-binding cassette transporter A1: Regulation of cholesterol efflux. Biochem. Soc. Trans. 2004, 32, 124–127. [Google Scholar] [CrossRef] [PubMed]
- Singaraja, R.R.; Visscher, H.; James, E.R.; Chroni, A.; Coutinho, J.M.; Brunham, L.R.; Kang, M.H.; Zannis, V.I.; Chimini, G.; Hayden, M.R.; et al. Specific mutations in ABCA1 have discrete effects on ABCA1 function and lipid phenotypes both in vivo and in vitro. Circ. Res. 2006, 99, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Brunham, L.R.; Singaraja, R.R.; Hayden, M.R. Variations on a gene: Rare and common variants in ABCA1 and their impact on HDL cholesterol levels and atherosclerosis. Annu. Rev. Nutr. 2006, 26, 105–129. [Google Scholar] [CrossRef] [PubMed]
- Zwarts, K.Y.; Clee, S.M.; Swinderman, A.H.; Engert, J.C.; Singaraja, R.; Loubser, O.; James, E.; Roomp, K.; Hudson, T.J.; Jukema, J.W.; et al. ABCA1 regulatory variants influence coronary artery disease independent of effects on plasma lipid levels. Clin. Genet. 2002, 61, 115–125. [Google Scholar] [CrossRef] [PubMed]
- Le Goff, W.; Dallinga-Thie, G.M. ABCG1: Not as good as expected? Atherosclerosis 2011, 219, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Lan, D.; Chen, W.; Matsuura, F.; Tall, A.R. ATP-binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high-density lipoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 9774–9779. [Google Scholar] [CrossRef] [PubMed]
- Tarling, E.J.; Edwards, P.A. ATP binding cassette transporter G1 (ABCG1) is an intracellular sterol transporter. Proc. Natl. Acad. Sci. USA 2011, 108, 19719–19724. [Google Scholar] [CrossRef] [PubMed]
- Favari, E.; Calabresi, L.; Adorni, M.P.; Jessup, W.; Simonelli, S.; Franceschini, G.; Bernini, F. Small discoidal pre-beta1 HDL particles are efficient acceptors of cell cholesterol via ABCA1 and ABCG1. Biochemistry 2009, 48, 11067–11074. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Matsuura, F.; Wang, N.; Bamberger, M.J.; Nguyen, T.; Rinninger, F.; Jiang, X.C.; Shear, C.L.; Tall, A.R. Inhibition of cholesteryl ester transfer protein by torcetrapib modestly increases macrophage cholesterol efflux to HDL. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Yvan-Charvet, L.; Kling, J.; Pagler, T.; Li, H.; Hubbard, B.; Fisher, T.; Sparrow, C.P.; Taggart, A.K.; Tall, A.R. Cholesterol efflux potential and antiinflammatory properties of high-density lipoprotein after treatment with niacin or anacetrapib. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 1430–1438. [Google Scholar] [CrossRef] [PubMed]
- Tall, A.R.; Yvan-Charvet, L.; Terasaka, N.; Pagler, T.; Wang, N. HDL, ABC transporters, and cholesterol efflux: Implications for the treatment of atherosclerosis. Cell Metab. 2008, 7, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Vergeer, M.; Korporaal, S.J.; Franssen, R.; Meurs, I.; Out, R.; Hovingh, G.K.; Hoekstra, M.; Sierts, J.A.; Dallinga-Thie, G.M.; Motazacker, M.M.; et al. Genetic variant of the scavenger receptor BI in humans. N. Engl. J. Med. 2011, 364, 136–145. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.; Pilon, A.; Albert, C.; Valle, M.; Hum, D.W.; Fruchart, J.C.; Najib, J.; Clavey, V.; Staels, B. Comparison of expression and regulation of the high-density lipoprotein receptor SR-BI and the low-density lipoprotein receptor in human adrenocortical carcinoma NCI-H295 cells. Eur. J. Biochem. 1999, 261, 481–491. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, N.; Julia, Z.; Villard, E.F.; El, K.P.; Duchene, E.; Chapman, M.J.; Fournier, N.; Le, G.W.; Guerin, M. Functionality of postprandial larger HDL2 particles is enhanced following CETP inhibition therapy. Atherosclerosis 2012, 221, 160–168. [Google Scholar] [CrossRef] [PubMed]
- Tardy, C.; Goffinet, M.; Boubekeur, N.; Ackermann, R.; Sy, G.; Bluteau, A.; Cholez, G.; Keyserling, C.; Lalwani, N.; Paolini, J.F.; et al. CER-001, a HDL-mimetic, stimulates the reverse lipid transport and atherosclerosis regression in high cholesterol diet-fed LDL-receptor deficient mice. Atherosclerosis 2014, 232, 110–118. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.K.; Ditmarsch, M.; Kallend, D.; Niesor, E.J.; Suchankova, G.; Upmanyu, R.; Nzures-Cabrera, J.; Lehnert, V.; Pauly-Evers, M.; Holme, I.; et al. The effect of cholesteryl ester transfer protein inhibition on lipids, lipoproteins, and markers of HDL function after an acute coronary syndrome: The dal-ACUTE randomized trial. Eur. Heart J. 2014, 35, 1792–1800. [Google Scholar] [CrossRef] [PubMed]
- Li, X.M.; Tang, W.H.; Mosior, M.K.; Huang, Y.; Wu, Y.; Matter, W.; Gao, V.; Schmitt, D.; Didonato, J.A.; Fisher, E.A.; et al. Paradoxical association of enhanced cholesterol efflux with increased incident cardiovascular risks. Arterioscler. Thromb. Vasc. Biol. 2013, 33, 1696–1705. [Google Scholar] [CrossRef] [PubMed]
- Attia, N.; Ramaharo, A.; Paul, J.L.; Cambillau, M.; Beaune, P.; Grynberg, A.; Simon, A.; Fournier, N. Enhanced removal of cholesterol from macrophage foam cells to serum from type IV hypertriglyceridemic subjects. Atherosclerosis 2008, 198, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Yassine, H.N.; Belopolskaya, A.; Schall, C.; Stump, C.S.; Lau, S.S.; Reaven, P.D. Enhanced cholesterol efflux to HDL through the ABCA1 transporter in hypertriglyceridemia of type 2 diabetes. Metab. Clin. Exp. 2014, 63, 727–734. [Google Scholar] [CrossRef] [PubMed]
- Assmann, G.; Herbert, P.N.; Fredrickson, D.S.; Forte, T. Isolation and characterization of an abnormal high density lipoprotein in Tangier Diesase. J. Clin. Investig. 1977, 60, 242–252. [Google Scholar] [CrossRef] [PubMed]
- Attie, A.D.; Hamon, Y.; Brooks-Wilson, A.R.; Gray-Keller, M.P.; MacDonald, M.L.; Rigot, V.; Tebon, A.; Zhang, L.H.; Mulligan, J.D.; Singaraja, R.R.; et al. Identification and functional analysis of a naturally occurring E89K mutation in the ABCA1 gene of the WHAM chicken. J. Lipid Res. 2002, 43, 1610–1617. [Google Scholar] [CrossRef] [PubMed]
- Brunham, L.R.; Kruit, J.K.; Pape, T.D.; Parks, J.S.; Kuipers, F.; Hayden, M.R. Tissue-specific induction of intestinal ABCA1 expression with a liver X receptor agonist raises plasma HDL cholesterol levels. Circ. Res. 2006, 99, 672–674. [Google Scholar] [CrossRef] [PubMed]
- De Haan, W.; Bhattacharjee, A.; Ruddle, P.; Kang, M.H.; Hayden, M.R. ABCA1 in adipocytes regulates adipose tissue lipid content, glucose tolerance, and insulin sensitivity. J. Lipid Res. 2014, 55, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Van Eck, M.; Bos, I.S.; Kaminski, W.E.; Orso, E.; Rothe, G.; Twisk, J.; Bottcher, A.; van Amersfoort, E.S.; Christiansen-Weber, T.A.; Fung-Leung, W.P.; et al. Leukocyte ABCA1 controls susceptibility to atherosclerosis and macrophage recruitment into tissues. Proc. Natl. Acad. Sci. USA 2002, 99, 6298–6303. [Google Scholar] [CrossRef] [PubMed]
- Iqbal, J.; Boutjdir, M.; Rudel, L.L.; Hussain, M.M. Intestine specific MTP deficiency with global ACAT2 gene ablation lowers acute cholesterol absorption with chylomicrons and high density lipoproteins. J. Lipid Res. 2014. [Google Scholar] [CrossRef]
- Hussain, M.M. Intestinal lipid absorption and lipoprotein formation. Curr. Opin. Lipidol. 2014, 25, 200–206. [Google Scholar] [CrossRef] [PubMed]
- Niesor, E.J.; Gauthamadasa, K.; Silva, R.A.; Suchankova, G.; Kallend, D.; Gylling, H.; Asztalos, B.; Damonte, E.; Rossomanno, S.; Abt, M.; et al. Xanthophylls, phytosterols and pre-beta1-HDL are differentially affected by Fenofibrate and Niacin HDL-raising in a cross-over study. Lipids 2013, 48, 1185–1196. [Google Scholar] [CrossRef] [PubMed]
- Nunes, V.S.; Leanca, C.C.; Panzoldo, N.B.; Parra, E.; Cazita, P.M.; Nakandakare, E.R.; de Faria, E.C.; Quintao, E.C. HDL-C concentration is related to markers of absorption and of cholesterol synthesis: Study in subjects with low vs. high HDL-C. Clin. Chim. Acta 2011, 412, 176–180. [Google Scholar] [CrossRef] [PubMed]
- Wilund, K.R.; Feeney, L.A.; Tomayko, E.J.; Weiss, E.P.; Hagberg, J.M. Effects of endurance exercise training on markers of cholesterol absorption and synthesis. Physiol. Res. 2009, 58, 545–552. [Google Scholar] [PubMed]
- Assmann, G.; Cullen, P.; Kannenberg, F.; Schulte, H. Relationship between phytosterol levels and components of the metabolic syndrome in the PROCAM study. Eur. J. Cardiovasc. Prev. Rehabil. 2007, 14, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Niesor, E.J.; Kallend, D.; Bentley, D.; Kastelein, J.J.; Kees, H.G.; Stroes, E.S. Treatment of low HDL-C subjects with the CETP modulator dalcetrapib increases plasma campesterol only in those without ABCA1 and/or ApoA1 mutations. Lipids 2014, 49, 1245–1249. [Google Scholar] [CrossRef] [PubMed]
- Matthan, N.R.; Giovanni, A.; Schaefer, E.J.; Brown, B.G.; Lichtenstein, A.H. Impact of simvastatin, niacin, and/or antioxidants on cholesterol metabolism in CAD patients with low HDL. J. Lipid Res. 2003, 44, 800–806. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.B. Plant sterols and stanols: Their role in health and disease. J. Clin. Lipidol. 2008, 2, S11–S19. [Google Scholar] [CrossRef] [PubMed]
- Robins, S.J.; Fasulo, J.M. High density lipoproteins, but not other lipoproteins, provide a vehicle for sterol transport to bile. J. Clin. Investig. 1997, 99, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Klimov, A.N.; Gurevich, V.S.; Nikiforova, A.A.; Shatilina, L.V.; Kuzmin, A.A.; Plavinsky, S.L.; Teryukova, N.P. Antioxidative activity of high density lipoproteins in vivo. Atherosclerosis 1993, 100, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Hama, S.Y.; Hough, G.P.; Subbanagounder, G.; Reddy, S.T.; Fogelman, A.M. A cell-free assay for detecting HDL that is dysfunctional in preventing the formation of or inactivating oxidized phospholipids. J. Lipid Res. 2001, 42, 1308–1317. [Google Scholar] [PubMed]
- Kontush, A.; Chapman, M.J. Antiatherogenic function of HDL particle subpopulations: Focus on antioxidative activities. Curr. Opin. Lipidol. 2010, 21, 312–318. [Google Scholar] [CrossRef] [PubMed]
- Kontush, A.; de Faria, E.C.; Chantepie, S.; Chapman, M.J. Antioxidative activity of HDL particle subspecies is impaired in hyperalphalipoproteinemia: Relevance of enzymatic and physicochemical properties. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Phuntuwate, W.; Suthisisang, C.; Koanantakul, B.; Chaloeiphap, P.; Mackness, B.; Mackness, M. Effect of fenofibrate therapy on paraoxonase1 status in patients with low HDL-C levels. Atherosclerosis 2008, 196, 122–128. [Google Scholar] [CrossRef] [PubMed]
- Tsimihodimos, V.; Kakafika, A.; Tambaki, A.P.; Bairaktari, E.; Chapman, M.J.; Elisaf, M.; Tselepis, A.D. Fenofibrate induces HDL-associated PAF-AH but attenuates enzyme activity associated with apoB-containing lipoproteins. J. Lipid Res. 2003, 44, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Nukuna, B.; Brennan, M.L.; Sun, M.; Goormastic, M.; Settle, M.; Schmitt, D.; Fu, X.; Thomson, L.; Fox, P.L.; et al. Apolipoprotein A-I is a selective target for myeloperoxidase-catalyzed oxidation and functional impairment in subjects with cardiovascular disease. J. Clin. Investig. 2004, 114, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Didonato, J.A.; Huang, Y.; Aulak, K.S.; Even-Or, O.; Gerstenecker, G.; Gogonea, V.; Wu, Y.; Fox, P.L.; Tang, W.H.; Plow, E.F.; et al. Function and distribution of apolipoprotein A1 in the artery wall are markedly distinct from those in plasma. Circulation 2013, 128, 1644–1655. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Didonato, J.A.; Levison, B.S.; Schmitt, D.; Li, L.; Wu, Y.; Buffa, J.; Kim, T.; Gerstenecker, G.S.; Gu, X.; et al. An abundant dysfunctional apolipoprotein A1 in human atheroma. Nat. Med. 2014, 20, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Schaefer, E.J.; Anthanont, P.; Asztalos, B.F. High-density lipoprotein metabolism, composition, function, and deficiency. Curr. Opin. Lipidol. 2014, 25, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Hirowatari, Y.; Yoshida, H.; Kurosawa, H.; Manita, D.; Tada, N. Automated measurement method for the determination of vitamin E in plasma lipoprotein classes. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Connor, S.L.; Johnson, E.J.; Klein, M.L.; Hughes, S.; Connor, W.E. Effect of dietary lutein and zeaxanthin on plasma carotenoids and their transport in lipoproteins in age-related macular degeneration. Am. J. Clin. Nutr. 2007, 85, 762–769. [Google Scholar] [PubMed]
- Goulinet, S.; Chapman, M.J. Plasma LDL and HDL subspecies are heterogenous in particle content of tocopherols and oxygenated and hydrocarbon carotenoids. Relevance to oxidative resistance and atherogenesis. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 786–796. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, J.H.; Navab, M.; Dwyer, K.M.; Hassan, K.; Sun, P.; Shircore, A.; Hama-Levy, S.; Hough, G.; Wang, X.; Drake, T.; et al. Oxygenated carotenoid lutein and progression of early atherosclerosis: The Los Angeles atherosclerosis study. Circulation 2001, 103, 2922–2927. [Google Scholar] [CrossRef] [PubMed]
- Reboul, E.; Trompier, D.; Moussa, M.; Klein, A.; Landrier, J.F.; Chimini, G.; Borel, P. ATP-binding cassette transporter A1 is significantly involved in the intestinal absorption of alpha- and gamma-tocopherol but not in that of retinyl palmitate in mice. Am. J. Clin. Nutr. 2009, 89, 177–184. [Google Scholar] [CrossRef] [PubMed]
- Hacquebard, M.; Vandenbranden, M.; Malaisse, W.J.; Ruysschaert, J.M.; Deckelbaum, R.J.; Carpentier, Y.A. Vitamin E transfer from lipid emulsions to plasma lipoproteins: Mediation by multiple mechanisms. Lipids 2008, 43, 663–671. [Google Scholar] [CrossRef] [PubMed]
- Kostner, G.M.; Oettl, K.; Jauhiainen, M.; Ehnholm, C.; Esterbauer, H.; Dieplinger, H. Human plasma phospholipid transfer protein accelerates exchange/transfer of alpha-tocopherol between lipoproteins and cells. Biochem. J. 1995, 305, 659–667. [Google Scholar] [PubMed]
- Tyssandier, V.; Choubert, G.; Grolier, P.; Borel, P. Carotenoids, mostly the xanthophylls, exchange between plasma lipoproteins. Int. J. Vitam. Nut. Res. 2002, 72, 300–308. [Google Scholar] [CrossRef]
- Nicod, N.; Parker, R.S. Vitamin E secretion by Caco-2 monolayers to APOA1, but not to HDL, is vitamer selective. J. Nutr. 2013, 143, 1565–1572. [Google Scholar] [CrossRef] [PubMed]
- Connor, W.E.; Duell, P.B.; Kean, R.; Wang, Y. The prime role of HDL to transport lutein into the retina: Evidence from HDL-deficient WHAM chicks having a mutant ABCA1 transporter. Investig. Ophthalmol. Vis. Sci. 2007, 48, 4226–4231. [Google Scholar] [CrossRef]
- Guyard-Dangremont, V.; Desrumaux, C.; Gambert, P.; Lallemant, C.; Lagrost, L. Phospholipid and cholesteryl ester transfer activities in plasma from 14 vertebrate species. Relation to atherogenesis susceptibility. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1998, 120, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Shichiri, M.; Takanezawa, Y.; Rotzoll, D.E.; Yoshida, Y.; Kokubu, T.; Ueda, K.; Tamai, H.; Arai, H. ATP-binding cassette transporter A1 is involved in hepatic alpha-tocopherol secretion. J. Nutr. Biochem. 2010, 21, 451–456. [Google Scholar] [CrossRef] [PubMed]
- Brunham, L.R.; Kruit, J.K.; Iqbal, J.; Fievet, C.; Timmins, J.M.; Pape, T.D.; Coburn, B.A.; Bissada, N.; Staels, B.; Groen, A.K.; et al. Intestinal ABCA1 directly contributes to HDL biogenesis in vivo. J. Clin. Investig. 2006, 116, 1052–1062. [Google Scholar] [CrossRef] [PubMed]
- Riwanto, M.; Landmesser, U. High density lipoproteins and endothelial functions: Mechanistic insights and alterations in cardiovascular disease. J. Lipid Res. 2013, 54, 3227–3243. [Google Scholar] [CrossRef] [PubMed]
- Navab, M.; Ananthramaiah, G.M.; Reddy, S.T.; van Lenten, B.J.; Ansell, B.J.; Fonarow, G.C.; Vahabzadeh, K.; Hama, S.; Hough, G.; Kamranpour, N.; et al. The oxidation hypothesis of atherogenesis: The role of oxidized phospholipids and HDL. J. Lipid Res. 2004, 45, 993–1007. [Google Scholar] [CrossRef] [PubMed]
- Nadeem, N.; Woodside, J.V.; Kelly, S.; Allister, R.; Young, I.S.; McEneny, J. The two faces of alpha- and gamma-tocopherols: An in vitro and ex vivo investigation into VLDL, LDL and HDL oxidation. J. Nutr. Biochem. 2012, 23, 845–851. [Google Scholar] [CrossRef]
- Attie, A.D. ABCA1: At the nexus of cholesterol, HDL and atherosclerosis. Trends Biochem. Sci. 2007, 32, 172–179. [Google Scholar] [CrossRef] [PubMed]
- Mardones, P.; Strobel, P.; Miranda, S.; Leighton, F.; Quinones, V.; Amigo, L.; Rozowski, J.; Krieger, M.; Rigotti, A. Alpha-tocopherol metabolism is abnormal in scavenger receptor class B type I (SR-BI)-deficient mice. J. Nutr. 2002, 132, 443–449. [Google Scholar] [PubMed]
- Chew, E.Y.; Clemons, T.E.; Sangiovanni, J.P.; Danis, R.P.; Ferris, F.L., III; Elman, M.J.; Antoszyk, A.N.; Ruby, A.J.; Orth, D.; Bressler, S.B.; et al. Secondary analyses of the effects of Lutein/Zeaxanthin on age-related macular degeneration progression: AREDS2 Report No. 3. JAMA Ophthalmol. 2014, 132, 142–149. [Google Scholar] [CrossRef] [PubMed]
- Dias, I.H.; Polidori, M.C.; Li, L.; Weber, D.; Stahl, W.; Nelles, G.; Grune, T.; Griffiths, H.R. Plasma levels of HDL and carotenoids are lower in Dementia patients with vascular comorbidities. J. Alzheimers Dis. 2014, 40, 399–408. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niesor, E.J. Will Lipidation of ApoA1 through Interaction with ABCA1 at the Intestinal Level Affect the Protective Functions of HDL? Biology 2015, 4, 17-38. https://doi.org/10.3390/biology4010017
Niesor EJ. Will Lipidation of ApoA1 through Interaction with ABCA1 at the Intestinal Level Affect the Protective Functions of HDL? Biology. 2015; 4(1):17-38. https://doi.org/10.3390/biology4010017
Chicago/Turabian StyleNiesor, Eric J. 2015. "Will Lipidation of ApoA1 through Interaction with ABCA1 at the Intestinal Level Affect the Protective Functions of HDL?" Biology 4, no. 1: 17-38. https://doi.org/10.3390/biology4010017
APA StyleNiesor, E. J. (2015). Will Lipidation of ApoA1 through Interaction with ABCA1 at the Intestinal Level Affect the Protective Functions of HDL? Biology, 4(1), 17-38. https://doi.org/10.3390/biology4010017