
DNA Methylation Analysis: Choosing the Right Method


Abstract
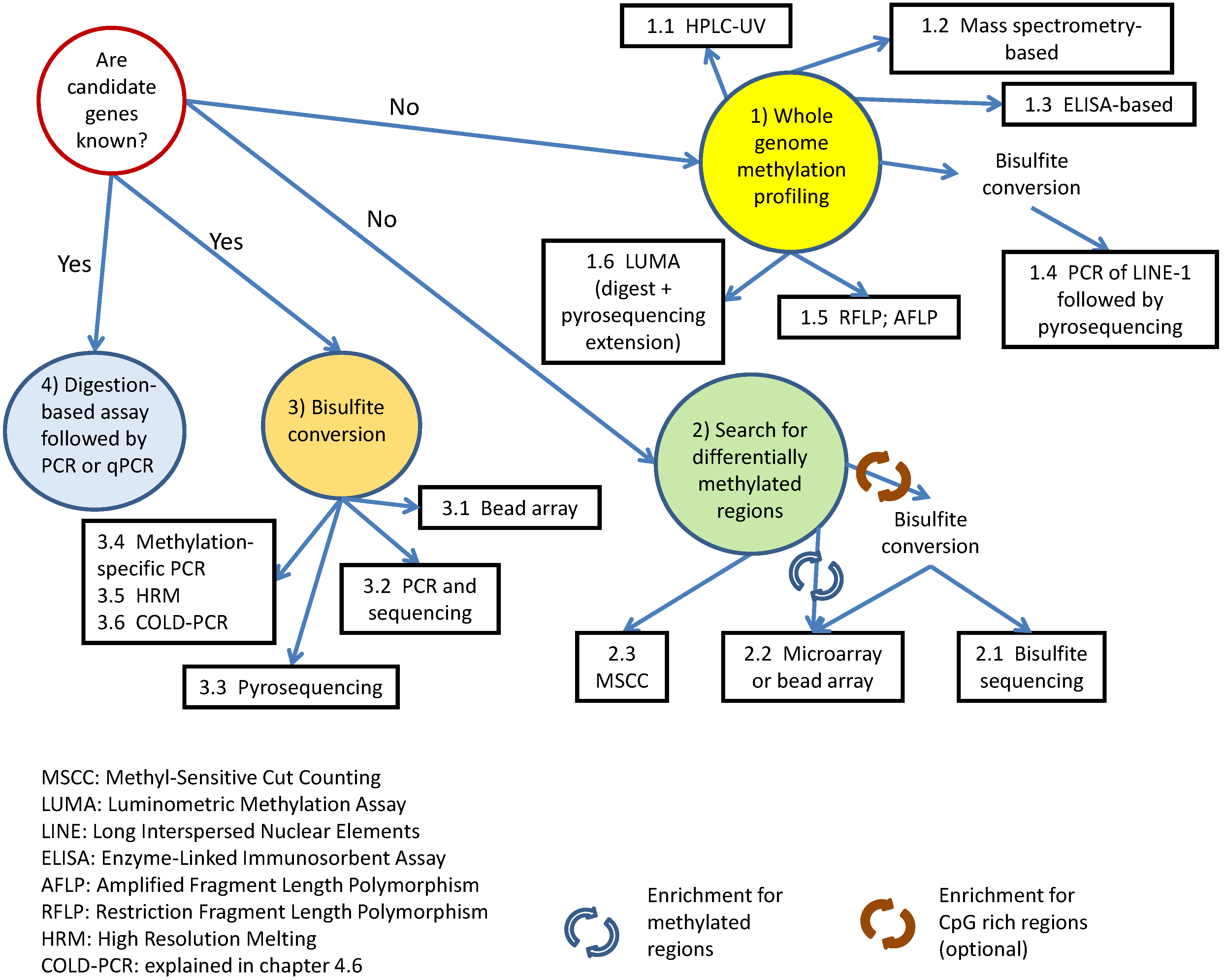
:1. Introduction
- The aims of the study (e.g., the discovery of de novo epigenetic changes or the investigation of known methylation sites of specific genes of interest);
- The amount and quality of the DNA sample(s) (e.g., Formalin-Fixed Paraffin-Embedded (FFPE) versus frozen tissue-derived DNA);
- The sensitivity and specificity requirements of the study;
- The robustness and simplicity of the method;
- The availability of bioinformatics software for analysis and interpretation of the data;
- The availability of specialized equipment and reagents;
- Cost.


2. Profiling Whole Genome Methylation
2.1. HPLC-UV
2.2. LC-MS/MS
2.3. ELISA-Based Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Company | Name of the Kit | Amount of DNA Required | Number of Citations |
|---|---|---|---|
| Cell Biolabs | Global DNA Methylation ELISA | 2 μg | 1 citation |
| Sigma-Aldrich | Imprint Methylated DNA Quantification kit (sandwich ELISA) | 100 ng | 14 citations |
| abcam | EpiSeeker methylated DNA Quantification Kit | 100 ng | None |
| Active Motif | Global DNA Methylation Assay — LINE-1 | 1 μg | New method |
| Zymo Research | 5-mC DNA ELISA Kit | 100 ng | 8 citations |
| Epigentek | MethylFlash Methylated DNA5-mC Quantification Kit (Colorimetric) | 100 ng | 88 citations |
| MethylFlash Methylated DNA5-mC Quantification Kit (Fluorometric) | 100 ng | 9 citations |
2.4. LINE-1 + Pyrosequencing
2.5. AFLP and RFLP
2.6. LUMA
2.7. Which Method to Use for Whole Genome Methylation Profiling?
3. Identification of Differentially-Methylated Regions
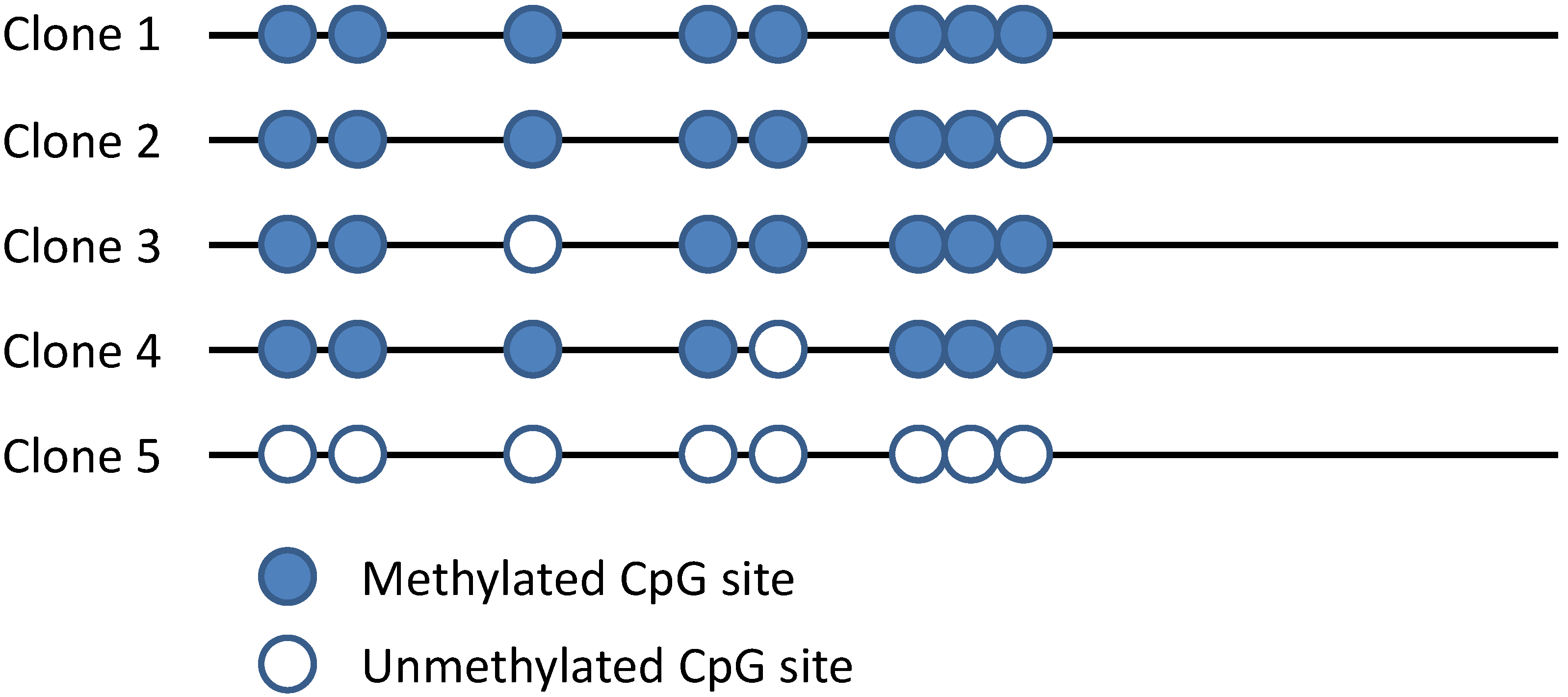
3.1. Bisulfite Sequencing
| Method | Availability as a Kit | Coverage | Sensitivity | Specificity | Amount of Starting Material | § Price | References |
|---|---|---|---|---|---|---|---|
| Methods for Whole Genome Profiling | Whole Genome Assessment without Identification of Differentially-Methylated Regions | ||||||
| Mass spectrometry (LC-MS/MS) | No | Whole genome assessment | *** detect 5% difference in methylation between samples | *** | 100 ng–1 μg | $80/sample (Millis Scientific) | [18,19,20,21] |
| LUMA (Luminometric-Based Assay) | No | Whole genome assessment | ** | ** | 250–500 ng | [34,38] | |
| LINE-1 + pyrosequencing | No | 17% of the whole genome | *** | ** | 50 ng | [39] | |
| HPLC-UV | No | Whole genome assessment | * | ** | 3–10 μg | [17] | |
| Methods for Identification of Differentially-Methylated Regions | |||||||
| # Enrichment of 5metC regions by pulldown with MBD protein (needs to be followed by NGS or microarray) | MethylCap kit (Diagenode) | ~80% of all 5–5 mCpG | *** | *** | 200 ng–1.2 μg | $550/48 rxns | [3,40] |
| MethylMiner Methylated DNA Enrichment Kit (Thermo Fisher Scientific) | * | * | 5 ng–1 μg | $485/25 rxns | |||
| # Enrichment for 5 mC regions by MBD2b/MBD3L1 proteins pulldown (MIRA-based assay) (needs to be followed by NGS or microarray) | MethylCollector Ultra (Active Motif) | No info | ** | ** | 1 ng–1 μg | $495/10 rxns | [40,41] |
| # Enrichment for 5 mC regions with antibodies (MeDIP) (needs to be followed by NGS) | MeDIP (Active Motif) | No info | No info | No info | 100 ng–1 μg | $395/10 rxns | [42,43] |
| MagMeDIP (Diagenode) | No info | No info | No info | 1.2 μg | $595/48 rxns; $325/10 rxns | [44] | |
| # Enrichment for CpG rich regions with RNA baits (needs to be followed by NGS) | SureSelect Human Methyl-Seq (Agilent) | 3.7 × 106 CpGs | *** | *** | 3 μg | ~$320 (baits); +$70 (library prep) | [5] |
| # Enrichment for CpG rich regions by hybridisation with bait oligonucleotides (needs to be followed by NGS) | SeqCap Epi CpGiant Enrichment Kit (Roche) | 5.5 × 106 CpGs | *** | *** | 1 μg | ~$450; + bisulfite conversion kit | [45,46] |
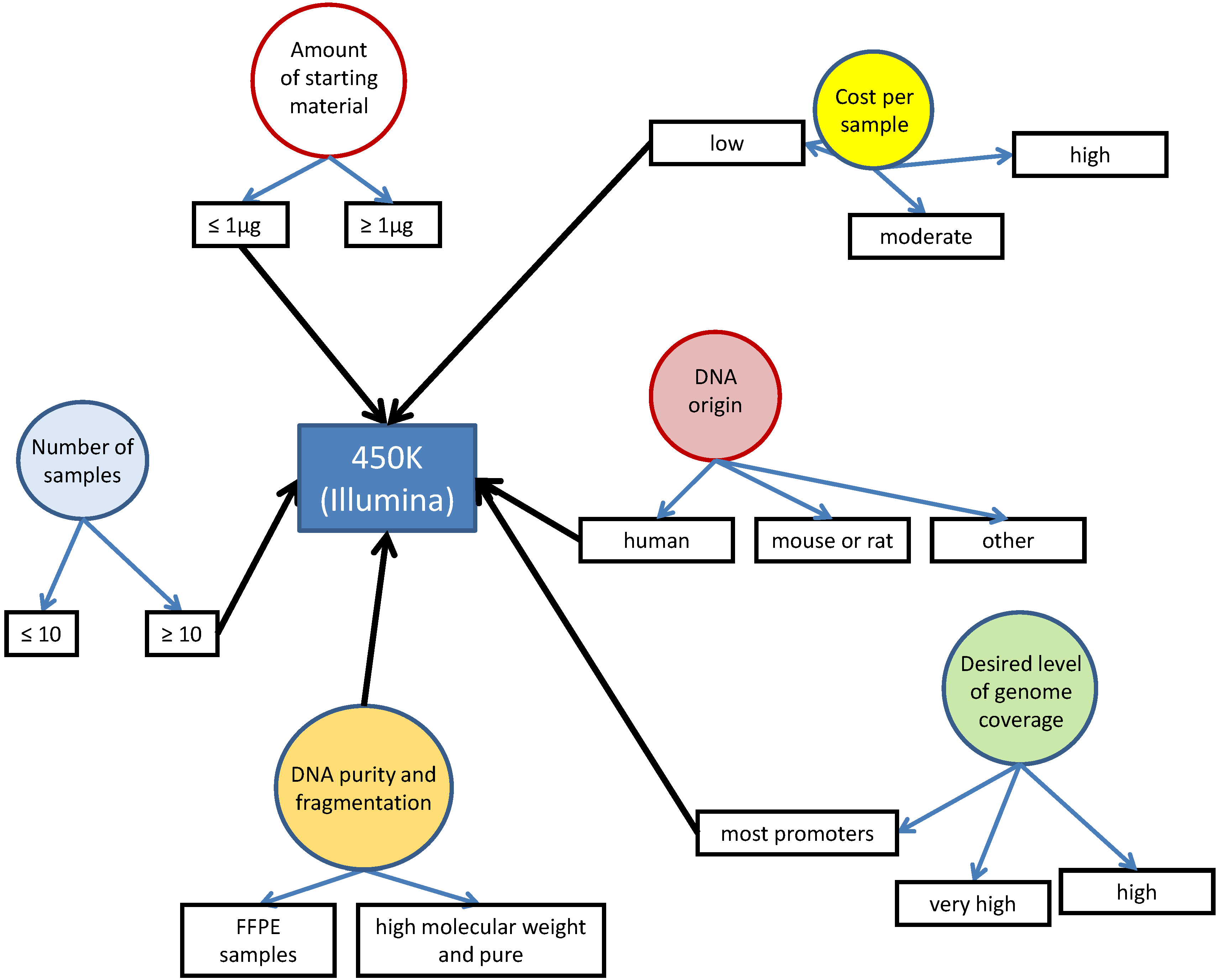
| HumanMethylation450 BeadChip array | Illumina | 482.000 CpG sites (99% of known genes) | *** | *** | 0.5–1 μg | ~$9000/24 samples (2 chips) | [47] |
| Sequencing of methylation-enriched fraction of the genome (after MeDIP or MIRA) | Illumina | varies, depending on the sample | varies, as number of reads correlates with the amount of DNA | Depends on the enrichment method | > 50 ng | $360/sample | |
| Whole genome bisulfite sequencing (WGBS) | Illumina | 100% | *** | 50–100 ng | $6000/sample | [48,49] | |
| Reduced representation bisulfite sequencing (RRBS) | Illumina | ~60% of promoters, 85% of CGI (~1.5 × 106 CpGs) | varies, as sequencing depth correlates with the amount of DNA (20X coverage is recommended) | *** | 1 μg | $2700–5000/sample (sequencing and library prep) | [50,51] |
| Methyl-MiniSeq (improved RRBS) | Zymoresearch | >85% coverage of all CpG islands and >80% of all gene promoters (~3 × 106 CpGs) | varies, as sequencing depth correlates with the amount of DNA (20X coverage is recommended) | 100 ng–5 μg | $2800 (all steps and bioinformatics included) | [44,52] | |
| Methyl-sensitive cut counting (MSCC) (needs to be followed by NGS) | No | 1% of all CpG | depends on the sequencing coverage | *** | 1–5 μg | [53] | |
| Improved MSCC (needs to be followed by NGS) | No | 30% of CpGs (~58% of CpG-rich regions) | *** | 1–5 μg | [54,55] | ||
| Low Throughput Methods | |||||||
| PCR-based (digestion followed by PCR) | OneStep qMethyl Kit (Zymoresearch) | Gene-specific 22 samples/ 96-well plate | *** | *** | ≥20 ng | [56] | |
| EpiTect II DNA methylation enzyme Kit (Qiagen) | 1–4 μg | [57] | |||||
3.2. Array or Bead Hybridization
3.3. Methyl-Sensitive Cut Counting: Endonuclease Digestion Followed by Sequencing

4. Methylation Status of Specific Genes of Interest
4.1. Bead Array
4.2. PCR and Sequencing
4.3. Pyrosequencing

4.4. Methylation-Specific PCR
4.5. PCR with High Resolution Melting
4.6. COLD-PCR for the Detection of Unmethylated Islands
5. Digestion-Based Assays
6. Enrichment Strategies in Search of Differentially-Methylated Loci
7. Detection of Hydroxymethyl Cytosine
8. Conclusions
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Olkhov-Mitsel, E.; Bapat, B. Strategies for discovery and validation of methylated and hydroxymethylated DNA biomarkers. Cancer Med. 2012, 1, 237–260. [Google Scholar] [CrossRef] [PubMed]
- Sant, K.E.; Nahar, M.S.; Dolinoy, D.C. DNA methylation screening and analysis. Methods Mol. Biol. 2012, 889, 385–406. [Google Scholar] [PubMed]
- Hsu, H.K.; Weng, Y.I.; Hsu, P.Y.; Huang, T.H.; Huang, Y.W. Detection of DNA methylation by medip and mbdcap assays: An overview of techniques. Methods Mol. Biol. 2014, 1105, 61–70. [Google Scholar] [PubMed]
- Hernandez, H.G.; Tse, M.Y.; Pang, S.C.; Arboleda, H.; Forero, D.A. Optimizing methodologies for pcr-based DNA methylation analysis. BioTechniques 2013, 55, 181–197. [Google Scholar] [PubMed]
- Sun, Z.; Cunningham, J.; Slager, S.; Kocher, J.P. Base resolution methylome profiling: Considerations in platform selection, data preprocessing and analysis. Epigenomics 2015, 7, 813–828. [Google Scholar] [CrossRef] [PubMed]
- Epigenie Informally Informative. Available online: http://epigenie.com/ (accessed on 20 December 2015).
- Protocol Online. Available online: http://www.protocol-online.org (accessed on 20 December 2015).
- https://www.scienceexchange.com.
- Find and Order Next-Generation Sequencing Services from Top Providers. Available online: https://genohub.com (accessed on 20 December 2015).
- Klamroth, R.; Holz, J.B.; Vercruysse, K.; Dreier, T.; Silence, K.; Antoine, I.; Stanssens, P.; Berlin, P.P.T. Results from a healthy volunteer, single-dose, phase 1 study investigating the safety, tolerability, and pharmacokinetics of the nanobody (tm) alx-0081 targeting von willebrand factor. Arterioscl. Throm. Vas. 2008, 28, E72–E72. [Google Scholar]
- Arakawa, Y.; Watanabe, M.; Inoue, N.; Sarumaru, M.; Hidaka, Y.; Iwatani, Y. Association of polymorphisms in dnmt1, dnmt3a, dnmt3b, mthfr and mtrr genes with global DNA methylation levels and prognosis of autoimmune thyroid disease. Clin. Exp. Immunol. 2012, 170, 194–201. [Google Scholar] [CrossRef] [PubMed]
- Kang, K.A.; Piao, M.J.; Kim, K.C.; Kang, H.K.; Chang, W.Y.; Park, I.C.; Keum, Y.S.; Surh, Y.J.; Hyun, J.W. Epigenetic modification of nrf2 in 5-fluorouracil-resistant colon cancer cells: Involvement of tet-dependent DNA demethylation. Cell Death Dis. 2014, 5, e1183. [Google Scholar] [CrossRef] [PubMed]
- Conti, A.; Kobets, T.; Escudero-Lourdes, C.; Montgomery, B.; Tryndyak, V.; Beland, F.A.; Doerge, D.R.; Pogribny, I.P. Dose- and time-dependent epigenetic changes in the livers of fisher 344 rats exposed to furan. Toxicol. Sci. 2014, 139, 371–380. [Google Scholar] [CrossRef] [PubMed]
- Gaglio, D.; Capitano, F.; Mastrodonato, A.; Minicocci, E.; Deiana, C.; Fragapane, P.; Camilloni, G.; Mele, A. Learning induced epigenetic modifications in the ventral striatum are necessary for long-term memory. Behav. Brain Res. 2014, 265, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Bujko, M.; Musialik, E.; Olbromski, R.; Przestrzelska, M.; Libura, M.; Pastwinska, A.; Juszczynski, P.; Zwierzchowski, L.; Baranowski, P.; Siedlecki, J.A. Repetitive genomic elements and overall DNA methylation changes in acute myeloid and childhood b-cell lymphoblastic leukemia patients. Int. J. Hematol. 2014, 100, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Calvo, X.; Nomdedeu, M.; Navarro, A.; Tejero, R.; Costa, D.; Munoz, C.; Pereira, A.; Pena, O.; Risueno, R.M.; Monzo, M.; et al. High levels of global DNA methylation are an independent adverse prognostic factor in a series of 90 patients with de novo myelodysplastic syndrome. Leuk. Res. 2014, 38, 874–881. [Google Scholar] [CrossRef] [PubMed]
- Kuo, K.C.; McCune, R.A.; Gehrke, C.W.; Midgett, R.; Ehrlich, M. Quantitative reversed-phase high performance liquid chromatographic determination of major and modified deoxyribonucleosides in DNA. Nucleic Acids Res. 1980, 8, 4763–4776. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; James, S.R.; Kazim, L.; Karpf, A.R. Specific method for the determination of genomic DNA methylation by liquid chromatography-electrospray ionization tandem mass spectrometry. Anal. Chem. 2005, 77, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Thuc, L.; Kim, K.P.; Fan, G.; Faull, K.F. A sensitive mass spectrometry method for simultaneous quantification of DNA methylation and hydroxymethylation levels in biological samples. Anal. Biochem. 2011, 412, 203–209. [Google Scholar]
- Liu, Z.; Wu, J.; Xie, Z.; Liu, S.; Fan-Havard, P.; Huang, T.H.; Plass, C.; Marcucci, G.; Chan, K.K. Quantification of regional DNA methylation by liquid chromatography/tandem mass spectrometry. Anal. Biochem. 2009, 391, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Quinlivan, E.P.; Gregory, J.F., 3rd. DNA methylation determination by liquid chromatography-tandem mass spectrometry using novel biosynthetic [u-15n]deoxycytidine and [u-15n]methyldeoxycytidine internal standards. Nucleic Acids Res. 2008, 36, e119. [Google Scholar] [CrossRef] [PubMed]
- So, M.Y.; Tian, Z.; Phoon, Y.S.; Sha, S.; Antoniou, M.N.; Zhang, J.; Wu, R.S.; Tan-Un, K.C. Gene expression profile and toxic effects in human bronchial epithelial cells exposed to zearalenone. PLoS ONE 2014, 9, e96404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohka, F.; Natsume, A.; Motomura, K.; Kishida, Y.; Kondo, Y.; Abe, T.; Nakasu, Y.; Namba, H.; Wakai, K.; Fukui, T.; et al. The global DNA methylation surrogate line-1 methylation is correlated with mgmt promoter methylation and is a better prognostic factor for glioma. PLoS ONE 2011, 6, e23332. [Google Scholar] [CrossRef] [PubMed]
- Ogino, S.; Nosho, K.; Kirkner, G.J.; Kawasaki, T.; Chan, A.T.; Schernhammer, E.S.; Giovannucci, E.L.; Fuchs, C.S. A cohort study of tumoral line-1 hypomethylation and prognosis in colon cancer. J. Natl. Cancer Inst. 2008, 100, 1734–1738. [Google Scholar] [CrossRef] [PubMed]
- Pattamadilok, J.; Huapai, N.; Rattanatanyong, P.; Vasurattana, A.; Triratanachat, S.; Tresukosol, D.; Mutirangura, A. Line-1 hypomethylation level as a potential prognostic factor for epithelial ovarian cancer. Int. J. Gynecol. Cancer 2008, 18, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.D.; Qu, J.H.; Chang, X.J.; Lu, Y.Y.; Bai, W.L.; Wang, H.; Xu, Z.X.; An, L.J.; Wang, C.P.; Zeng, Z.; et al. Hypomethylation of long interspersed nuclear element-1 promoter is associated with poor outcomes for curative resected hepatocellular carcinoma. Liver Int. 2014, 34, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Hur, K.; Cejas, P.; Feliu, J.; Moreno-Rubio, J.; Burgos, E.; Boland, C.R.; Goel, A. Hypomethylation of long interspersed nuclear element-1 (line-1) leads to activation of proto-oncogenes in human colorectal cancer metastasis. Gut 2014, 63, 635–646. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Utsunomiya, T.; Ikemoto, T.; Yamada, S.; Morine, Y.; Imura, S.; Arakawa, Y.; Takasu, C.; Ishikawa, D.; Imoto, I.; et al. Hypomethylation of long interspersed nuclear element-1 (line-1) is associated with poor prognosis via activation of c-met in hepatocellular carcinoma. Ann. Surg. Oncol. 2014, 21, S729–S735. [Google Scholar] [CrossRef] [PubMed]
- Aung, H.T.; Harrison, D.K.; Findlay, I.; Mattick, J.S.; Martin, N.G.; Carroll, B.J. Stringent programming of DNA methylation in humans. Twin Res. Hum. Genet. 2010, 13, 405–411. [Google Scholar] [CrossRef] [PubMed]
- Jaligot, E.; Beule, T.; Rival, A. Methylation-sensitive rflps: Characterisation of two oil palm markers showing somaclonal variation-associated polymorphism. Theor. Appl. Genet. 2002, 104, 1263–1269. [Google Scholar] [PubMed]
- Jorda, M.; Rodriguez, J.; Frigola, J.; Peinado, M.A. Analysis of DNA methylation by amplification of intermethylated sites (aims). Methods Mol. Biol. 2009, 507, 107–116. [Google Scholar] [PubMed]
- Yamamoto, F.; Yamamoto, M.; Soto, J.L.; Kojima, E.; Wang, E.N.; Perucho, M.; Sekiya, T.; Yamanaka, H. Notl-msell methylation-sensitive amplied fragment length polymorhism for DNA methylation analysis of human cancers. Electrophoresis 2001, 22, 1946–1956. [Google Scholar] [CrossRef]
- Xu, M.L.; Li, X.Q.; Korban, S.S. Aflp-based detection of DNA methylation. Plant Mol. Biol. Rep. 2000, 18, 361–368. [Google Scholar] [CrossRef]
- Karimi, M.; Johanssona, S.; Stachc, D.; Corcorand, M.; Grandérd, D.; Schallingb, M.; Bakalkina, G.; Lykoc, F.; Larssonb, C.; Ekström, T.J. LUMA (LUminometric Methylation Assay) — A high throughput method to the analysis of genomic DNA methylation. Exp. Cell Res. 2006, 312, 1989–1995. [Google Scholar] [PubMed]
- Lisanti, S.; Omar, W.A.; Tomaszewski, B.; de Prins, S.; Jacobs, G.; Koppen, G.; Mathers, J.C.; Langie, S.A. Comparison of methods for quantification of global DNA methylation in human cells and tissues. PLoS ONE 2013, 8, e79044. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Demirci, F.; Mahalingam, G.; Demirci, C.; Nakano, M.; Meyers, B.C. An integrated workflow for DNA methylation analysis. J. Genet. Genomics 2013, 40, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Miura, F.; Ito, T. Highly sensitive targeted methylome sequencing by post-bisulfite adaptor tagging. DNA Res. 2014, 22, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Karimi, M.; Johansson, S.; Ekstrom, T.J. Using luma a luminometric-based assay for global DNA-methylation. Epigenetics 2006, 1, 45–48. [Google Scholar] [PubMed]
- Estecio, M.R.H.; Gharibyan, V.; Shen, L.L.; Ibrahim, A.E.K.; Doshi, K.; He, R.; Jelinek, J.; Yang, A.S.; Yan, P.S.; Huang, T.H.M.; et al. Line-1 hypomethylation in cancer is highly variable and inversely correlated with microsatellite instability. PLoS ONE 2007, 2. [Google Scholar] [CrossRef] [PubMed]
- De Meyer, T.; Mampaey, E.; Vlemmix, M.; Denil, S.; Trooskens, G.; Renard, J.P.; de Keulenaer, S.; Dehan, P.; Menschaert, G.; van Criekinge, W. Quality evaluation of methyl binding domain based kits for enrichment DNA-methylation sequencing. PLoS ONE 2013, 8, e59068. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tompkins, J.D.; Hall, C.; Chen, V.C.; Li, A.X.; Wu, X.; Hsu, D.; Couture, L.A.; Riggs, A.D. Epigenetic stability, adaptability, and reversibility in human embryonic stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 12544–12549. [Google Scholar] [CrossRef] [PubMed]
- Baker-Andresen, D.; Flavell, C.R.; Li, X.; Bredy, T.W. Activation of bdnf signaling prevents the return of fear in female mice. Learn. Mem. 2013, 20, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Edelkraut, U.; Hoffmann, A.; Daniel, G.; Spengler, D. Zac1 regulates astroglial differentiation of neural stem cells through socs3. Stem Cells 2013, 31, 1621–1632. [Google Scholar] [CrossRef] [PubMed]
- Gotze, S.; Schumacher, E.C.; Kordes, C.; Haussinger, D. Epigenetic changes during hepatic stellate cell activation. PLoS ONE 2015, 10, e0128745. [Google Scholar] [CrossRef] [PubMed]
- Allum, F.; Shao, X.; Guenard, F.; Simon, M.M.; Busche, S.; Caron, M.; Lambourne, J.; Lessard, J.; Tandre, K.; Hedman, A.K.; et al. Characterization of functional methylomes by next-generation capture sequencing identifies novel disease-associated variants. Nat. Commun. 2015, 6, 7211. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Suzuki, M.; Wendt, J.; Patterson, N.; Eichten, S.R.; Hermanson, P.J.; Green, D.; Jeddeloh, J.; Richmond, T.; Rosenbaum, H.; et al. Post-conversion targeted capture of modified cytosines in mammalian and plant genomes. Nucleic Acids Res. 2015, 43, e81. [Google Scholar] [CrossRef] [PubMed]
- Marabita, F.; Almgren, M.; Lindholm, M.E.; Ruhrmann, S.; Fagerstrom-Billai, F.; Jagodic, M.; Sundberg, C.J.; Ekstrom, T.J.; Teschendorff, A.E.; Tegner, J.; et al. An evaluation of analysis pipelines for DNA methylation profiling using the illumina humanmethylation450 beadchip platform. Epigenetics 2013, 8, 333–346. [Google Scholar] [CrossRef] [PubMed]
- Ziller, M.J.; Hansen, K.D.; Meissner, A.; Aryee, M.J. Coverage recommendations for methylation analysis by whole-genome bisulfite sequencing. Nat. Methods 2015, 12, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.D.; Mueller, M.; Game, L.; Aitman, T.J. Single nucleotide analysis of cytosine methylation by whole-genome shotgun bisulfite sequencing. Curr. Protoc. Mol. Biol. 2012. [Google Scholar] [CrossRef]
- Lee, Y.K.; Jin, S.; Duan, S.; Lim, Y.C.; Ng, D.P.; Lin, X.M.; Yeo, G.; Ding, C. Improved reduced representation bisulfite sequencing for epigenomic profiling of clinical samples. Biol. Proced. Online 2014, 16, 1. [Google Scholar] [CrossRef] [PubMed]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef] [PubMed]
- Ashktorab, H.; Daremipouran, M.; Goel, A.; Varma, S.; Leavitt, R.; Sun, X.; Brim, H. DNA methylome profiling identifies novel methylated genes in african american patients with colorectal neoplasia. Epigenetics 2014, 9, 503–512. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.U.; Ma, D.K.; Mo, H.; Ball, M.P.; Jang, M.H.; Bonaguidi, M.A.; Balazer, J.A.; Eaves, H.L.; Xie, B.; Ford, E.; et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011, 14, 1345–1351. [Google Scholar] [CrossRef] [PubMed]
- Colaneri, A.; Staffa, N.; Fargo, D.C.; Gao, Y.; Wang, T.Y.; Peddada, S.D.; Birnbaumer, L. Expanded methyl-sensitive cut counting reveals hypomethylation as an epigenetic state that highlights functional sequences of the genome. Proc. Natl. Acad. Sci. USA 2011, 108, 9715–9720. [Google Scholar] [CrossRef] [PubMed]
- Colaneri, A.C.; Jones, A.M. Genome-wide quantitative identification of DNA differentially methylated sites in arabidopsis seedlings growing at different water potential. PLoS ONE 2013, 8. [Google Scholar] [CrossRef] [PubMed]
- Rao, J.S.; Keleshian, V.L.; Klein, S.; Rapoport, S.I. Epigenetic modifications in frontal cortex from alzheimer’s disease and bipolar disorder patients. Transl. Psychiatry 2012, 2, e132. [Google Scholar] [CrossRef] [PubMed]
- Al-Moghrabi, N.; Nofel, A.; Al-Yousef, N.; Madkhali, S.; Bin Amer, S.M.; Alaiya, A.; Shinwari, Z.; Al-Tweigeri, T.; Karakas, B.; Tulbah, A.; et al. The molecular significance of methylated brca1 promoter in white blood cells of cancer-free females. BMC Cancer 2014, 14, 830. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Rodger, E.J.; Stockwell, P.A.; Weeks, R.J.; Morison, I.M. Technical considerations for reduced representation bisulfite sequencing with multiplexed libraries. J. Biomed. Biotechnol. 2012, 2012, 741542. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.L.; Bhagwate, A.V.; Baheti, S.; Smalley, R.L.; Hilker, C.A.; Sun, Z.; Cunningham, J.M. DNA methylation profiling: Comparison of genome-wide sequencing methods and the infinium human methylation 450 bead chip. Epigenomics 2015, 7, 1287–1302. [Google Scholar] [CrossRef] [PubMed]
- Flusberg, B.A.; Webster, D.R.; Lee, J.H.; Travers, K.J.; Olivares, E.C.; Clark, T.A.; Korlach, J.; Turner, S.W. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat. Methods 2010, 7, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Laszlo, A.H.; Derrington, I.M.; Brinkerhoff, H.; Langford, K.W.; Nova, I.C.; Samson, J.M.; Bartlett, J.J.; Pavlenok, M.; Gundlach, J.H. Detection and mapping of 5-methylcytosine and 5-hydroxymethylcytosine with nanopore mspa. Proc. Natl. Acad. Sci. USA 2013, 110, 18904–18909. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, J.; Wescoe, Z.L.; Abu-Shumays, R.; Vivian, J.T.; Baatar, B.; Karplus, K.; Akeson, M. Error rates for nanopore discrimination among cytosine, methylcytosine, and hydroxymethylcytosine along individual DNA strands. Proc. Natl. Acad. Sci. USA 2013, 110, 18910–18915. [Google Scholar] [CrossRef] [PubMed]
- Bibikova, M.; Fan, J.B. Goldengate assay for DNA methylation profiling. Methods Mol. Biol. 2009, 507, 149–163. [Google Scholar] [PubMed]
- Wilhelm-Benartzi, C.S.; Koestler, D.C.; Karagas, M.R.; Flanagan, J.M.; Christensen, B.C.; Kelsey, K.T.; Marsit, C.J.; Houseman, E.A.; Brown, R. Review of processing and analysis methods for DNA methylation array data. Br. J. Cancer 2013, 109, 1394–1402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, X.; Wang, Y.W.; Zhang, M.Q.; Gazdar, A.F. DNA methylation data analysis and its application to cancer research. Epigenomics 2013, 5, 301–316. [Google Scholar] [CrossRef] [PubMed]
- Bock, C. Analysing and interpreting DNA methylation data. Nat. Rev. Genet. 2012, 13, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.A.; Lemire, M.; Choufani, S.; Butcher, D.T.; Grafodatskaya, D.; Zanke, B.W.; Gallinger, S.; Hudson, T.J.; Weksberg, R. Discovery of cross-reactive probes and polymorphic cpgs in the illumina infinium humanmethylation450 microarray. Epigenetics 2013, 8, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Yao, J.; Polyak, K. Methylation-specific digital karyotyping. Nat. Protoc. 2006, 1, 1621–1636. [Google Scholar] [CrossRef] [PubMed]
- Ball, M.P.; Li, J.B.; Gao, Y.; Lee, J.H.; LeProust, E.M.; Park, I.H.; Xie, B.; Daley, G.Q.; Church, G.M. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat. Biotechnol. 2009, 27, 361–368. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Jing, Q.; Lia, D.; Pascual, M.; McLellan, A.; Greally, J.M. Optimized design and data analysis of tag-based cytosine methylation assays. Genome Biol. 2010, 11, R36. [Google Scholar] [CrossRef] [PubMed]
- Matsumura, H.; Yoshida, K.; Luo, S.J.; Kimura, E.; Fujibe, T.; Albertyn, Z.; Barrero, R.A.; Kruger, D.H.; Kahl, G.; Schroth, G.P.; et al. High-throughput supersage for digital gene expression analysis of multiple samples using next generation sequencing. PLoS ONE 2010, 5. [Google Scholar] [CrossRef] [PubMed]
- Zawada, A.M.; Rogacev, K.S.; Hummel, B.; Grun, O.S.; Friedrich, A.; Rotter, B.; Winter, P.; Geisel, J.; Fliser, D.; Heine, G.H. Supertag methylation-specific digital karyotyping reveals uremia-induced epigenetic dysregulation of atherosclerosis-related genes. Circ. Cardiovasc. Genet. 2012, 5, 611–620. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Gao, F.; Li, N.; Li, S.; Yin, G.; Tian, G.; Jia, S.; Wang, K.; Zhang, X.; Yang, H.; et al. An improved method for genome wide DNA methylation profiling correlated to transcription and genomic instability in two breast cancer cell lines. BMC Genom. 2009, 10, 223. [Google Scholar] [CrossRef] [PubMed]
- Tarasova, G.V.; Nayakshina, T.N.; Degtyarev, S.K. Substrate specificity of new methyl-directed DNA endonuclease glai. BMC Mol. Biol. 2008, 9, 7. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.N.; Young, G.P.; Ho, T.; Molloy, P.L. Sensitive and selective amplification of methylated DNA sequences using helper-dependent chain reaction in combination with a methylation-dependent restriction enzymes. Nucleic Acids Res. 2013, 41. [Google Scholar] [CrossRef] [PubMed]
- Cohen-Karni, D.; Xu, D.; Apone, L.; Fomenkov, A.; Sun, Z.; Davis, P.J.; Kinney, S.R.; Yamada-Mabuchi, M.; Xu, S.Y.; Davis, T.; et al. The mspji family of modification-dependent restriction endonucleases for epigenetic studies. Proc. Natl. Acad. Sci. USA 2011, 108, 11040–11045. [Google Scholar] [CrossRef] [PubMed]
- High-Throughput DNA Methylation Profiling with VeraCode® Technology. Available online: http://www.illumina.com/content/dam/illumina-marketing/documents/products/datasheets/datasheet_veracode_methylation.pdf (accessed on 20 December 2015).
- Methprimer. Available online: http://www.urogene.org/methprimer (accessed on 20 December 2015).
- Kurdyukov, S.; Mathesius, U.; Nolan, K.E.; Sheahan, M.B.; Goffard, N.; Carroll, B.J.; Rose, R.J. The 2ha line of medicago truncatula has characteristics of an epigenetic mutant that is weakly ethylene insensitive. Bmc Plant Biol. 2014, 14, 174. [Google Scholar] [CrossRef] [PubMed]
- Mahapatra, S.; Klee, E.W.; Young, C.Y.; Sun, Z.; Jimenez, R.E.; Klee, G.G.; Tindall, D.J.; Donkena, K.V. Global methylation profiling for risk prediction of prostate cancer. Clin. Cancer Res. 2012, 18, 2882–2895. [Google Scholar] [CrossRef] [PubMed]
- Herman, J.G.; Graff, J.R.; Myohanen, S.; Nelkin, B.D.; Baylin, S.B. Methylation-specific pcr: A novel pcr assay for methylation status of cpg islands. Proc. Natl. Acad. Sci. USA 1996, 93, 9821–9826. [Google Scholar] [CrossRef] [PubMed]
- Wojdacz, T.K.; Dobrovic, A.; Hansen, L.L. Methylation-sensitive high-resolution melting. Nat. Protoc. 2008, 3, 1903–1908. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Rizaldos, E.; Milbury, C.A.; Karatza, E.; Chen, C.C.; Makrigiorgos, G.M.; Merewood, A. Cold-pcr amplification of bisulfite-converted DNA allows the enrichment and sequencing of rare un-methylated genomic regions. PLoS ONE 2014, 9, e94103. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, S.; Kitamoto, S.; Yamada, N.; Houjou, I.; Sugai, T.; Nakamura, S.; Arisaka, Y.; Takaori, K.; Higashi, M.; Yonezawa, S. The application of methylation specific electrophoresis (mse) to DNA methylation analysis of the 5′ cpg island of mucin in cancer cells. BMC Cancer 2012, 12, 67. [Google Scholar] [CrossRef] [PubMed]
- Cedar, H.; Solage, A.; Glaser, G.; Razin, A. Direct detection of methylated cytosine in DNA by use of the restriction enzyme mspi. Nucleic Acids Res. 1979, 6, 2125–2132. [Google Scholar] [CrossRef] [PubMed]
- Takamiya, T.; Hosobuchi, S.; Asai, K.; Nakamura, E.; Tomioka, K.; Kawase, M.; Kakutani, T.; Paterson, A.H.; Murakami, Y.; Okuizumi, H. Restriction landmark genome scanning method using isoschizomers (mspi/hpaii) for DNA methylation analysis. Electrophoresis 2006, 27, 2846–2856. [Google Scholar] [CrossRef] [PubMed]
- Cai, W.; Mao, F.; Teng, H.; Cai, T.; Zhao, F.; Wu, J.; Sun, Z.S. Mbridge: An accurate and cost-effective method for profiling DNA methylome at single-base resolution. J. Mol. Cell Biol. 2015, 7, 299–313. [Google Scholar] [CrossRef] [PubMed]
- Harris, R.A.; Wang, T.; Coarfa, C.; Nagarajan, R.P.; Hong, C.; Downey, S.L.; Johnson, B.E.; Fouse, S.D.; Delaney, A.; Zhao, Y.; et al. Comparison of sequencing-based methods to profile DNA methylation and identification of monoallelic epigenetic modifications. Nat. Biotechnol. 2010, 28, 1097–1105. [Google Scholar] [CrossRef] [PubMed]
- Rauch, T.; Pfeifer, G.P. Methylated-cpg island recovery assay: A new technique for the rapid detection of methylated-cpg islands in cancer. Lab. Investig. 2005, 85, 1172–1180. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Greally, J.M. Genome-wide DNA methylation analysis using massively parallel sequencing technologies. Semin. Hematol. 2013, 50, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Oda, M.; Glass, J.L.; Thompson, R.F.; Mo, Y.; Olivier, E.N.; Figueroa, M.E.; Selzer, R.R.; Richmond, T.A.; Zhang, X.; Dannenberg, L.; et al. High-resolution genome-wide cytosine methylation profiling with simultaneous copy number analysis and optimization for limited cell numbers. Nucleic Acids Res. 2009, 37, 3829–3839. [Google Scholar] [CrossRef] [PubMed]
- Brunner, A.L.; Johnson, D.S.; Kim, S.W.; Valouev, A.; Reddy, T.E.; Neff, N.F.; Anton, E.; Medina, C.; Nguyen, L.; Chiao, E.; et al. Distinct DNA methylation patterns characterize differentiated human embryonic stem cells and developing human fetal liver. Genome Res. 2009, 19, 1044–1056. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, S.I.; Iseli, C.; Sharp, A.J.; Robyr, D.; Rougemont, J.; Gehrig, C.; Farinelli, L.; Antonarakis, S.E. Detection of genomic variation by selection of a 9 mb DNA region and high throughput sequencing. PLoS ONE 2009, 4, e6659. [Google Scholar] [CrossRef] [PubMed]
- Okou, D.T.; Steinberg, K.M.; Middle, C.; Cutler, D.J.; Albert, T.J.; Zwick, M.E. Microarray-based genomic selection for high-throughput resequencing. Nat. Methods 2007, 4, 907–909. [Google Scholar] [CrossRef] [PubMed]
- Newburger, D.E.; Natsoulis, G.; Grimes, S.; Bell, J.M.; Davis, R.W.; Batzoglou, S.; Ji, H.P. The human oligogenome resource: A database of oligonucleotide capture probes for resequencing target regions across the human genome. Nucleic Acids Res. 2012, 40, D1137–D1143. [Google Scholar] [CrossRef] [PubMed]
- Harakalova, M.; Mokry, M.; Hrdlickova, B.; Renkens, I.; Duran, K.; van Roekel, H.; Lansu, N.; van Roosmalen, M.; de Bruijn, E.; Nijman, I.J.; et al. Multiplexed array-based and in-solution genomic enrichment for flexible and cost-effective targeted next-generation sequencing. Nat. Protoc. 2011, 6, 1870–1886. [Google Scholar] [CrossRef] [PubMed]
- Brooks, R.; Rose, R.J.; Sheahan, M.B.; Kurdyukov, S. Whole genome methylation scanning based on phi29 polymerase amplification. Biochemistry 2011, 76, 999–1002. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurdyukov, S.; Bullock, M. DNA Methylation Analysis: Choosing the Right Method. Biology 2016, 5, 3. https://doi.org/10.3390/biology5010003
Kurdyukov S, Bullock M. DNA Methylation Analysis: Choosing the Right Method. Biology. 2016; 5(1):3. https://doi.org/10.3390/biology5010003
Chicago/Turabian StyleKurdyukov, Sergey, and Martyn Bullock. 2016. "DNA Methylation Analysis: Choosing the Right Method" Biology 5, no. 1: 3. https://doi.org/10.3390/biology5010003
APA StyleKurdyukov, S., & Bullock, M. (2016). DNA Methylation Analysis: Choosing the Right Method. Biology, 5(1), 3. https://doi.org/10.3390/biology5010003