Identification and Characterization of microRNAs and Their Predicted Functions in Biomineralization in the Pearl Oyster (Pinctada fucata)

 , , , and
, , , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics
2.2. Small RNA Isolation and cDNA Library Construction
2.3. Small RNA Sequence Analysis
2.4. Analysis of Differentially Expressed miRNAs among Tissues
2.5. Target Analysis for miRNAs and Biomineralization-Related Genes
2.6. Validation of P. fucata miRNAs by Stem-Loop qRT-PCR
2.7. Data Accessibility
3. Results
3.1. Overview of Small RNA Library Sequencing
3.2. Identification of Known and Novel miRNAs
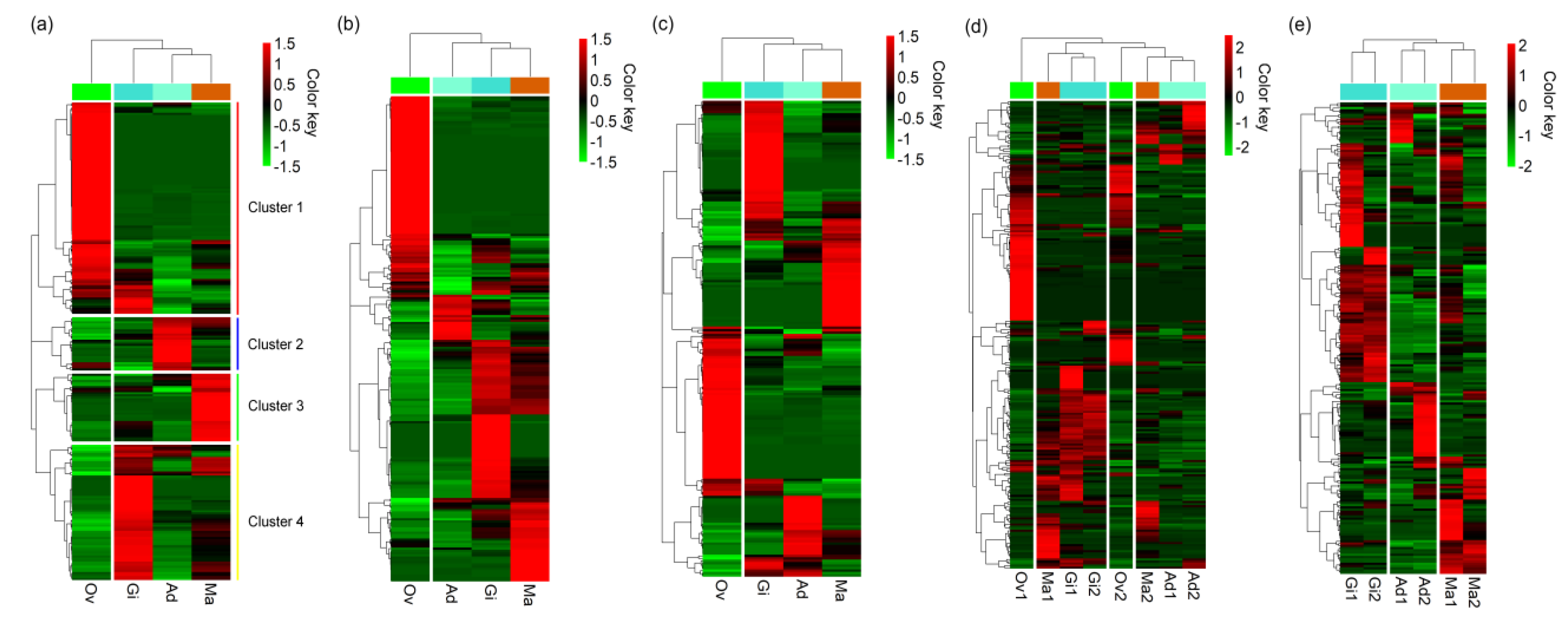
3.3. Expression Levels of miRNAs in P. fucata
3.4. Validation of miRNA by Stem-Loop qRT-PCR
3.5. Functional Prediction of miRNA in Biomineralization
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Takeuchi, T.; Kawashima, T.; Koyanagi, R.; Gyoja, F.; Tanaka, M.; Ikuta, T.; Shoguchi, E.; Fujiwara, M.; Shinzato, C.; Hisata, K.; et al. Draft genome of the pearl oyster Pinctada fucata: A platform for understanding bivalve biology. DNA Res. 2012, 19, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, T.; Koyanagi, R.; Gyoja, F.; Kanda, M.; Hisata, K.; Fujie, M.; Goto, H.; Yamasaki, S.; Nagai, K.; Morino, Y.; et al. Bivalve-specific gene expansion in the pearl oyster genome: Implications of adaptation to a sessile lifestyle. Zool. Lett. 2016, 2, 3. [Google Scholar] [CrossRef] [PubMed]
- Samata, T.; Hayashi, N.; Kono, M.; Hasegawac, K.; Horitad, C.; Akerad, S. A new matrix protein family related to the nacreous layer formation of Pinctada fucata. FEBS Lett. 1999, 462, 225–229. [Google Scholar] [CrossRef]
- Suzuki, M.; Murayama, E.; Inoue, H.; Ozaki, N.; Tohse, H.; Kogure, T.; Nagasawa, H. Characterization of Prismalin-14, a novel matrix protein from the prismatic layer of the Japanese pearl oyster (Pinctada fucata). Biochem. J. 2004, 382, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Yano, M.; Nagai, K.; Morimoto, K.; Miyamoto, H. Shematrin: A family of glycine-rich structural proteins in the shell of the pearl oyster Pinctada fucata. Comp. Biochem. Phys. B 2006, 144, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.L.; Liu, S.F.; Ge, Y.J.; Liu, J.; Wang, X.Y.; Xie, L.P.; Zhang, R.Q.; Wang, Z. Identification and characterization of a biomineralization related gene PFMG1 highly expressed in the mantle of Pinctada fucata. Biochemistry 2007, 46, 844–851. [Google Scholar] [CrossRef] [PubMed]
- Takagi, R.; Miyashita, T. Prismin: A new matrix protein family in the Japanese pearl oyster (Pinctada fucata) involved in prismatic layer formation. Zool. Sci. 2010, 27, 416–426. [Google Scholar] [CrossRef]
- Fang, D.; Xu, G.; Hu, Y.; Pan, C.; Xie, L.P.; Zhang, R.Q. Identification of genes directly involved in shell formation and their functions in pearl oyster, Pinctada fucata. PLoS ONE 2011, 6, e21860. [Google Scholar] [CrossRef]
- Kinoshita, S.; Wang, N.; Inoue, H.; Maeyama, K.; Okamoto, K.; Nagai, K.; Kondo, H.; Hirono, I.; Asakawa, S.; Watabe, S. Deep sequencing of ESTs from nacreous and prismatic layer producing tissues and a screen for novel shell formation-related genes in the pearl oyster. PLoS ONE 2011, 6, e21238. [Google Scholar] [CrossRef]
- Joubert, C.; Piquemal, D.; Marie, B.; Manchon, L.; Pierrat, F.; Zanella-Cleon, I.; Cochennec-Laureau, N.; Gueguen, Y.; Montagnani, C. Transcriptome and proteome analysis of Pinctada margaritifera calcifying mantle and shell: Focus on biomineralization. BMC Genom. 2010, 11, 613. [Google Scholar] [CrossRef]
- Berland, S.; Marie, A.; Duplat, D.; Milet, C.; Sire, J.Y.; Bedouet, L. Coupling proteomics and transcriptomics for the identification of novel and variant forms of mollusk shell proteins: A study with P. margaritifera. Chembiochem 2011, 12, 950–961. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Li, S.; Kong, J.; Liu, Y.; Wang, T.; Xie, L.; Zhang, R.Q. In-depth proteomic analysis of shell matrix proteins of Pinctada fucata. Sci. Rep. 2015, 5, 17269. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, H.; Endo, H.; Hashimoto, N.; Limura, K.; Isowa, Y.; Kinoshita, S.; Kotaki, T.; Masaoka, T.; Miki, T.; Nakayama, S.; et al. The diversity of shell matrix proteins: Genome-wide investigation of the pearl oyster, Pinctada fucata. Zool. Sci. 2013, 30, 801–816. [Google Scholar] [CrossRef] [PubMed]
- Du, X.D.; Fan, G.Y.; Jiao, Y.; Zhang, H.; Guo, X.M.; Huang, R.L.; Zheng, Z.; Bian, C.; Deng, Y.W.; Wang, Q.H.; et al. The pearl oyster Pinctada fucata martensii genome and multi-omic analyses provide insights into biomineralization. Gigascience 2017, 6, gix059. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, M.; Saruwatari, K.; Kogure, T.; Yamamoto, Y.; Nishimura, T.; Kato, T.; Nagasawa, H. An acidic matrix protein, Pif, is a key macromolecule for nacre formation. Science 2009, 325, 1388–1390. [Google Scholar] [CrossRef]
- Fang, D.; Pan, C.; Lin, H.; Lin, Y.; Zhang, G.Y.; Wang, H.Z.; He, M.X.; Xie, L.P.; Zhang, R.Q. Novel basic protein, PfN23, functions as key macromolecule during nacre formation. J. Biol. Chem. 2012, 287, 15776–15785. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.Q.; Cao, X.J.; Tian, X.C.; Wang, W.M. High-throughput sequencing identifies microRNAs from posterior intestine of loach (Misgurnus anguillicaudatus) and their response to intestinal air-breathing inhibition. PLoS ONE 2016, 11, e0149123. [Google Scholar] [CrossRef]
- Di, L.G.; Calin, G.A.; Croce, C.M. MicroRNAs: Fundamental facts and involvement in human diseases. Birth Defects Res. C 2006, 78, 180–189. [Google Scholar]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Urbich, C.; Kuehbacher, A.; Dimmeler, S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res. 2008, 79, 581–588. [Google Scholar] [CrossRef] [Green Version]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. MicroRNA-directed phasing during transacting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Bushati, N.; Cohen, S.M. MicroRNA functions. Annu. Rev. Cell Dev. Biol. 2007, 23, 175–205. [Google Scholar] [CrossRef] [PubMed]
- Sugatani, T.; Hruska, K.A. MicroRNA-223 is a key factor in osteoclast differentiation. J. Cell. Biochem. 2007, 101, 996–999. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.; Chen, C.; Chen, P.; Xie, H.; Luo, X. MicroRNAs and their roles in osteoclast differentiation. Front. Med. 2011, 5, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Kang, I.H.; Jeong, B.C.; Hur, S.W.; Choi, H.; Choi, S.H.; Ryu, J.H.; Hwang, Y.C.; Koh, J.T. MicroRNA-302a stimulates osteoblastic differentiation by repressing COUP-TFII expression. J. Cell. Physiol. 2015, 230, 911–921. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, B.M.; Heimberg, A.M.; Moy, V.N.; Sperling, E.A.; Holstein, T.W.; Heber, S.; Peterson, K.J. The deep evolution of metazoan microRNAs. Evol. Dev. 2009, 11, 50–68. [Google Scholar] [CrossRef]
- Jiao, Y.; Zheng, Z.; Du, X.D.; Wang, Q.H.; Huang, R.L.; Deng, Y.W.; Shi, S.L.; Zhao, X.X. Identification and characterization of miRNAs in pearl oyster Pinctada martensii by solexa sequencing. Mar. Biotechnol. 2014, 16, 54–62. [Google Scholar] [CrossRef]
- Zhou, Z.; Wang, L.; Song, L.; Liu, R.; Zhang, H.; Huang, M.; Chen, H. The identification and characteristics of immune-related microRNAs in haemocytes of oyster Crassostrea gigas. PLoS ONE 2014, 9, e88397. [Google Scholar] [CrossRef]
- Zhao, X.L.; Yu, H.; Kong, L.F.; Liu, S.K.; Li, Q. High throughput sequencing of small RNAs transcriptomes in two Crassostrea oysters identifies microRNAs involved in osmotic stress response. Sci. Rep. 2016, 6, 22687. [Google Scholar] [CrossRef]
- Kenny, N.J.; Namigai, E.K.; Marletaz, F.; Hui, J.H.; Shimeld, S.M. Draft genome assemblies and predicted microRNA complements of the intertidal lophotrochozoans Patella vulgata (Mollusca, Patellogastropoda) and Spirobranchus (Pomatoceros) lamarcki (Annelida, Serpulida). Mar. Genom. 2015, 24, 139–146. [Google Scholar] [CrossRef]
- Zheng, Z.; Jiao, Y.; Du, X.D.; Tian, Q.L.; Wang, Q.H.; Huang, R.L.; Deng, Y.W. Computational prediction of candidate miRNAs and their potential functions in biomineralization in pearl oyster Pinctada martensii. Saudi J. Biol. Sci. 2016, 23, 372–378. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Zheng, Z.; Tian, R.R.; Du, X.D.; Wang, Q.H.; Huang, R.L. Pm-miR-2305, participates in nacre formation by targeting pearlin in pearl oyster Pinctada martensii. Int. J. Mol. Sci. 2015, 16, 21442–21453. [Google Scholar] [CrossRef] [PubMed]
- Szymanski, M.; Erdmann, V.A.; Barciszewski, J. Noncoding RNAs database (ncRNAdb). Nucleic Acids Res. 2007, 35, D162–D164. [Google Scholar] [CrossRef] [PubMed]
- Kalvari, I.; Argasinska, J.; Quinones-Olvera, N.; Nawrocki, E.P.; Rivas, E.; Eddy, S.R.; Bateman, A.; Finn, R.D.; Petrov, A.I. Rfam 13.0: Shifting to a genome-centric resource for non-coding RNA families. Nucleic Acids Res. 2018, 46, D335–D342. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed]
- Kozomara, A.; Birgaoanu, M.; Griffiths-Jones, S. MiRBase: From microRNA sequences to function. Nucleic Acids Res. 2019, 47, D155–D162. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, M.R.; Mackowiak, S.D.; Li, N.; Chen, W.; Rajewsky, N. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res. 2012, 40, 37–52. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2017, 41, D36–D42. [Google Scholar] [CrossRef]
- Rehmsmeier, M.; Steffen, P.; Hochsmann, M.; Giegerich, R. Fast and effective prediction of microRNA/target duplexes. RNA 2014, 10, 1507–1517. [Google Scholar] [CrossRef]
- Enright, A.J.; John, B.; Gaul, U.; Tuschl, T.; Sander, C.; Marks, D.S. MicroRNA targets in Drosophila. Genome Biol. 2003, 5, R1. [Google Scholar] [CrossRef] [PubMed]
- Loher, P.; Rigoutsos, I. Interactive exploration of RNA22 microRNA target predictions. Bioinformatics 2012, 28, 3322–3323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of micro-RNAs by stem-loop RT-PCR. Nucleic Acids Res. 2005, 33, e179. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2011, 29, e45. [Google Scholar] [CrossRef] [PubMed]
- Obernosterer, G.; Leuschner, P.J.; Alenius, M.; Martinez, J. Post-transcriptional regulation of microRNA expression. RNA 2006, 12, 1161–1167. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, S.Q.; Ichikawa, Y.; Igarashi, Y.; Yoshitake, K.; Kinoshita, S.; Omori, F.; Maeyama, K.; Nagai, K.; Watabe, S.; Asakawa, S. Piwi-interacting RNA (piRNA) expression patterns in pearl oyster (Pinctada fucata) somatic tissues. Sci. Rep. 2019, 9, 247. [Google Scholar] [CrossRef] [PubMed]
- Sun, K.; Jee, D.; de Navas, L.F.; Duan, H.; Lai, E.C. Multiple in vivo biological processes are mediated by functionally redundant activities of drosophila mir-279 and mir-996. PLoS Genet. 2015, 11, e1005245. [Google Scholar] [CrossRef] [PubMed]
- Cayirlioglu, P.; Kadow, I.G.; Zhan, X.; Okamura, K.; Suh, G.S.; Gunning, D.; Lai, E.C.; Zipursky, S.L. Hybrid neurons in a microRNA mutant are putative evolutionary intermediates in insect CO2 sensory systems. Science 2008, 319, 1256–1260. [Google Scholar] [CrossRef]
- Luo, W.; Sehgal, A. Regulation of circadian behavioral output via a microRNA-JAK/STAT circuit. Cell 2012, 148, 765–779. [Google Scholar] [CrossRef]
- Henson, B.J.; Bhattacharjee, S.; O’Dee, D.M.; Feingold, E.; Gollin, S.M. Decreased expression of miR-125b and miR-100 in oral cancer cells contributes to malignancy. Gene Chromosome Cancer 2009, 48, 569–582. [Google Scholar] [CrossRef] [Green Version]
- Giangreco, A.A.; Vaishnav, A.; Wagner, D.; Finelli, A.; Fleshner, N.; Van der Kwast, T.; Vieth, R.; Nonn, L. Tumor suppressor microRNAs, miR-100 and -125b, are regulated by 1,25-dihydroxyvitamin D in primary prostate cells and in patient tissue. Cancer Prev. Res. 2013, 6, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.D.; Esquela-Kerscher, A.; Stefani, G.; Byrom, M.; Kelnar, K.; Ovcharenko, D.; Wilson, M.; Wang, X.; Shelton, J.; Shingara, J.; et al. The let-7 microRNA represses cell proliferation pathways in human cells. Cancer Res. 2007, 67, 7713–7722. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, T.; Tanaka, K.; Kawano, M.; Itonaga, I.; Tsumura, H. Tumor-suppressive microRNA-let-7a inhibits cell proliferation via targeting of E2F2 in osteosarcoma cells. Int. J. Oncol. 2015, 46, 1543–1550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajewsky, N. MicroRNA target predictions in animals. Nat. Genet. 2006, 38, S8–S13. [Google Scholar] [CrossRef] [PubMed]
- Witkos, T.M.; Koscianska, E.; Krzyzosiak, W.J. Practical aspects of microRNA target prediction. Curr. Mol. Med. 2011, 1, 93–109. [Google Scholar] [CrossRef]
- Peterson, S.M.; Thompson, J.A.; Ufkin, M.L.; Sathyanarayana, P.; Liaw, L.; Congdon, C.B. Common features of microRNA target prediction tools. Front. Genet. 2014, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubota, K.; Tsuchihashi, Y.; Kogure, T.; Maeyama, K.; Hattori, F.; Kinoshita, S.; Sakuda, S.; Nagasawa, H.; Yoshimura, E.; Suzuki, M. Structural and functional analyses of a TIMP and MMP in the ligament of Pinctada fucata. J. Struct. Biol. 2017, 199, 216–224. [Google Scholar] [CrossRef]
- Gao, J.; Chen, Y.; Yang, Y.; Liang, J.; Xie, J.; Liu, J.; Li, S.G.; Zheng, G.L.; Xie, L.P.; Zhang, R.Q. The transcription factor Pf-POU3F4 regulates the expression of the matrix protein genes Aspein and Prismalin-14 in pearl oyster (Pinctada fucata). FEBS J. 2016, 283, 1962–1978. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Small RNA | Stem-Loop Primer | qRT-PCR Forward Primer |
|---|---|---|
| let-7a | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAACTATAC | ACACTCCAGCTGGGTGAGGTAGTAGGTTGT |
| miR-1493 | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGACTGATGT | ACACTCCAGCTGGGAGAACTGTGTATGGAC |
| miR-1990c-3p | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGGCAAGTAG | ACACTCCAGCTGGGCGGGACTACGTCAACT |
| miR-1993 | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCTCGTGA | ACACTCCAGCTGGGTATTATGCTGTTATTC |
| miR-279 | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGGGATGAGT | ACACTCCAGCTGGGTGACTAGATCCACAC |
| miR-876 | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGTCACGGAT | ACACTCCAGCTGGGTGGATTTCCCAAGAT |
| miR-9a-3p | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCCTCCGGT | ACACTCCAGCTGGGATAAAGCTAGGTTAC |
| miR-183 | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGCCGTGAAT | ACACTCCAGCTGGGAATGGCACTGGTAGAAT |
| miR-200a | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGGACATCTT | ACACTCCAGCTGGGTAATACTGTCAGGTAAA |
| novel-1 | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGGCGGAATC | ACACTCCAGCTGGGAGGCGAGCCTAAACGA |
| novel-3 | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGGCAGGTAG | ACACTCCAGCTGGGTGCCGTCACAAGGACT |
| novel-10 | CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGGATACTGG | ACACTCCAGCTGGGTGCACCAAACTAATGCC |
| miRNA reverse primer | TGGTGTCGTGGAGTCG | |
| U6 forward primer | TTGCTTCGGCGGTACATATA | |
| U6 reverse primer | ATTTGCGTGTCATCCTTGC |
| Libraries | Ad1 | Ad2 | Gi1 | Gi2 | Ov1 | Ov2 | Ma1 | Ma2 | Total |
|---|---|---|---|---|---|---|---|---|---|
| Raw reads | 1.96 | 5.14 | 13.59 | 5.74 | 6.74 | 3.04 | 5.57 | 8.54 | 50.32 |
| Step 1 | 1.58 | 4.61 | 11.36 | 5.38 | 5.63 | 2.7 | 3.64 | 6.82 | 41.72 |
| Step 2 | 1.57 | 4.61 | 11.35 | 5.37 | 5.63 | 2.7 | 3.64 | 6.81 | 41.68 |
| Step 3 | 1.33 | 3.59 | 9.42 | 4.31 | 3.97 | 1.92 | 3.07 | 5.65 | 33.26 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, S.; Ichikawa, Y.; Yoshitake, K.; Kinoshita, S.; Igarashi, Y.; Omori, F.; Maeyama, K.; Nagai, K.; Watabe, S.; Asakawa, S. Identification and Characterization of microRNAs and Their Predicted Functions in Biomineralization in the Pearl Oyster (Pinctada fucata). Biology 2019, 8, 47. https://doi.org/10.3390/biology8020047
Huang S, Ichikawa Y, Yoshitake K, Kinoshita S, Igarashi Y, Omori F, Maeyama K, Nagai K, Watabe S, Asakawa S. Identification and Characterization of microRNAs and Their Predicted Functions in Biomineralization in the Pearl Oyster (Pinctada fucata). Biology. 2019; 8(2):47. https://doi.org/10.3390/biology8020047
Chicago/Turabian StyleHuang, Songqian, Yuki Ichikawa, Kazutoshi Yoshitake, Shigeharu Kinoshita, Yoji Igarashi, Fumito Omori, Kaoru Maeyama, Kiyohito Nagai, Shugo Watabe, and Shuichi Asakawa. 2019. "Identification and Characterization of microRNAs and Their Predicted Functions in Biomineralization in the Pearl Oyster (Pinctada fucata)" Biology 8, no. 2: 47. https://doi.org/10.3390/biology8020047
APA StyleHuang, S., Ichikawa, Y., Yoshitake, K., Kinoshita, S., Igarashi, Y., Omori, F., Maeyama, K., Nagai, K., Watabe, S., & Asakawa, S. (2019). Identification and Characterization of microRNAs and Their Predicted Functions in Biomineralization in the Pearl Oyster (Pinctada fucata). Biology, 8(2), 47. https://doi.org/10.3390/biology8020047