Soluble Epoxide Hydrolase Inhibition in Liver Diseases: A Review of Current Research and Knowledge Gaps

 , ,
, ,
Abstract
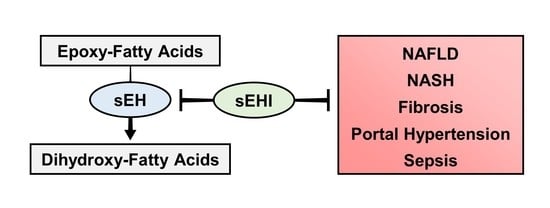
:
1. Introduction
2. Methodology
3. sEH Inhibition in Metabolic Syndrome and Non-Alcoholic Fatty Liver Disease
4. sEH Inhibition in Hepatic Fibrosis and Portal Hypertension
5. sEH Inhibition in Sepsis Models
6. Molecular Mechanisms of sEHI-Mediated Protection against Liver Diseases
7. Future Directions and Knowledge Gaps
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morisseau, C.; Hammock, B.D. Impact of soluble epoxide hydrolase and epoxyeicosanoids on human health. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 37–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, G.T.; Harrison, A.; Lewis, S.E. Cyclodiene epoxide ring hydration by microsomes from mammalian liver and houseflies. Biochem. Pharmacol. 1970, 19, 255–273. [Google Scholar] [CrossRef]
- Beetham, J.K.; Grant, D.; Arand, M.; Garbarino, J.; Kiyosue, T.; Pinot, F.; Oesch, F.; Belknap, W.R.; Shinozaki, K.; Hammock, B.D. Gene evolution of epoxide hydrolases and recommended nomenclature. DNA Cell Biol. 1995, 14, 61–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, T.R.; Aronov, P.A.; Jones, P.D.; Tanaka, H.; Arand, M.; Hammock, B.D. Identification of two epoxide hydrolases in Caenorhabditis elegans that metabolize mammalian lipid signaling molecules. Arch. Biochem. Biophys. 2008, 472, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, P.; Leeson, C.; Zhi, X.; Leng, F.; Pierce, R.H.; Henry, M.S.; Rein, K.S. Characterization of an epoxide hydrolase from the Florida red tide dinoflagellate, Karenia brevis. Phytochemistry 2016, 122, 11–21. [Google Scholar] [CrossRef] [PubMed]
- van Loo, B.; Kingma, J.; Arand, M.; Wubbolts, M.G.; Janssen, D.B. Diversity and biocatalytic potential of epoxide hydrolases identified by genome analysis. Appl. Environ. Microbiol. 2006, 72, 2905–2917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeldin, D.C. Epoxygenase pathways of arachidonic acid metabolism. J. Biol. Chem. 2001, 276, 36059–36062. [Google Scholar] [CrossRef] [Green Version]
- Node, K.; Huo, Y.; Ruan, X.; Yang, B.; Spiecker, M.; Ley, K.; Zeldin, D.C.; Liao, J.K. Anti-inflammatory properties of cytochrome P450 epoxygenase-derived eicosanoids. Science (New York, N.Y.) 1999, 285, 1276–1279. [Google Scholar] [CrossRef] [Green Version]
- Morin, C.; Sirois, M.; Echave, V.; Gomes, M.M.; Rousseau, E. EET displays anti-inflammatory effects in TNF-alpha stimulated human bronchi: Putative role of CPI-17. Am. J. Respir. Cell Mol. Biol. 2008, 38, 192–201. [Google Scholar] [CrossRef]
- Morin, C.; Sirois, M.; Echave, V.; Albadine, R.; Rousseau, E. 17,18-epoxyeicosatetraenoic acid targets PPARgamma and p38 mitogen-activated protein kinase to mediate its anti-inflammatory effects in the lung: Role of soluble epoxide hydrolase. Am. J. Respir. Cell Mol. Biol. 2010, 43, 564–575. [Google Scholar] [CrossRef]
- Imig, J.D. Epoxyeicosanoids in hypertension. Physiol. Res. 2019, 68, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Gilroy, D.W.; Edin, M.L.; De Maeyer, R.P.; Bystrom, J.; Newson, J.; Lih, F.B.; Stables, M.; Zeldin, D.C.; Bishop-Bailey, D. CYP450-derived oxylipins mediate inflammatory resolution. Proc. Natl. Acad. Sci. USA 2016, 113, E3240–E3249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Panigrahy, D.; Kalish, B.T.; Huang, S.; Bielenberg, D.R.; Le, H.D.; Yang, J.; Edin, M.L.; Lee, C.R.; Benny, O.; Mudge, D.K.; et al. Epoxyeicosanoids promote organ and tissue regeneration. Proc. Natl. Acad. Sci. USA 2013, 110, 13528–13533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thompson, D.A.; Hammock, B.D. Dihydroxyoctadecamonoenoate esters inhibit the neutrophil respiratory burst. J. Biosci. 2007, 32, 279–291. [Google Scholar] [CrossRef]
- Spector, A.A. Arachidonic acid cytochrome P450 epoxygenase pathway. J. Lipid Res. 2009, 50, S52–S56. [Google Scholar] [CrossRef] [Green Version]
- Kramer, J.; Proschak, E. Phosphatase activity of soluble epoxide hydrolase. Prostaglandins Other Lipid Mediat. 2017, 133, 88–92. [Google Scholar] [CrossRef]
- Moghaddam, M.F.; Grant, D.F.; Cheek, J.M.; Greene, J.F.; Williamson, K.C.; Hammock, B.D. Bioactivation of leukotoxins to their toxic diols by epoxide hydrolase. Nat. Med. 1997, 3, 562–566. [Google Scholar] [CrossRef]
- Lee, K.S.; Liu, J.Y.; Wagner, K.M.; Pakhomova, S.; Dong, H.; Morisseau, C.; Fu, S.H.; Yang, J.; Wang, P.; Ulu, A.; et al. Optimized inhibitors of soluble epoxide hydrolase improve in vitro target residence time and in vivo efficacy. J. Med. Chem. 2014, 57, 7016–7030. [Google Scholar] [CrossRef]
- Li, D.; Liu, Y.; Zhang, X.; Lv, H.; Pang, W.; Sun, X.; Gan, L.M.; Hammock, B.D.; Ai, D.; Zhu, Y. Inhibition of soluble epoxide hydrolase alleviated atherosclerosis by reducing monocyte infiltration in Ldlr(-/-) mice. J. Mol. Cell. Cardiol. 2016, 98, 128–137. [Google Scholar] [CrossRef] [Green Version]
- Gurung, A.B.; Mayengbam, B.; Bhattacharjee, A. Discovery of novel drug candidates for inhibition of soluble epoxide hydrolase of arachidonic acid cascade pathway implicated in atherosclerosis. Comput. Biol. Chem. 2018, 74, 1–11. [Google Scholar] [CrossRef]
- Bettaieb, A.; Koike, S.; Chahed, S.; Zhao, Y.; Bachaalany, S.; Hashoush, N.; Graham, J.; Fatima, H.; Havel, P.J.; Gruzdev, A.; et al. Podocyte-specific soluble epoxide hydrolase deficiency in mice attenuates acute kidney injury. FEBS J. 2017, 284, 1970–1986. [Google Scholar] [CrossRef] [PubMed]
- Klocke, J.; Ulu, A.; Wu, K.; Rudolph, B.; Dragun, D.; Gollasch, M.; Schunck, W.H.; Hammock, B.D.; Riemekasten, G.; Enghard, P. Prophylactic inhibition of soluble epoxide hydrolase delays onset of nephritis and ameliorates kidney damage in NZB/W F1 mice. Sci. Rep. 2019, 9, 8993. [Google Scholar] [CrossRef] [Green Version]
- Chiang, C.W.; Lee, H.T.; Tarng, D.C.; Kuo, K.L.; Cheng, L.C.; Lee, T.S. Genetic deletion of soluble epoxide hydrolase attenuates inflammation and fibrosis in experimental obstructive nephropathy. Mediat. Inflamm 2015, 2015, 693260. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.P.; Yang, S.H.; Lee, H.Y.; Kim, B.; Cho, J.Y.; Paik, J.H.; Oh, Y.J.; Kim, D.K.; Lim, C.S.; Kim, Y.S. Soluble epoxide hydrolase activity determines the severity of ischemia-reperfusion injury in kidney. PLoS ONE 2012, 7, e37075. [Google Scholar] [CrossRef] [Green Version]
- Li, P.S.; Tao, W.; Yang, L.Q.; Shu, Y.S. Effect of Soluble Epoxide Hydrolase in Hyperoxic Acute Lung Injury in Mice. Inflammation 2018, 41, 1065–1072. [Google Scholar] [CrossRef]
- Tao, W.; Li, P.S.; Yang, L.Q.; Ma, Y.B. Effects of a Soluble Epoxide Hydrolase Inhibitor on Lipopolysaccharide-Induced Acute Lung Injury in Mice. PLoS ONE 2016, 11, e0160359. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.P.; Li, B.; Shuai, T.K.; Zhu, L.; Li, Y.M. Deletion of soluble epoxide hydrolase attenuates mice Hyperoxic acute lung injury. BMC Anesthesiol. 2018, 18, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Liu, T.; Duan, J.X.; Li, P.; Sun, G.Y.; Liu, Y.P.; Zhang, J.; Dong, L.; Lee, K.S.S.; Hammock, B.D.; et al. Soluble Epoxide Hydrolase Inhibitor Attenuates Lipopolysaccharide-Induced Acute Lung Injury and Improves Survival in Mice. Shock 2017, 47, 638–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Yang, J.; Zhang, J.; Wang, Y.; Hwang, S.H.; Qi, W.; Wan, D.; Kim, D.; Sun, J.; Sanidad, K.Z.; et al. Lipidomic profiling reveals soluble epoxide hydrolase as a therapeutic target of obesity-induced colonic inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, 5283–5288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Yang, A.L.; Liao, J.; Li, H.; Dong, H.; Chung, Y.T.; Bai, H.; Matkowskyj, K.A.; Hammock, B.D.; Yang, G.Y. Soluble epoxide hydrolase gene deficiency or inhibition attenuates chronic active inflammatory bowel disease in IL-10(-/-) mice. Dig. Dis. Sci. 2012, 57, 2580–2591. [Google Scholar] [CrossRef]
- Reisdorf, W.C.; Xie, Q.; Zeng, X.; Xie, W.; Rajpal, N.; Hoang, B.; Burgert, M.E.; Kumar, V.; Hurle, M.R.; Rajpal, D.K.; et al. Preclinical evaluation of EPHX2 inhibition as a novel treatment for inflammatory bowel disease. PLoS ONE 2019, 14, e0215033. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Kodani, S.; Hammock, B.D. Stabilized epoxygenated fatty acids regulate inflammation, pain, angiogenesis and cancer. Prog. Lipid Res. 2014, 53, 108–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, R.; Sun, L.; Liao, J.; Li, H.; You, X.; Xu, D.; Yang, J.; Hwang, S.H.; Jones, R.D.; Hammock, B.; et al. Inhibition of Pancreatic Carcinoma Growth Through Enhancing omega-3 Epoxy Polyunsaturated Fatty Acid Profile by Inhibition of Soluble Epoxide Hydrolase. Anticancer Res. 2019, 39, 3651–3660. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Cai, H.; Song, J.; Chang, Q. The effects of sEH inhibitor on depression-like behavior and neurogenesis in male mice. J. Neurosci. Res. 2017, 95, 2483–2492. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, K. Role of Soluble Epoxide Hydrolase in Metabolism of PUFAs in Psychiatric and Neurological Disorders. Front. Pharmacol. 2019, 10, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, Q. Soluble Epoxide Hydrolase Inhibitor: A Novel Potential Therapeutic or Prophylactic Drug for Psychiatric Disorders. Front. Pharmacol. 2019, 10, 420. [Google Scholar] [CrossRef]
- Hashimoto, K. Soluble epoxide hydrolase: A new therapeutic target for depression. Expert Opin. Ther. Targets 2016, 20, 1149–1151. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Tang, Y.; Yu, J.; Dong, R.; Yang, Y.; Fu, M.; Luo, J.; Hu, S.; Wang, D.W.; Tu, L.; et al. sEH Inhibitor TPPU Ameliorates Cecal Ligation and Puncture-Induced Sepsis by Regulating Macrophage Functions. Shock 2019, 52, 76–771. [Google Scholar] [CrossRef]
- Yang, L.; Cheriyan, J.; Gutterman, D.D.; Mayer, R.J.; Ament, Z.; Griffin, J.L.; Lazaar, A.L.; Newby, D.E.; Tal-Singer, R.; Wilkinson, I.B. Mechanisms of Vascular Dysfunction in COPD and Effects of a Novel Soluble Epoxide Hydrolase Inhibitor in Smokers. Chest 2017, 151, 555–563. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Dang, H.; Li, D.; Pang, W.; Hammock, B.D.; Zhu, Y. Inhibition of soluble epoxide hydrolase attenuates high-fat-diet-induced hepatic steatosis by reduced systemic inflammatory status in mice. PLoS ONE 2012, 7, e39165. [Google Scholar] [CrossRef] [Green Version]
- Bettaieb, A.; Nagata, N.; AbouBechara, D.; Chahed, S.; Morisseau, C.; Hammock, B.D.; Haj, F.G. Soluble epoxide hydrolase deficiency or inhibition attenuates diet-induced endoplasmic reticulum stress in liver and adipose tissue. J. Biol. Chem. 2013, 288, 14189–14199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez-Vicario, C.; Alcaraz-Quiles, J.; Garcia-Alonso, V.; Rius, B.; Hwang, S.H.; Titos, E.; Lopategi, A.; Hammock, B.D.; Arroyo, V.; Claria, J. Inhibition of soluble epoxide hydrolase modulates inflammation and autophagy in obese adipose tissue and liver: Role for omega-3 epoxides. Proc. Natl. Acad. Sci. USA 2015, 112, 536–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, C.C.; Zhang, C.Y.; Duan, J.X.; Guan, X.X.; Yang, H.H.; Jiang, H.L.; Hammock, B.D.; Hwang, S.H.; Zhou, Y.; Guan, C.X.; et al. PTUPB ameliorates high-fat diet-induced non-alcoholic fatty liver disease via inhibiting NLRP3 inflammasome activation in mice. Biochem. Biophys. Res. Commun. 2020, 523, 1020–1026. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.H.; Zheng, L.; Gui, L.; Lin, J.Y.; Zhu, Y.M.; Deng, W.S.; Luo, M. Soluble epoxide hydrolase inhibition with t-TUCB alleviates liver fibrosis and portal pressure in carbon tetrachloride-induced cirrhosis in rats. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 118–125. [Google Scholar] [CrossRef]
- Yao, L.; Cao, B.; Cheng, Q.; Cai, W.; Ye, C.; Liang, J.; Liu, W.; Tan, L.; Yan, M.; Li, B.; et al. Inhibition of soluble epoxide hydrolase ameliorates hyperhomocysteinemia-induced hepatic steatosis by enhancing beta-oxidation of fatty acid in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G527–G538. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Lazaar, A.L.; Yang, L.; Boardley, R.L.; Goyal, N.S.; Robertson, J.; Baldwin, S.J.; Newby, D.E.; Wilkinson, I.B.; Tal-Singer, R.; Mayer, R.J.; et al. Pharmacokinetics, pharmacodynamics and adverse event profile of GSK2256294, a novel soluble epoxide hydrolase inhibitor. Br. J. Clin. Pharmacol. 2016, 81, 971–979. [Google Scholar] [CrossRef] [Green Version]
- Iyer, A.; Kauter, K.; Alam, M.A.; Hwang, S.H.; Morisseau, C.; Hammock, B.D.; Brown, L. Pharmacological inhibition of soluble epoxide hydrolase ameliorates diet-induced metabolic syndrome in rats. Exp. Diabetes Res. 2012, 2012, 758614. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Xu, R.; Zhang, S.; Wang, Y.; Wang, P.; Edin, M.L.; Zeldin, D.C.; Wang, D.W. CYP2J2 overexpression attenuates nonalcoholic fatty liver disease induced by high-fat diet in mice. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E97–E110. [Google Scholar] [CrossRef] [Green Version]
- Mangels, N.; Awwad, K.; Wettenmann, A.; Dos Santos, L.R.; Fromel, T.; Fleming, I. The soluble epoxide hydrolase determines cholesterol homeostasis by regulating AMPK and SREBP activity. Prostaglandins Other Lipid. Mediat. 2016, 125, 30–39. [Google Scholar] [CrossRef]
- Harris, T.R.; Bettaieb, A.; Kodani, S.; Dong, H.; Myers, R.; Chiamvimonvat, N.; Haj, F.G.; Hammock, B.D. Inhibition of soluble epoxide hydrolase attenuates hepatic fibrosis and endoplasmic reticulum stress induced by carbon tetrachloride in mice. Toxicol. Appl. Pharmacol. 2015, 286, 102–111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deng, W.; Zhu, Y.; Lin, J.; Zheng, L.; Zhang, C.; Luo, M. Inhibition of soluble epoxide hydrolase lowers portal hypertension in cirrhotic rats by ameliorating endothelial dysfunction and liver fibrosis. Prostaglandins Other Lipid Mediat. 2017, 131, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Fife, K.L.; Liu, Y.; Schmelzer, K.R.; Tsai, H.J.; Kim, I.H.; Morisseau, C.; Hammock, B.D.; Kroetz, D.L. Inhibition of soluble epoxide hydrolase does not protect against endotoxin-mediated hepatic inflammation. J. Pharmacol. Exp. Ther. 2008, 327, 707–715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paschos, P.; Paletas, K. Non alcoholic fatty liver disease and metabolic syndrome. Hippokratia 2009, 13, 9–19. [Google Scholar] [PubMed]
- Maurice, J.; Manousou, P. Non-alcoholic fatty liver disease. Clin. Med. (Lond.) 2018, 18, 245–250. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A. Non-alcoholic fatty liver disease. BMC Med. 2017, 15, 45. [Google Scholar] [CrossRef] [Green Version]
- Hagstrom, H.; Elfwen, O.; Hultcrantz, R.; Stal, P. Steatohepatitis Is Not Associated with an Increased Risk for Fibrosis Progression in Nonalcoholic Fatty Liver Disease. Gastroenterol. Res. Pract. 2018, 2018, 1942648. [Google Scholar] [CrossRef] [Green Version]
- Armstrong, M.J.; Adams, L.A.; Canbay, A.; Syn, W.K. Extrahepatic complications of nonalcoholic fatty liver disease. Hepatology 2014, 59, 1174–1197. [Google Scholar] [CrossRef]
- Yao, L.; Wang, C.; Zhang, X.; Peng, L.; Liu, W.; Zhang, X.; Liu, Y.; He, J.; Jiang, C.; Ai, D.; et al. Hyperhomocysteinemia activates the aryl hydrocarbon receptor/CD36 pathway to promote hepatic steatosis in mice. Hepatology 2016, 64, 92–105. [Google Scholar] [CrossRef]
- Dai, H.; Wang, W.; Tang, X.; Chen, R.; Chen, Z.; Lu, Y.; Yuan, H. Association between homocysteine and non-alcoholic fatty liver disease in Chinese adults: A cross-sectional study. Nutr. J. 2016, 15, 102. [Google Scholar] [CrossRef] [Green Version]
- Luan, J.; Ju, D. Inflammasome: A Double-Edged Sword in Liver Diseases. Front. Immunol. 2018, 9, 2201. [Google Scholar] [CrossRef] [PubMed]
- Henkel, A.; Green, R.M. The unfolded protein response in fatty liver disease. Semin. Liver Dis. 2013, 33, 321–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebeaupin, C.; Vallee, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Komatsu, M. Autophagy in the liver: Functions in health and disease. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 170–184. [Google Scholar] [CrossRef] [PubMed]
- Grumati, P.; Dikic, I.; Stolz, A. ER-phagy at a glance. J. Cell. Sci. 2018, 131, jcs217364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, J.L.; Cuervo, A.M. Liver autophagy: Much more than just taking out the trash. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 187–200. [Google Scholar] [CrossRef] [Green Version]
- Kisseleva, T.; Brenner, D.A. Mechanisms of fibrogenesis. Exp. Biol. Med. (Maywood) 2008, 233, 109–122. [Google Scholar] [CrossRef]
- Iwaisako, K.; Jiang, C.; Zhang, M.; Cong, M.; Moore-Morris, T.J.; Park, T.J.; Liu, X.; Xu, J.; Wang, P.; Paik, Y.H.; et al. Origin of myofibroblasts in the fibrotic liver in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E3297–E3305. [Google Scholar] [CrossRef] [Green Version]
- El-Swefy, S.; Hassanen, S.I. Improvement of hepatic fibrosis by leukotriene inhibition in cholestatic rats. Ann. Hepatol. 2009, 8, 41–49. [Google Scholar] [CrossRef]
- Kim, S.M.; Park, K.C.; Kim, H.G.; Han, S.J. Effect of selective cyclooxygenase-2 inhibitor meloxicam on liver fibrosis in rats with ligated common bile ducts. Hepatol. Res. 2008, 38, 800–809. [Google Scholar] [CrossRef]
- Node, K.; Ruan, X.L.; Dai, J.; Yang, S.X.; Graham, L.; Zeldin, D.C.; Liao, J.K. Activation of Galpha s mediates induction of tissue-type plasminogen activator gene transcription by epoxyeicosatrienoic acids. J. Biol. Chem. 2001, 276, 15983–15989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gracia-Sancho, J.; Laleman, W. Mechanisms of portal hypertension: Bench to bedside. Clin. Liver Dis. (Hoboken) 2016, 8, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Rockey, D.C.; Chung, J.J. Reduced nitric oxide production by endothelial cells in cirrhotic rat liver: Endothelial dysfunction in portal hypertension. Gastroenterology 1998, 114, 344–351. [Google Scholar] [CrossRef]
- Hercule, H.C.; Schunck, W.H.; Gross, V.; Seringer, J.; Leung, F.P.; Weldon, S.M.; da Costa Goncalves, A.; Huang, Y.; Luft, F.C.; Gollasch, M. Interaction between P450 eicosanoids and nitric oxide in the control of arterial tone in mice. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 54–60. [Google Scholar] [CrossRef] [Green Version]
- Gotts, J.E.; Matthay, M.A. Sepsis: Pathophysiology and clinical management. BMJ 2016, 353, i1585. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, J.; Navasa, M.; Gomez, J.; Colmenero, J.; Vila, J.; Arroyo, V.; Rodes, J. Bacterial infections in cirrhosis: Epidemiological changes with invasive procedures and norfloxacin prophylaxis. Hepatology 2002, 35, 140–148. [Google Scholar] [CrossRef]
- Borzio, M.; Salerno, F.; Piantoni, L.; Cazzaniga, M.; Angeli, P.; Bissoli, F.; Boccia, S.; Colloredo-Mels, G.; Corigliano, P.; Fornaciari, G.; et al. Bacterial infection in patients with advanced cirrhosis: A multicentre prospective study. Dig. Liver Dis. 2001, 33, 41–48. [Google Scholar] [CrossRef]
- Schmelzer, K.R.; Kubala, L.; Newman, J.W.; Kim, I.H.; Eiserich, J.P.; Hammock, B.D. Soluble epoxide hydrolase is a therapeutic target for acute inflammation. Proc. Natl. Acad. Sci. USA 2005, 102, 9772–9777. [Google Scholar] [CrossRef] [Green Version]
- Buras, J.A.; Holzmann, B.; Sitkovsky, M. Animal models of sepsis: Setting the stage. Nat. Rev. Drug Discov. 2005, 4, 854–865. [Google Scholar] [CrossRef]
- Wang, W.; Yang, J.; Edin, M.L.; Wang, Y.; Luo, Y.; Wan, D.; Yang, H.; Song, C.Q.; Xue, W.; Sanidad, K.Z.; et al. Targeted Metabolomics Identifies the Cytochrome P450 Monooxygenase Eicosanoid Pathway as a Novel Therapeutic Target of Colon Tumorigenesis. Cancer Res. 2019, 79, 1822–1830. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, Y.; Schmelzer, K.; Lee, T.S.; Fang, X.; Zhu, Y.; Spector, A.A.; Gill, S.; Morisseau, C.; Hammock, B.D.; et al. The antiinflammatory effect of laminar flow: The role of PPARgamma, epoxyeicosatrienoic acids, and soluble epoxide hydrolase. Proc. Natl. Acad. Sci. USA 2005, 102, 16747–16752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liss, K.H.; Finck, B.N. PPARs and nonalcoholic fatty liver disease. Biochimie 2017, 136, 65–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, D.Y.; Davis, B.B.; Wang, Z.H.; Zhao, S.P.; Wasti, B.; Liu, Z.L.; Li, N.; Morisseau, C.; Chiamvimonvat, N.; Hammock, B.D. A potent soluble epoxide hydrolase inhibitor, t-AUCB, acts through PPARgamma to modulate the function of endothelial progenitor cells from patients with acute myocardial infarction. Int. J. Cardiol. 2013, 167, 1298–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hildreth, K.; Overby, H.; Kodani, S.; Morisseau, C.; Hammock, B.; Bettaieb, A.; Zhao, L. Soluble Epoxide Hydrolase Inhibitor t-AUCB Promotes Murine Brown Adipogenesis: Role of PPAR Gamma and PPAR Alpha (P21-069-19). Curr. Dev. Nutr. 2019, 3, nzz041.P21-069-19. [Google Scholar] [CrossRef] [Green Version]
- Dai, N.; Zhao, C.; Kong, Q.; Li, D.; Cai, Z.; Wang, M. Vascular repair and anti-inflammatory effects of soluble epoxide hydrolase inhibitor. Exp. Ther Med. 2019, 17, 3580–3588. [Google Scholar] [CrossRef] [Green Version]
- Fang, X.; Hu, S.; Xu, B.; Snyder, G.D.; Harmon, S.; Yao, J.; Liu, Y.; Sangras, B.; Falck, J.R.; Weintraub, N.L.; et al. 14,15-Dihydroxyeicosatrienoic acid activates peroxisome proliferator-activated receptor-alpha. Am. J. Physiol. Heart. Circ. Physiol. 2006, 290, H55–H63. [Google Scholar] [CrossRef]
- Fang, X.; Hu, S.; Watanabe, T.; Weintraub, N.L.; Snyder, G.D.; Yao, J.; Liu, Y.; Shyy, J.Y.; Hammock, B.D.; Spector, A.A. Activation of peroxisome proliferator-activated receptor alpha by substituted urea-derived soluble epoxide hydrolase inhibitors. J. Pharmacol. Exp. Ther. 2005, 314, 260–270. [Google Scholar] [CrossRef] [Green Version]
- Khadge, S.; Sharp, J.G.; Thiele, G.M.; McGuire, T.R.; Klassen, L.W.; Duryee, M.J.; Britton, H.C.; Dafferner, A.J.; Beck, J.; Black, P.N.; et al. Dietary omega-3 and omega-6 polyunsaturated fatty acids modulate hepatic pathology. J. Nutr. Biochem. 2018, 52, 92–102. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef]
- Koerner, I.P.; Zhang, W.; Cheng, J.; Parker, S.; Hurn, P.D.; Alkayed, N.J. Soluble epoxide hydrolase: Regulation by estrogen and role in the inflammatory response to cerebral ischemia. Front. Biosci. 2008, 13, 2833–2841. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.M.; Sun, D.; Kandhi, S.; Froogh, G.; Zhuge, J.; Huang, W.; Hammock, B.D.; Huang, A. Estrogen-dependent epigenetic regulation of soluble epoxide hydrolase via DNA methylation. Proc. Natl. Acad. Sci. USA 2018, 115, 613–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, A.; Sun, D. Sexually Dimorphic Regulation of EET Synthesis and Metabolism: Roles of Estrogen. Front. Pharmacol. 2018, 9, 1222. [Google Scholar] [CrossRef] [PubMed]
- Osna, N.A.; Donohue, T.M., Jr.; Kharbanda, K.K. Alcoholic Liver Disease: Pathogenesis and Current Management. Alcohol Res. 2017, 38, 147–161. [Google Scholar] [PubMed]
- Hirschfield, G.M.; Heathcote, E.J.; Gershwin, M.E. Pathogenesis of cholestatic liver disease and therapeutic approaches. Gastroenterology 2010, 139, 1481–1496. [Google Scholar] [CrossRef] [PubMed]
- Rocken, C.; Carl-McGrath, S. Pathology and pathogenesis of hepatocellular carcinoma. Dig. Dis. 2001, 19, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Przybyla-Zawislak, B.D.; Srivastava, P.K.; Vazquez-Matias, J.; Mohrenweiser, H.W.; Maxwell, J.E.; Hammock, B.D.; Bradbury, J.A.; Enayetallah, A.E.; Zeldin, D.C.; Grant, D.F. Polymorphisms in human soluble epoxide hydrolase. Mol. Pharmacol. 2003, 64, 482–490. [Google Scholar] [CrossRef] [Green Version]
- Morisseau, C.; Wecksler, A.T.; Deng, C.; Dong, H.; Yang, J.; Lee, K.S.; Kodani, S.D.; Hammock, B.D. Effect of soluble epoxide hydrolase polymorphism on substrate and inhibitor selectivity and dimer formation. J. Lipid Res. 2014, 55, 1131–1138. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, C.E.; Shuey, M.M.; Milne, G.L.; Gilbert, K.; Hui, N.; Yu, C.; Luther, J.M.; Brown, N.J. Arg287Gln variant of EPHX2 and epoxyeicosatrienoic acids are associated with insulin sensitivity in humans. Prostaglandins Other Lipid Mediat. 2014, 113–115, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Fornage, M.; Boerwinkle, E.; Doris, P.A.; Jacobs, D.; Liu, K.; Wong, N.D. Polymorphism of the soluble epoxide hydrolase is associated with coronary artery calcification in African-American subjects: The Coronary Artery Risk Development in Young Adults (CARDIA) study. Circulation 2004, 109, 335–339. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.R.; Pretorius, M.; Schuck, R.N.; Burch, L.H.; Bartlett, J.; Williams, S.M.; Zeldin, D.C.; Brown, N.J. Genetic variation in soluble epoxide hydrolase (EPHX2) is associated with forearm vasodilator responses in humans. Hypertension 2011, 57, 116–122. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
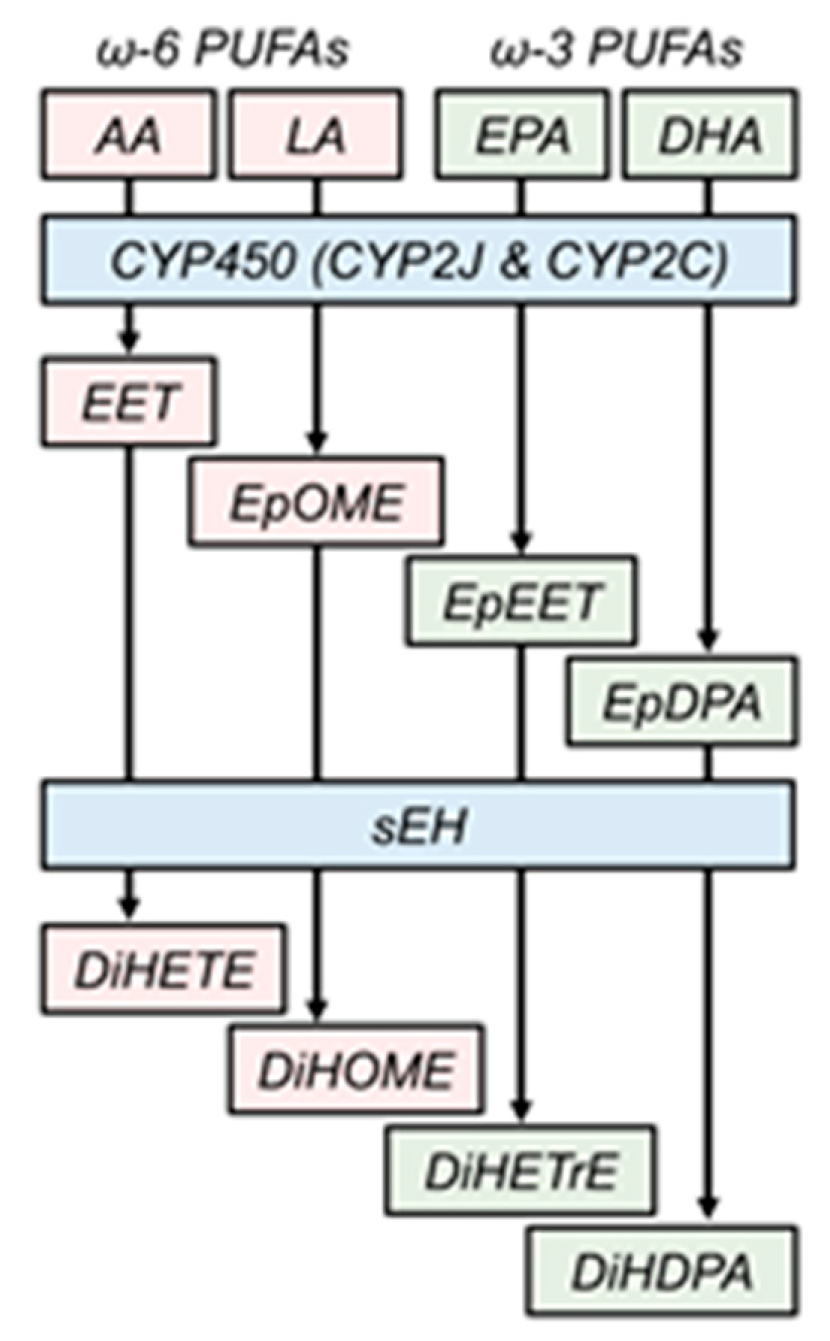{kind=link}
{kind=link}
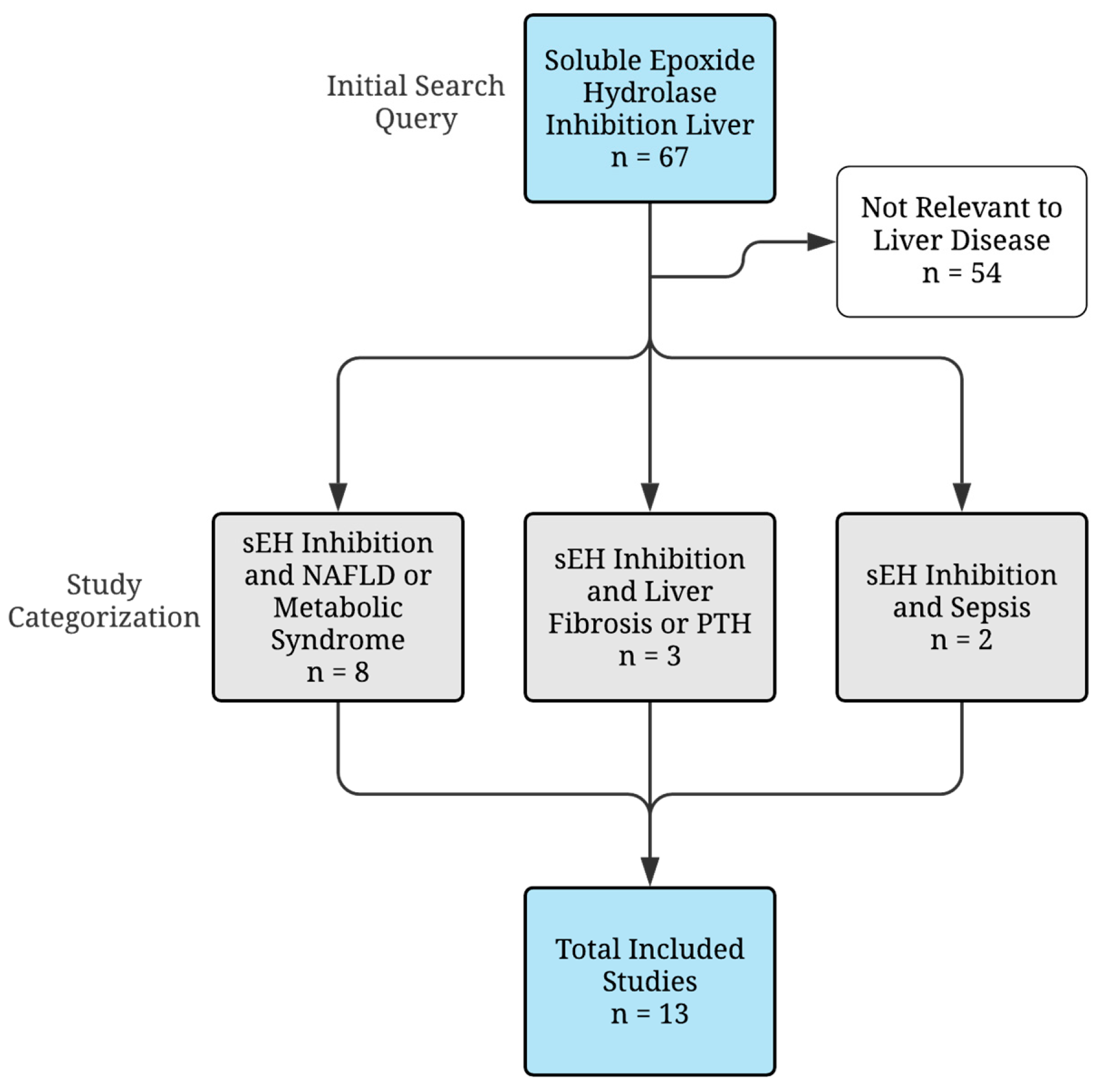{kind=link}
{kind=link}
| Authors [Ref] | Inhibitor | Disease | Model | Result | Molecular Mechanisms | |
|---|---|---|---|---|---|---|
| In Vivo | In Vitro | |||||
| Iyer et al. [48] | t-AUCB | NAFLD | HFHC Diet, Rat | ↓ Insulin Resistance ↓ Hypertension ↓ Steatosis ↓ Liver Hypertrophy | ↓ Cholesterol ↓ GTT Glucose AUC | N/A |
| Liu et al. [40] | t-AUCB | NAFLD | HFD, Mouse | ↓ Steatosis ↑ sEH Activity | ↓ Plasma Inflammatory Cytokines ↓ Adipose Macrophage Infiltration | N/A |
| Bettaieb et al. [41] | TUPS | NAFLD | HFD, Mouse | ↓ Hepatic/Adipose ER Stress ↓ Cell Death, in vitro ↑ Insulin Signaling, in vitro ↑ sEH Protein | ↓ BiP, XBP1, CHOP ↓ Caspase 3, cJUN, JNK, p38 | EpOMEs and EETs in HepG2 cells: ↑ phospho-IR, phospho-AKT |
| Lopez-Vicario et al. [42] | t-TUCB | NAFLD | HFD, Mouse | ↑ Brown Fat ↑ Hepatic Autophagy ↓ Steatosis ↑ sEH Protein | ↑ IL10, RELMα, CD206, MGL1 ↑ M2 Polarization | 14,15-EET, 19,20-EpDPA, and 17,18-EpETE in Primary Hepatocytes: ↓ Lipid accumulation ↓ phospho-eIF2α, phospho-IRE1α ↑ LC3II:LC3I ratio |
| Sun et al. [43] | PTUPB | NAFLD | HFD, Mouse | ↓ Body/Liver Weight ↓ Liver Injury and Steatosis ↓ Fibrosis ↓ Inflammation ↑ sEH Protein | ↓ NLRP3 Inflammasome Activation ↓ Inflammatory Cytokines ↓ COX2 Expression | N/A |
| Chen et al. [49] | N/A | NAFLD | HFD, Mouse | ↓ Steatosis ↓ Inflammation ↓ Oxidative Stress | ↓ NFκB ↓ JNK ↑ SOD, GPX | 14,15-EET in HepG2 cells: ↓ NFκB, TNFα, IL1β, IL6 14,15-EET in RAW264.7 cells: ↓ TNFα, IL1β, IL6 |
| Yao et al. [45] | TPPU | NAFLD | HMD, Mouse | ↓ Steatosis ↑ sEH Protein | ↑ Fatty Acid β-Oxidation Genes ↑ PPARα Activation | sEH Inhibition and 11,12-EET in Primary Hepatocytes: ↑ PPARα Activation |
| Mangels et al. [50] | t-AUCB | Metabolic Syndrome | Mouse | ↓ Cholesterol | ↑ AMPK Activation ↓ SREBP1 ↓ HMG CoA Reductase | 12,13-EpOME in vitro: ↑ phospho-AMPK ↓ HMG CoA Reductase |
| Harris et al. [51] | TPPU | Liver Fibrosis | CCl4, Mouse | ↓ Fibrosis ↓ ER Stress | ↑ Metalloproteases ↓ Col1a2/Col3a1 mRNA ↓ JNK, Caspase 3 | N/A |
| Zhang et al. [44] | t-TUCB | Liver Fibrosis | CCl4, Rat | ↓ Fibrosis ↓ Portal Hypertension ↓ Inflammation ↓ Oxidative Stress ↑ sEH Protein | ↓ TGFb ↓ Smad ↓ NFkB ↑ Metalloproteases ↑ SOD, GSH | N/A |
| Deng et al. [52] | t-TUCB | Portal Hypertension | CCl4, Rat | ↓ Portal Pressure ↓ Liver Fibrosis ↓ Liver Endothelial Dysfunction ↑ sEH Protein | ↑ p-eNOS ↑ NO ↓ Caveolin 1 ↓ NFkB | N/A |
| Fife et al. [53] | AUDA | Sepsis | LPS, Mouse | n.s. Inflammation ↑ sEH Activity by Lipidome | ↓ iNOS | N/A |
| Chen et al. [38] | TPPU | Sepsis | Cecal Ligation, Puncture, Mouse | ↑ Survival ↓ Organ Damage ↓ Systemic Inflammation | ↑ MAPK Signaling ↑ Macrophage Phagocytosis ↓ Inflammatory Cytokines ↓ ALT/AST, BUN ↓ Bacterial CFU’s | 14,15-EET in vitro: ↓ TNFα, IL1β, IL6 ↑ IL10 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Warner, J.; Hardesty, J.; Zirnheld, K.; McClain, C.; Warner, D.; Kirpich, I. Soluble Epoxide Hydrolase Inhibition in Liver Diseases: A Review of Current Research and Knowledge Gaps. Biology 2020, 9, 124. https://doi.org/10.3390/biology9060124
Warner J, Hardesty J, Zirnheld K, McClain C, Warner D, Kirpich I. Soluble Epoxide Hydrolase Inhibition in Liver Diseases: A Review of Current Research and Knowledge Gaps. Biology. 2020; 9(6):124. https://doi.org/10.3390/biology9060124
Chicago/Turabian StyleWarner, Jeffrey, Josiah Hardesty, Kara Zirnheld, Craig McClain, Dennis Warner, and Irina Kirpich. 2020. "Soluble Epoxide Hydrolase Inhibition in Liver Diseases: A Review of Current Research and Knowledge Gaps" Biology 9, no. 6: 124. https://doi.org/10.3390/biology9060124
APA StyleWarner, J., Hardesty, J., Zirnheld, K., McClain, C., Warner, D., & Kirpich, I. (2020). Soluble Epoxide Hydrolase Inhibition in Liver Diseases: A Review of Current Research and Knowledge Gaps. Biology, 9(6), 124. https://doi.org/10.3390/biology9060124