
Secondary Mechanisms of Neurotrauma: A Closer Look at the Evidence

 , and
, and 
Abstract
:1. Introduction
2. ER Stress
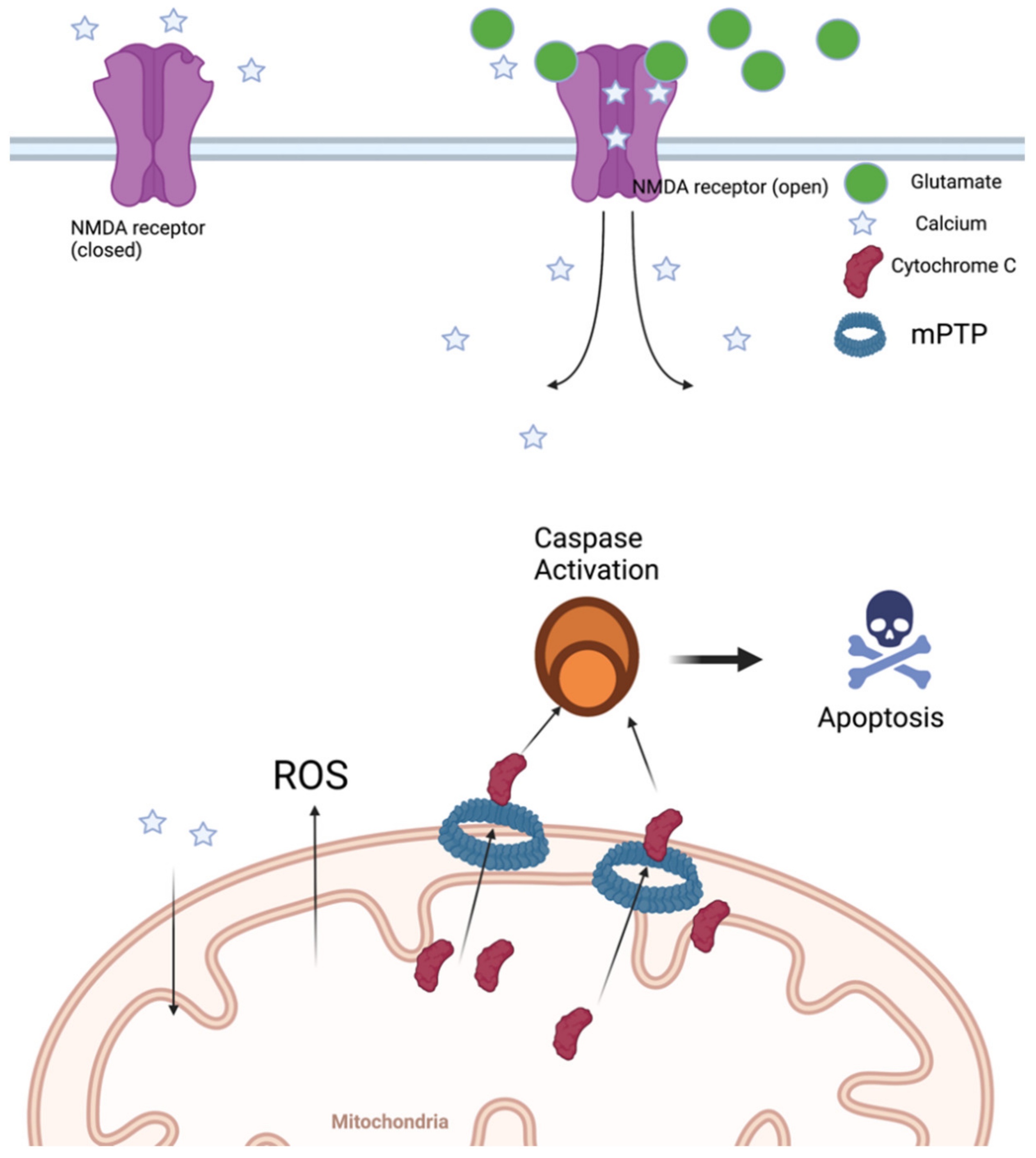
3. Mitochondrial Dysfunction
4. Oxidative Stress
5. Emerging Treatments
5.1. ER Stress
5.2. Mitochondrial Dysfunction
5.3. Antioxidant Therapy
5.4. Immunoglobulin
5.5. Cell-Based Therapy
5.6. MSC-Exosomes
5.7. CCR5 Antagonists
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Smith, C. Neurotrauma. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; Volume 145, pp. 115–132. ISBN 978-0-12-802395-2. [Google Scholar]
- Menon, D.K.; Schwab, K.; Wright, D.W.; Maas, A.I. Position Statement: Definition of Traumatic Brain Injury. Arch. Phys. Med. Rehabil. 2010, 91, 1637–1640. [Google Scholar] [CrossRef] [PubMed]
- Chang, W.-T.W.; Badjatia, N. Neurotrauma. Emerg. Med. Clin. N. Am. 2014, 32, 889–905. [Google Scholar] [CrossRef] [PubMed]
- Kempuraj, D.; Thangavel, R.; Kempuraj, D.D.; Ahmed, M.E.; Selvakumar, G.P.; Raikwar, S.P.; Zaheer, S.A.; Iyer, S.S.; Govindarajan, R.; Chandrasekaran, P.N.; et al. Neuroprotective effects of flavone luteolin in neuroinflammation and neurotrauma. BioFactors 2021, 47, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Long, B.; Koyfman, A. Secondary Gains. Emerg. Med. Clin. N. Am. 2018, 36, 107–133. [Google Scholar] [CrossRef]
- Swadron, S.P.; LeRoux, P.; Smith, W.S.; Weingart, S.D. Emergency Neurological Life Support: Traumatic Brain Injury. Neurocrit. Care 2012, 17, 112–121. [Google Scholar] [CrossRef]
- Smith, D.H.; Hicks, R.; Povlishock, J.T. Therapy development for diffuse axonal injury. J. Neurotrauma 2013, 30, 307–323. [Google Scholar] [CrossRef] [Green Version]
- Oakes, S.A.; Papa, F.R. The Role of Endoplasmic Reticulum Stress in Human Pathology. Annu. Rev. Pathol. Mech. Dis. 2015, 10, 173–194. [Google Scholar] [CrossRef] [Green Version]
- Shi, M.; Chai, Y.; Zhang, J.; Chen, X. Endoplasmic Reticulum Stress-Associated Neuronal Death and Innate Immune Response in Neurological Diseases. Front. Immunol. 2022, 12, 794580. [Google Scholar] [CrossRef]
- Curry, D.J.; Wright, D.A.; Lee, R.C.; Kang, U.J.; Frim, D.M. Surfactant poloxamer 188—Related decreases in inflammation and tissue damage after experimental brain injury in rats. J. Neurosurg. Pediatr. 2004, 101, 91–96. [Google Scholar] [CrossRef] [Green Version]
- McAllister, T.W. Neurobiological consequences of traumatic brain injury. Dialogues Clin. Neurosci. 2011, 13, 287–300. [Google Scholar] [CrossRef]
- Mietto, B.S.; Mostacada, K.; Martinez, A.M.B. Neurotrauma and Inflammation: CNS and PNS Responses. Mediat. Inflamm. 2015, 2015, 251204. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. From endoplasmic-reticulum stress to the inflammatory response. Nature 2008, 454, 455–462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centers for Disease Control and Prevention (CDC). CDC grand rounds: Reducing severe traumatic brain injury in the United States. MMWR Morb. Mortal. Wkly. Rep. 2013, 62, 549–552. [Google Scholar]
- Osier, N.D.; Carlson, S.W.; DeSana, A.; Dixon, C.E. Chronic Histopathological and Behavioral Outcomes of Experimental Traumatic Brain Injury in Adult Male Animals. J. Neurotrauma 2015, 32, 1861–1882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, H.S.; Meyers, C.A.; Grossman, R.G.; Sarwar, M. Ventricular Enlargement after Closed Head Injury. Arch. Neurol. 1981, 38, 623–629. [Google Scholar] [CrossRef]
- Meyers, C.A.; Levin, H.S.; Eisenberg, H.M.; Guinto, F.C. Early versus late lateral ventricular enlargement following closed head injury. J. Neurol. Neurosurg. Psychiatry 1983, 46, 1092–1097. [Google Scholar] [CrossRef] [Green Version]
- Johnson, S.C.; Bigler, E.D.; Burr, R.B.; Blatter, D.D. White matter atrophy, ventricular dilation, and intellectual functioning following traumatic brain injury. Neuropsychology 1994, 8, 307–315. [Google Scholar] [CrossRef]
- Knoblach, S.M.; Alroy, D.A.; Nikolaeva, M.; Cernak, I.; Stoica, B.A.; Faden, A.I. Caspase Inhibitor z-DEVD-fmk Attenuates Calpain and Necrotic Cell Death in Vitro and after Traumatic Brain Injury. J. Cereb. Blood Flow Metab. 2004, 24, 1119–1132. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Chen, L.; Gao, X.; Luo, B.; Chen, J. Moderate Traumatic Brain Injury Triggers Rapid Necrotic Death of Immature Neurons in the Hippocampus. J. Neuropathol. Exp. Neurol. 2012, 71, 348–359. [Google Scholar] [CrossRef] [Green Version]
- Fox, G.B.; Fan, L.; Levasseur, R.A.; Faden, A.I. Sustained Sensory/Motor and Cognitive Deficits With Neuronal Apoptosis Following Controlled Cortical Impact Brain Injury in the Mouse. J. Neurotrauma 1998, 15, 599–614. [Google Scholar] [CrossRef]
- Chen, S.-F.; Richards, H.K.; Smielewski, P.; Johnström, P.; Salvador, R.; Pickard, J.D.; Harris, N.G. Relationship between Flow-Metabolism Uncoupling and Evolving Axonal Injury after Experimental Traumatic Brain Injury. J. Cereb. Blood Flow Metab. 2004, 24, 1025–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, E.D.; Sullivan, P.G.; Gibson, T.R.; Pavel, K.M.; Thompson, B.M.; Scheff, S.W. Spatial and Temporal Characteristics of Neurodegeneration after Controlled Cortical Impact in Mice: More than a Focal Brain Injury. J. Neurotrauma 2005, 22, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Lyman, M.; Lloyd, D.G.; Ji, X.; Vizcaychipi, M.P.; Ma, D. Neuroinflammation: The role and consequences. Neurosci. Res. 2014, 79, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Katsnelson, A.; De Strooper, B.; Zoghbi, H.Y. Neurodegeneration: From cellular concepts to clinical applications. Sci. Transl. Med. 2016, 8, 364ps18. [Google Scholar] [CrossRef]
- Ismail, H.; Shakkour, Z.; Tabet, M.; Abdelhady, S.; Kobaisi, A.; Abedi, R.; Nasrallah, L.; Pintus, G.; Al-Dhaheri, Y.; Mondello, S.; et al. Traumatic Brain Injury: Oxidative Stress and Novel Anti-Oxidants Such as Mitoquinone and Edaravone. Antioxidants 2020, 9, 943. [Google Scholar] [CrossRef]
- Iurlaro, R.; Muñoz-Pinedo, C. Cell death induced by endoplasmic reticulum stress. FEBS J. 2016, 283, 2640–2652. [Google Scholar] [CrossRef] [Green Version]
- Deslauriers, A.M.; Afkhami-Goli, A.; Paul, A.M.; Bhat, R.K.; Acharjee, S.; Ellestad, K.K.; Noorbakhsh, F.; Michalak, M.; Power, C. Neuroinflammation and Endoplasmic Reticulum Stress Are Coregulated by Crocin To Prevent Demyelination and Neurodegeneration. J. Immunol. 2011, 187, 4788–4799. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Huang, Y.; Cheng, Y.; Tan, Y.; Wu, F.; Wu, J.; Shi, H.; Zhang, H.; Yu, X.; Gao, H.; et al. Endoplasmic reticulum stress-induced neuronal inflammatory response and apoptosis likely plays a key role in the development of diabetic encephalopathy. Oncotarget 2016, 7, 78455–78472. [Google Scholar] [CrossRef] [Green Version]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Bailes, J.E.; Huber, J.D.; Rosen, C.L. Linking Traumatic Brain Injury to Chronic Traumatic Encephalopathy: Identification of Potential Mechanisms Leading to Neurofibrillary Tangle Development. J. Neurotrauma 2014, 31, 1129–1138. [Google Scholar] [CrossRef] [Green Version]
- Read, A.; Schröder, M. The Unfolded Protein Response: An Overview. Biology 2021, 10, 384. [Google Scholar] [CrossRef]
- Cao, S.S.; Kaufman, R.J. Unfolded protein response. Curr. Biol. 2012, 22, R622–R626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, M.; Wilkinson, S. ER homeostasis and autophagy. Essays Biochem. 2017, 61, 625–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindholm, D.; Wootz, H.; Korhonen, L. ER stress and neurodegenerative diseases. Cell Death Differ. 2006, 13, 385–392. [Google Scholar] [CrossRef]
- Larner, S.F.; Hayes, R.L.; Wang, K.K.W. Unfolded Protein Response after Neurotrauma. J. Neurotrauma 2006, 23, 807–829. [Google Scholar] [CrossRef] [PubMed]
- Paschen, W.; Mengesdorf, T. Endoplasmic reticulum stress response and neurodegeneration. Cell Calcium 2005, 38, 409–415. [Google Scholar] [CrossRef]
- Schröder, M.; Kaufman, R.J. The Mammalian Unfolded Protein Response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Paschen, W.; Frandsen, A. Endoplasmic reticulum dysfunction—A common denominator for cell injury in acute and degenerative diseases of the brain?: ER dysfunction and neurodegeneration. J. Neurochem. 2008, 79, 719–725. [Google Scholar] [CrossRef]
- Breckenridge, D.G.; Germain, M.; Mathai, J.P.; Nguyen, M.; Shore, G.C. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene 2003, 22, 8608–8618. [Google Scholar] [CrossRef] [Green Version]
- Sokka, A.-L.; Putkonen, N.; Mudo, G.; Pryazhnikov, E.; Reijonen, S.; Khiroug, L.; Belluardo, N.; Lindholm, D.; Korhonen, L. Endoplasmic Reticulum Stress Inhibition Protects against Excitotoxic Neuronal Injury in the Rat Brain. J. Neurosci. 2007, 27, 901–908. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wang, J.; Gao, X.; Wu, Y.; Gu, G.; Shi, M.; Chai, Y.; Yue, S.; Zhang, J. Tauroursodeoxycholic acid prevents ER stress-induced apoptosis and improves cerebral and vascular function in mice subjected to subarachnoid hemorrhage. Brain Res. 2020, 1727, 146566. [Google Scholar] [CrossRef]
- Bence, N.F.; Sampat, R.M.; Kopito, R.R. Impairment of the ubiquitin-proteasome system by protein aggregation. Science 2001, 292, 1552–1555. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Soda, M.; Inoue, H.; Hattori, N.; Mizuno, Y.; Takahashi, R. An Unfolded Putative Transmembrane Polypeptide, which Can Lead to Endoplasmic Reticulum Stress, Is a Substrate of Parkin. Cell 2001, 105, 891–902. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Zhao, Z.; Kumar, A.; Lipinski, M.M.; Loane, D.J.; Stoica, B.A.; Faden, A.I. Endoplasmic Reticulum Stress and Disrupted Neurogenesis in the Brain Are Associated with Cognitive Impairment and Depressive-Like Behavior after Spinal Cord Injury. J. Neurotrauma 2016, 33, 1919–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A. Physiology and Pathophysiology of the Calcium Store in the Endoplasmic Reticulum of Neurons. Physiol. Rev. 2005, 85, 201–279. [Google Scholar] [CrossRef] [PubMed]
- LaFerla, F.M. Calcium dyshomeostasis and intracellular signalling in Alzheimer’s disease. Nat. Rev. Neurosci. 2002, 3, 862–872. [Google Scholar] [CrossRef]
- Katayama, T.; Imaizumi, K.; Manabe, T.; Hitomi, J.; Kudo, T.; Tohyama, M. Induction of neuronal death by ER stress in Alzheimer’s disease. J. Chem. Neuroanat. 2004, 28, 67–78. [Google Scholar] [CrossRef]
- Lucke-Wold, B.P.; Turner, R.C.; Logsdon, A.F.; Nguyen, L.; Bailes, J.E.; Lee, J.M.; Robson, M.J.; Omalu, B.I.; Huber, J.D.; Rosen, C.L. Endoplasmic reticulum stress implicated in chronic traumatic encephalopathy. J. Neurosurg. 2016, 124, 687–702. [Google Scholar] [CrossRef] [Green Version]
- Larner, S.F.; Hayes, R.L.; McKinsey, D.M.; Pike, B.R.; Wang, K.K.W. Increased expression and processing of caspase-12 after traumatic brain injury in rats: Caspase-12 induction after TBI. J. Neurochem. 2003, 88, 78–90. [Google Scholar] [CrossRef]
- Stutzbach, L.D.; Xie, S.X.; Naj, A.C.; Albin, R.; Gilman, S.; PSP Genetics Study Group; Lee, V.M.Y.; Trojanowski, J.Q.; Devlin, B.; Schellenberg, G.D. The unfolded protein response is activated in disease-affected brain regions in progressive supranuclear palsy and Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 31. [Google Scholar] [CrossRef] [Green Version]
- Nishitoh, H.; Kadowaki, H.; Nagai, A.; Maruyama, T.; Yokota, T.; Fukutomi, H.; Noguchi, T.; Matsuzawa, A.; Takeda, K.; Ichijo, H. ALS-linked mutant SOD1 induces ER stress- and ASK1-dependent motor neuron death by targeting Derlin-1. Genes Dev. 2008, 22, 1451–1464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kikuchi, H.; Almer, G.; Yamashita, S.; Guegan, C.; Nagai, M.; Xu, Z.; Sosunov, A.A.; McKhann, G.M.; Przedborski, S. Spinal cord endoplasmic reticulum stress associated with a microsomal accumulation of mutant superoxide dismutase-1 in an ALS model. Proc. Natl. Acad. Sci. USA 2006, 103, 6025–6030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, T.; Trinh, M.A.; Wexler, A.J.; Bourbon, C.; Gatti, E.; Pierre, P.; Cavener, D.R.; Klann, E. Suppression of eIF2α kinases alleviates Alzheimer’s disease–related plasticity and memory deficits. Nat. Neurosci. 2013, 16, 1299–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angelova, P.R.; Abramov, A.Y. Role of mitochondrial ROS in the brain: From physiology to neurodegeneration. FEBS Lett. 2018, 592, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.-R.; Zhang, X.-N.; Chen, Z. Mitochondrial transport serves as a mitochondrial quality control strategy in axons: Implications for central nervous system disorders. CNS Neurosci. Ther. 2019, 25, 876–886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, T.; Vespa, P.M.; Prins, M.L. Alternative substrate metabolism depends on cerebral metabolic state following traumatic brain injury. Exp. Neurol. 2020, 329, 113289. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Highton, D.; Kolyva, C.; Tachtsidis, I.; Elwell, C.E.; Smith, M. Hyperoxia results in increased aerobic metabolism following acute brain injury. J. Cereb. Blood Flow Metab. 2017, 37, 2910–2920. [Google Scholar] [CrossRef]
- Stefani, M.A.; Modkovski, R.; Hansel, G.; Zimmer, E.R.; Kopczynski, A.; Muller, A.P.; Strogulski, N.R.; Rodolphi, M.S.; Carteri, R.K.; Schmidt, A.P.; et al. Elevated glutamate and lactate predict brain death after severe head trauma. Ann. Clin. Transl. Neurol. 2017, 4, 392–402. [Google Scholar] [CrossRef]
- Liao, R.; Wood, T.R.; Nance, E. Nanotherapeutic modulation of excitotoxicity and oxidative stress in acute brain injury. Nanobiomedicine 2020, 7, 1849543520970819. [Google Scholar] [CrossRef]
- Zhang, L.; Wang, H.; Zhou, X.; Mao, L.; Ding, K.; Hu, Z. Role of mitochondrial calcium uniporter-mediated Ca2+ and iron accumulation in traumatic brain injury. J. Cell. Mol. Med. 2019, 23, 2995–3009. [Google Scholar] [CrossRef] [Green Version]
- Vespa, P.; Bergsneider, M.; Hattori, N.; Wu, H.-M.; Huang, S.-C.; Martin, N.A.; Glenn, T.C.; McArthur, D.L.; Hovda, D.A. Metabolic Crisis without Brain Ischemia is Common after Traumatic Brain Injury: A Combined Microdialysis and Positron Emission Tomography Study. J. Cereb. Blood Flow Metab. 2005, 25, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carvajal, F.J.; Mattison, H.A.; Cerpa, W. Role of NMDA Receptor-Mediated Glutamatergic Signaling in Chronic and Acute Neuropathologies. Neural Plast. 2016, 2016, e2701526. [Google Scholar] [CrossRef] [PubMed]
- Luo, P.; Li, X.; Wu, X.; Dai, S.; Yang, Y.; Xu, H.; Jing, D.; Rao, W.; Xu, H.; Gao, X.; et al. Preso regulates NMDA receptor-mediated excitotoxicity via modulating nitric oxide and calcium responses after traumatic brain injury. Cell Death Dis. 2019, 10, 496. [Google Scholar] [CrossRef] [PubMed]
- Kumar Sahel, D.; Kaira, M.; Raj, K.; Sharma, S.; Singh, S. Mitochondrial dysfunctioning and neuroinflammation: Recent highlights on the possible mechanisms involved in Traumatic Brain Injury. Neurosci. Lett. 2019, 710, 134347. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Chapter 11—Excitotoxicity. In Stress: Physiology, Biochemistry, and Pathology; Fink, G., Ed.; Academic Press: New York, NY, USA, 2019; pp. 125–134. ISBN 978-0-12-813146-6. [Google Scholar]
- Giorgi, C.; Baldassari, F.; Bononi, A.; Bonora, M.; De Marchi, E.; Marchi, S.; Missiroli, S.; Patergnani, S.; Rimessi, A.; Suski, J.M.; et al. Mitochondrial Ca2+ and apoptosis. Cell Calcium 2012, 52, 36–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millet, A.; Bouzat, P.; Trouve-Buisson, T.; Batandier, C.; Pernet-Gallay, K.; Gaide-Chevronnay, L.; Barbier, E.L.; Debillon, T.; Fontaine, E.; Payen, J.-F. Erythropoietin and Its Derivates Modulate Mitochondrial Dysfunction after Diffuse Traumatic Brain Injury. J. Neurotrauma 2016, 33, 1625–1633. [Google Scholar] [CrossRef]
- Springer Targeting the Mitochondrial Permeability Transition Pore in Traumatic Central Nervous System Injury. Available online: https://www.nrronline.org/article.asp?issn=1673-5374;year=2018;volume=13;issue=8;spage=1338;epage=1341;aulast=Springer (accessed on 28 January 2022).
- Vringer, E.; Tait, S.W.G. Mitochondria and Inflammation: Cell Death Heats Up. Front. Cell Dev. Biol. 2019, 7, 100. [Google Scholar] [CrossRef]
- Kalkavan, H.; Green, D.R. MOMP, cell suicide as a BCL-2 family business. Cell Death Differ. 2018, 25, 46–55. [Google Scholar] [CrossRef]
- Tait, S.W.G.; Green, D.R. Mitochondrial Regulation of Cell Death. Cold Spring Harb. Perspect. Biol. 2013, 5, a008706. [Google Scholar] [CrossRef] [Green Version]
- Adapted from “Coronavirus Replication Cycle”, by BioRender.com. 2020. Available online: https://app.biorender.com/biorender-templates (accessed on 22 February 2022).
- Ljubisavljevic, S. Oxidative Stress and Neurobiology of Demyelination. Mol. Neurobiol. 2016, 53, 744–758. [Google Scholar] [CrossRef]
- Da Silva Meirelles, L.; Simon, D.; Regner, A. Neurotrauma: The Crosstalk between Neurotrophins and Inflammation in the Acutely Injured Brain. Int. J. Mol. Sci. 2017, 18, 1082. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, J.; Wang, H.; Shen, R.; Fang, J.; Yang, Y.; Dai, W.; Zhu, Y.; Zhou, M. Mitochondrial-targeted antioxidant MitoQ provides neuroprotection and reduces neuronal apoptosis in experimental traumatic brain injury possibly via the Nrf2-ARE pathway. Am. J. Transl. Res. 2018, 10, 1887–1899. [Google Scholar] [PubMed]
- Bains, M.; Hall, E.D. Antioxidant therapies in traumatic brain and spinal cord injury. Biochim. Biophys. Acta BBA—Mol. Basis Dis. 2012, 1822, 675–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guemez-Gamboa, A.; Estrada-Sánchez, A.M.; Montiel, T.; Páramo, B.; Massieu, L.; Morán, J. Activation of NOX2 by the Stimulation of Ionotropic and Metabotropic Glutamate Receptors Contributes to Glutamate Neurotoxicity In Vivo Through the Production of Reactive Oxygen Species and Calpain Activation. J. Neuropathol. Exp. Neurol. 2011, 70, 1020–1035. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Sakakima, H.; Dhammu, T.S.; Shunmugavel, A.; Im, Y.-B.; Gilg, A.G.; Singh, A.K.; Singh, I. S-Nitrosoglutathione reduces oxidative injury and promotes mechanisms of neurorepair following traumatic brain injury in rats. J. Neuroinflamm. 2011, 8, 78. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-C.; Wee, H.-Y.; Hu, C.-Y.; Chio, C.-C.; Kuo, J.-R. The Effects of Memantine on Glutamic Receptor-Associated Nitrosative Stress in a Traumatic Brain Injury Rat Model. World Neurosurg. 2018, 112, e719–e731. [Google Scholar] [CrossRef]
- Logsdon, A.F.; Turner, R.C.; Lucke-Wold, B.P.; Robson, M.J.; Naser, Z.J.; Smith, K.E.; Matsumoto, R.R.; Huber, J.D.; Rosen, C.L. Altering endoplasmic reticulum stress in a model of blast-induced traumatic brain injury controls cellular fate and ameliorates neuropsychiatric symptoms. Front. Cell. Neurosci. 2014, 8, 421. [Google Scholar] [CrossRef] [Green Version]
- Lucke-Wold, B.P.; Logsdon, A.F.; Turner, R.C.; Huber, J.D.; Rosen, C.L. Endoplasmic Reticulum Stress Modulation as a Target for Ameliorating Effects of Blast Induced Traumatic Brain Injury. J. Neurotrauma 2017, 34, S-62–S-70. [Google Scholar] [CrossRef] [Green Version]
- Tan, H.-P.; Guo, Q.; Hua, G.; Chen, J.-X.; Liang, J.-C. Inhibition of endoplasmic reticulum stress alleviates secondary injury after traumatic brain injury. Neural Regen. Res. 2018, 13, 827. [Google Scholar] [CrossRef]
- Rubovitch, V.; Barak, S.; Rachmany, L.; Goldstein, R.B.; Zilberstein, Y.; Pick, C.G. The Neuroprotective Effect of Salubrinal in a Mouse Model of Traumatic Brain Injury. NeuroMolecular Med. 2015, 17, 58–70. [Google Scholar] [CrossRef]
- Donnelly, N.; Gorman, A.M.; Gupta, S.; Samali, A. The eIF2α kinases: Their structures and functions. Cell. Mol. Life Sci. CMLS 2013, 70, 3493–3511. [Google Scholar] [CrossRef] [PubMed]
- Dash, P.K.; Hylin, M.J.; Hood, K.N.; Orsi, S.A.; Zhao, J.; Redell, J.B.; Tsvetkov, A.S.; Moore, A.N. Inhibition of Eukaryotic Initiation Factor 2 Alpha Phosphatase Reduces Tissue Damage and Improves Learning and Memory after Experimental Traumatic Brain Injury. J. Neurotrauma 2015, 32, 1608–1620. [Google Scholar] [CrossRef] [Green Version]
- Hood, K.N.; Zhao, J.; Redell, J.B.; Hylin, M.J.; Harris, B.; Perez, A.; Moore, A.N.; Dash, P.K. Endoplasmic Reticulum Stress Contributes to the Loss of Newborn Hippocampal Neurons after Traumatic Brain Injury. J. Neurosci. Off. J. Soc. Neurosci. 2018, 38, 2372–2384. [Google Scholar] [CrossRef] [PubMed]
- Ruiz, A.; Zuazo, J.; Ortiz-Sanz, C.; Luchena, C.; Matute, C.; Alberdi, E. Sephin1 Protects Neurons against Excitotoxicity Independently of the Integrated Stress Response. Int. J. Mol. Sci. 2020, 21, 6088. [Google Scholar] [CrossRef] [PubMed]
- Vaccaro, A.; Patten, S.A.; Aggad, D.; Julien, C.; Maios, C.; Kabashi, E.; Drapeau, P.; Parker, J.A. Pharmacological reduction of ER stress protects against TDP-43 neuronal toxicity in vivo. Neurobiol. Dis. 2013, 55, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.G.; Lee, J.H.; Hong, S.H.; Lee, H.N.; Kim, C.M.; Kim, S.Y.; Yoon, K.J.; Oh, B.J.; Kim, J.H.; Jung, S.Y.; et al. Tauroursodeoxycholic Acid, a Bile Acid, Promotes Blood Vessel Repair by Recruiting Vasculogenic Progenitor Cells. Stem Cells 2015, 33, 792–805. [Google Scholar] [CrossRef] [PubMed]
- Battson, M.L.; Lee, D.M.; Jarrell, D.K.; Hou, S.; Ecton, K.E.; Phan, A.B.; Gentile, C.L. Tauroursodeoxycholic Acid Reduces Arterial Stiffness and Improves Endothelial Dysfunction in Type 2 Diabetic Mice. J. Vasc. Res. 2017, 54, 280–287. [Google Scholar] [CrossRef] [PubMed]
- Kochanek, P.M.; Jackson, T.C.; Ferguson, N.M.; Carlson, S.W.; Simon, D.W.; Brockman, E.C.; Ji, J.; Bayir, H.; Poloyac, S.M.; Wagner, A.K.; et al. Emerging Therapies in Traumatic Brain Injury. Semin. Neurol. 2015, 35, 83–100. [Google Scholar] [CrossRef] [Green Version]
- Ma, G.; Liu, C.; Hashim, J.; Conley, G.; Morriss, N.; Meehan, W.P.; Qiu, J.; Mannix, R. Memantine Mitigates Oligodendrocyte Damage after Repetitive Mild Traumatic Brain Injury. Neuroscience 2019, 421, 152–161. [Google Scholar] [CrossRef]
- Bernardi, P. The permeability transition pore. Control points of a cyclosporin A-sensitive mitochondrial channel involved in cell death. Biochim. Biophys. Acta BBA—Bioenerg. 1996, 1275, 5–9. [Google Scholar] [CrossRef] [Green Version]
- Halestrap, A.P.; Connern, C.P.; Griffiths, E.J.; Kerr, P.M. Cyclosporin A binding to mitochondrial cyclophilin inhibits the permeability transition pore and protects hearts from ischaemia/reperfusion injury. Mol. Cell. Biochem. 1997, 174, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Nicolli, A.; Basso, E.; Petronilli, V.; Bernardi, P. Two modes of activation of the permeability transition pore: The role of mitochondrial cyclophilin. Mol. Cell. Biochem. 1997, 174, 181–184. [Google Scholar] [CrossRef]
- Springer, J.E.; Visavadiya, N.P.; Sullivan, P.G.; Hall, E.D. Post-Injury Treatment with NIM811 Promotes Recovery of Function in Adult Female Rats after Spinal Cord Contusion: A Dose-Response Study. J. Neurotrauma 2018, 35, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.G.; Rabchevsky, A.G.; Waldmeier, P.C.; Springer, J.E. Mitochondrial permeability transition in CNS trauma: Cause or effect of neuronal cell death? J. Neurosci. Res. 2005, 79, 231–239. [Google Scholar] [CrossRef]
- Miyamoto, K.; Ohtaki, H.; Dohi, K.; Tsumuraya, T.; Song, D.; Kiriyama, K.; Satoh, K.; Shimizu, A.; Aruga, T.; Shioda, S. Therapeutic Time Window for Edaravone Treatment of Traumatic Brain Injury in Mice. BioMed Res. Int. 2013, 2013, 379206. [Google Scholar] [CrossRef]
- Wang, G.-H.; Jiang, Z.-L.; Li, Y.-C.; Li, X.; Shi, H.; Gao, Y.-Q.; Vosler, P.S.; Chen, J. Free-Radical Scavenger Edaravone Treatment Confers Neuroprotection Against Traumatic Brain Injury in Rats. J. Neurotrauma 2011, 28, 2123–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itoh, T.; Satou, T.; Nishida, S.; Tsubaki, M.; Hashimoto, S.; Ito, H. The Novel Free Radical Scavenger, Edaravone, Increases Neural Stem Cell Number Around the Area of Damage Following Rat Traumatic Brain Injury. Neurotox. Res. 2009, 16, 378–389. [Google Scholar] [CrossRef]
- Chandran, R.; Mehta, S.L.; Vemuganti, R. Antioxidant Combo Therapy Protects White Matter after Traumatic Brain Injury. NeuroMolecular Med. 2021, 23, 344–347. [Google Scholar] [CrossRef]
- Ünal, İ.; Çalışkan-Ak, E.; Üstündağ, Ü.V.; Ateş, P.S.; Alturfan, A.A.; Altinoz, M.A.; Elmaci, I.; Emekli-Alturfan, E. Neuroprotective effects of mitoquinone and oleandrin on Parkinson’s disease model in zebrafish. Int. J. Neurosci. 2020, 130, 574–582. [Google Scholar] [CrossRef]
- Pinho, B.R.; Duarte, A.I.; Canas, P.M.; Moreira, P.I.; Murphy, M.P.; Oliveira, J.M.A. The interplay between redox signalling and proteostasis in neurodegeneration: In vivo effects of a mitochondria-targeted antioxidant in Huntington’s disease mice. Free Radic. Biol. Med. 2020, 146, 372–382. [Google Scholar] [CrossRef]
- Miquel, E.; Cassina, A.; Martínez-Palma, L.; Souza, J.M.; Bolatto, C.; Rodríguez-Bottero, S.; Logan, A.; Smith, R.A.J.; Murphy, M.P.; Barbeito, L.; et al. Neuroprotective effects of the mitochondria-targeted antioxidant MitoQ in a model of inherited amyotrophic lateral sclerosis. Free Radic. Biol. Med. 2014, 70, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Young, M.L.; Franklin, J.L. The mitochondria-targeted antioxidant MitoQ inhibits memory loss, neuropathology, and extends lifespan in aged 3xTg-AD mice. Mol. Cell. Neurosci. 2019, 101, 103409. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, A.; Chandran, K.; Kalivendi, S.V.; Joseph, J.; Antholine, W.E.; Hillard, C.J.; Kanthasamy, A.; Kanthasamy, A.; Kalyanaraman, B. Neuroprotection by a mitochondria-targeted drug in a Parkinson’s disease model. Free Radic. Biol. Med. 2010, 49, 1674–1684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, A.; Shahpasand, K.; Mannix, R.; Qiu, J.; Moncaster, J.; Chen, C.-H.; Yao, Y.; Lin, Y.-M.; Driver, J.A.; Sun, Y.; et al. Antibody against early driver of neurodegeneration cis P-tau blocks brain injury and tauopathy. Nature 2015, 523, 431–436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajmohan, R.; Reddy, P.H. Amyloid Beta and Phosphorylated Tau Accumulations Cause Abnormalities at Synapses of Alzheimer’s disease Neurons. J. Alzheimers Dis. JAD 2017, 57, 975–999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kshirsagar, S.; Sawant, N.; Morton, H.; Reddy, A.P.; Reddy, P.H. Mitophagy enhancers against phosphorylated Tau-induced mitochondrial and synaptic toxicities in Alzheimer disease. Pharmacol. Res. 2021, 174, 105973. [Google Scholar] [CrossRef]
- John, A.; Reddy, P.H. Synaptic Basis of Alzheimer’s Disease: Focus on Synaptic Amyloid Beta, P-Tau and Mitochondria. Ageing Res. Rev. 2021, 65, 101208. [Google Scholar] [CrossRef]
- Alavi Naini, S.M.; Soussi-Yanicostas, N. Tau Hyperphosphorylation and Oxidative Stress, a Critical Vicious Circle in Neurodegenerative Tauopathies? Oxid. Med. Cell. Longev. 2015, 2015, 151979. [Google Scholar] [CrossRef] [Green Version]
- Albayram, O.; Kondo, A.; Mannix, R.; Smith, C.; Tsai, C.-Y.; Li, C.; Herbert, M.K.; Qiu, J.; Monuteaux, M.; Driver, J.; et al. Cis P-tau is induced in clinical and preclinical brain injury and contributes to post-injury sequelae. Nat. Commun. 2017, 8, 1000. [Google Scholar] [CrossRef]
- Lu, G.; Li, T.; Ye, H.; Liu, S.; Zhang, P.; Wang, W. D-dimer in the diagnosis of periprosthetic joint infection: A systematic review and meta-analysis. J. Orthop. Surg. 2020, 15, 265. [Google Scholar] [CrossRef]
- Kim, N.; Wang, B.; Koikawa, K.; Nezu, Y.; Qiu, C.; Lee, T.H.; Zhou, X.Z. Inhibition of death-associated protein kinase 1 attenuates cis P-tau and neurodegeneration in traumatic brain injury. Prog. Neurobiol. 2021, 203, 102072. [Google Scholar] [CrossRef] [PubMed]
- Mohsenian Sisakht, A.; Karamzade-Ziarati, N.; Jahanbakhshi, A.; Shahpasand, K.; Aghababaei, S.; Ahmadvand, O.; Azar, M.; Fattahi, A.; Zamanzadeh, S. Pathogenic cis p-tau levels in CSF reflects severity of traumatic brain injury. Neurol. Res. 2022, 3, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Ren, X.; Wang, Y.; Li, J.; Guo, T.; Li, F.; Zhao, Z. Traumatic brain injury research and expression of caveolin-1 and its relationship with disease prognosis. Pak. J. Pharm. Sci. 2017, 30, 997–1000. [Google Scholar] [PubMed]
- Chang, C.-F.; Chen, S.-F.; Lee, T.-S.; Lee, H.-F.; Chen, S.-F.; Shyue, S.-K. Caveolin-1 Deletion Reduces Early Brain Injury after Experimental Intracerebral Hemorrhage. Am. J. Pathol. 2011, 178, 1749–1761. [Google Scholar] [CrossRef]
- Bucci, M.; Gratton, J.-P.; Rudic, R.D.; Acevedo, L.; Roviezzo, F.; Cirino, G.; Sessa, W.C. In vivo delivery of the caveolin-1 scaffolding domain inhibits nitric oxide synthesis and reduces inflammation. Nat. Med. 2000, 6, 1362–1367. [Google Scholar] [CrossRef]
- Lajoie, P.; Goetz, J.G.; Dennis, J.W.; Nabi, I.R. Lattices, rafts, and scaffolds: Domain regulation of receptor signaling at the plasma membrane. J. Cell Biol. 2009, 185, 381–385. [Google Scholar] [CrossRef] [Green Version]
- Badaut, J.; Ajao, D.O.; Sorensen, D.W.; Fukuda, A.M.; Pellerin, L. Caveolin expression changes in the neurovascular unit after juvenile traumatic brain injury: Signs of blood–brain barrier healing? Neuroscience 2015, 285, 215–226. [Google Scholar] [CrossRef] [Green Version]
- Förstermann, U.; Xia, N.; Li, H. Roles of Vascular Oxidative Stress and Nitric Oxide in the Pathogenesis of Atherosclerosis. Circ. Res. 2017, 120, 713–735. [Google Scholar] [CrossRef]
- Ren, X.; Ren, L.; Wei, Q.; Shao, H.; Chen, L.; Liu, N. Advanced glycation end-products decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Cardiovasc. Diabetol. 2017, 16, 52. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Shao, A.; Xu, W.; Wu, H.; Deng, Y. Advance of Stem Cell Treatment for Traumatic Brain Injury. Front. Cell. Neurosci. 2019, 13, 301. [Google Scholar] [CrossRef] [Green Version]
- Guo, S.; Zhen, Y.; Wang, A. Transplantation of bone mesenchymal stem cells promotes angiogenesis and improves neurological function after traumatic brain injury in mouse. Neuropsychiatr. Dis. Treat. 2017, 13, 2757–2765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schepici, G.; Silvestro, S.; Bramanti, P.; Mazzon, E. Traumatic Brain Injury and Stem Cells: An Overview of Clinical Trials, the Current Treatments and Future Therapeutic Approaches. Medicina 2020, 56, 137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Liu, Y.; Yan, K.; Chen, L.; Chen, X.-R.; Li, P.; Chen, F.-F.; Jiang, X.-D. Anti-inflammatory and immunomodulatory mechanisms of mesenchymal stem cell transplantation in experimental traumatic brain injury. J. Neuroinflamm. 2013, 10, 871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Islam, M.N.; Das, S.R.; Emin, M.T.; Wei, M.; Sun, L.; Westphalen, K.; Rowlands, D.J.; Quadri, S.K.; Bhattacharya, S.; Bhattacharya, J. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat. Med. 2012, 18, 759–765. [Google Scholar] [CrossRef] [Green Version]
- Jackson, M.V.; Morrison, T.J.; Doherty, D.F.; McAuley, D.F.; Matthay, M.A.; Kissenpfennig, A.; O’Kane, C.M.; Krasnodembskaya, A.D. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells Dayt. Ohio 2016, 34, 2210–2223. [Google Scholar] [CrossRef] [Green Version]
- Cui, L.; Nitzsche, F.; Pryazhnikov, E.; Tibeykina, M.; Tolppanen, L.; Rytkönen, J.; Huhtala, T.; Mu, J.; Khiroug, L.; Boltze, J.; et al. Integrin α4 Overexpression on Rat Mesenchymal Stem Cells Enhances Transmigration and Reduces Cerebral Embolism After Intracarotid Injection. Stroke 2017, 48, 2895–2900. [Google Scholar] [CrossRef]
- Večerić-Haler, Ž.; Cerar, A.; Perše, M. (Mesenchymal) Stem Cell-Based Therapy in Cisplatin-Induced Acute Kidney Injury Animal Model: Risk of Immunogenicity and Tumorigenicity. Stem Cells Int. 2017, 2017, e7304643. [Google Scholar] [CrossRef] [Green Version]
- Wang Pre-Clinical Study of Human Umbilical Cord Mesenchymal Stem Cell Transplantation for the Treatment of Traumatic Brain Injury: Safety Evaluation from Immunogenic and Oncogenic Perspectives. Available online: https://www.nrronline.org/article.asp?issn=1673-5374;year=2022;volume=17;issue=2;spage=354;epage=361;aulast=Wang (accessed on 28 January 2022).
- Kalluri, R.; LeBleu, V.S. The biology, function, and biomedical applications of exosomes. Science 2020, 367, eaau6977. [Google Scholar] [CrossRef]
- Ghosh, S.; Garg, S.; Ghosh, S. Cell-Derived Exosome Therapy: A Novel Approach to Treat Post-traumatic Brain Injury Mediated Neural Injury. ACS Chem. Neurosci. 2020, 11, 2045–2047. [Google Scholar] [CrossRef]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.K.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; El Oakley, R.M.; et al. Exosome secreted by MSC reduces myocardial ischemia/reperfusion injury. Stem Cell Res. 2010, 4, 214–222. [Google Scholar] [CrossRef] [Green Version]
- Sun, C.-K.; Chen, C.-H.; Chang, C.-L.; Chiang, H.-J.; Sung, P.-H.; Chen, K.-H.; Chen, Y.-L.; Chen, S.-Y.; Kao, G.-S.; Chang, H.-W.; et al. Melatonin treatment enhances therapeutic effects of exosomes against acute liver ischemia-reperfusion injury. Am. J. Transl. Res. 2017, 9, 1543–1560. [Google Scholar] [PubMed]
- Xin, H.; Li, Y.; Cui, Y.; Yang, J.J.; Zhang, Z.G.; Chopp, M. Systemic Administration of Exosomes Released from Mesenchymal Stromal Cells Promote Functional Recovery and Neurovascular Plasticity After Stroke in Rats. J. Cereb. Blood Flow Metab. 2013, 33, 1711–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, C.; Zhao, L.; Zhang, L.; Bao, Q.; Li, L. Mesenchymal stem cell-based cell-free strategies: Safe and effective treatments for liver injury. Stem Cell Res. Ther. 2020, 11, 377. [Google Scholar] [CrossRef] [PubMed]
- Buono, L.; Scalabrin, S.; De Iuliis, M.; Tanzi, A.; Grange, C.; Tapparo, M.; Nuzzi, R.; Bussolati, B. Mesenchymal Stem Cell-Derived Extracellular Vesicles Protect Human Corneal Endothelial Cells from Endoplasmic Reticulum Stress-Mediated Apoptosis. Int. J. Mol. Sci. 2021, 22, 4930. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Li, J.; Ma, B.; Li, N.; Wang, S.; Sun, Z.; Xue, C.; Han, Q.; Wei, J.; Zhao, R.C. MSC-derived exosomes promote recovery from traumatic brain injury via microglia/macrophages in rat. Aging 2020, 12, 18274–18296. [Google Scholar] [CrossRef] [PubMed]
- Liao, Z.; Luo, R.; Li, G.; Song, Y.; Zhan, S.; Zhao, K.; Hua, W.; Zhang, Y.; Wu, X.; Yang, C. Exosomes from mesenchymal stem cells modulate endoplasmic reticulum stress to protect against nucleus pulposus cell death and ameliorate intervertebral disc degeneration in vivo. Theranostics 2019, 9, 4084–4100. [Google Scholar] [CrossRef]
- Zhang, Y.; Chopp, M.; Meng, Y.; Katakowski, M.; Xin, H.; Mahmood, A.; Xiong, Y. Effect of exosomes derived from multipluripotent mesenchymal stromal cells on functional recovery and neurovascular plasticity in rats after traumatic brain injury. J. Neurosurg. 2015, 122, 856–867. [Google Scholar] [CrossRef] [Green Version]
- Ni, H.; Yang, S.; Siaw-Debrah, F.; Hu, J.; Wu, K.; He, Z.; Yang, J.; Pan, S.; Lin, X.; Ye, H.; et al. Exosomes Derived From Bone Mesenchymal Stem Cells Ameliorate Early Inflammatory Responses Following Traumatic Brain Injury. Front. Neurosci. 2019, 13, 14. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.M.; Dennahy, I.S.; Bhatti, U.F.; Halaweish, I.; Xiong, Y.; Chang, P.; Nikolian, V.C.; Chtraklin, K.; Brown, J.; Zhang, Y.; et al. Mesenchymal Stem Cell-Derived Exosomes Provide Neuroprotection and Improve Long-Term Neurologic Outcomes in a Swine Model of Traumatic Brain Injury and Hemorrhagic Shock. J. Neurotrauma 2019, 36, 54–60. [Google Scholar] [CrossRef]
- Thomi, G.; Surbek, D.; Haesler, V.; Joerger-Messerli, M.; Schoeberlein, A. Exosomes derived from umbilical cord mesenchymal stem cells reduce microglia-mediated neuroinflammation in perinatal brain injury. Stem Cell Res. Ther. 2019, 10, 105. [Google Scholar] [CrossRef]
- Kranjc, M.K.; Novak, M.; Pestell, R.G.; Lah, T.T. Cytokine CCL5 and receptor CCR5 axis in glioblastoma multiforme. Radiol. Oncol. 2019, 53, 397–406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morganti, J.M.; Riparip, L.-K.; Chou, A.; Liu, S.; Gupta, N.; Rosi, S. Age exacerbates the CCR2/5-mediated neuroinflammatory response to traumatic brain injury. J. Neuroinflamm. 2016, 13, 80. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oppermann, M. Chemokine receptor CCR5: Insights into structure, function, and regulation. Cell. Signal. 2004, 16, 1201–1210. [Google Scholar] [CrossRef]
- Scurci, I.; Martins, E.; Hartley, O. CCR5: Established paradigms and new frontiers for a ‘celebrity’ chemokine receptor. Cytokine 2018, 109, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Alard, J.-E.; Dueymes, M.; Mageed, R.A.; Saraux, A.; Youinou, P.; Jamin, C. Mitochondrial heat shock protein (HSP) 70 synergizes with HSP60 in transducing endothelial cell apoptosis induced by anti-HSP60 autoantibody. FASEB J. 2009, 23, 2772–2779. [Google Scholar] [CrossRef]
- Lai, Y.; Stange, C.; Wisniewski, S.R.; Adelson, P.D.; Janesko-Feldman, K.L.; Brown, D.S.; Kochanek, P.M.; Clark, R.S.B. Mitochondrial Heat Shock Protein 60 Is Increased in Cerebrospinal Fluid following Pediatric Traumatic Brain Injury. Dev. Neurosci. 2006, 28, 336–341. [Google Scholar] [CrossRef]
- Haruwaka, K.; Ikegami, A.; Tachibana, Y.; Ohno, N.; Konishi, H.; Hashimoto, A.; Matsumoto, M.; Kato, D.; Ono, R.; Kiyama, H.; et al. Dual microglia effects on blood brain barrier permeability induced by systemic inflammation. Nat. Commun. 2019, 10, 5816. [Google Scholar] [CrossRef] [Green Version]
- Ciechanowska, A.; Popiolek-Barczyk, K.; Pawlik, K.; Ciapała, K.; Oggioni, M.; Mercurio, D.; De Simoni, M.-G.; Mika, J. Changes in macrophage inflammatory protein-1 (MIP-1) family members expression induced by traumatic brain injury in mice. Immunobiology 2020, 225, 151911. [Google Scholar] [CrossRef]
- Joy, M.T.; Assayag, E.B.; Shabashov-Stone, D.; Liraz-Zaltsman, S.; Mazzitelli, J.; Arenas, M.; Abduljawad, N.; Kliper, E.; Korczyn, A.D.; Thareja, N.S.; et al. CCR5 Is a Therapeutic Target for Recovery after Stroke and Traumatic Brain Injury. Cell 2019, 176, 1143–1157.e13. [Google Scholar] [CrossRef] [Green Version]
- Liraz-Zaltsman, S.; Friedman-Levi, Y.; Shabashov-Stone, D.; Gincberg, G.; Atrakcy-Baranes, D.; Joy, M.T.; Carmichael, S.T.; Silva, A.J.; Shohami, E. Chemokine Receptors CC Chemokine Receptor 5 and C-X-C Motif Chemokine Receptor 4 Are New Therapeutic Targets for Brain Recovery after Traumatic Brain Injury. J. Neurotrauma 2021, 38, 2003–2017. [Google Scholar] [CrossRef]
- Victoria, E.C.G.; de Toscano, E.C.B.; de Cardoso, A.C.S.; da Silva, D.G.; de Miranda, A.S.; da Barcelos, L.S.; Sugimoto, M.A.; Sousa, L.P.; de Lima, I.V.A.; de Oliveira, A.C.P.; et al. Knockdown of C-C Chemokine Receptor 5 (CCR5) is Protective Against Cerebral Ischemia and Reperfusion Injury. Curr. Neurovasc. Res. 2017, 14, 125–131. [Google Scholar] [CrossRef] [PubMed]
- Friedman-Levi, Y.; Liraz-Zaltsman, S.; Shemesh, C.; Rosenblatt, K.; Kesner, E.L.; Gincberg, G.; Carmichael, S.T.; Silva, A.J.; Shohami, E. Pharmacological blockers of CCR5 and CXCR4 improve recovery after traumatic brain injury. Exp. Neurol. 2021, 338, 113604. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Therapies | Potential Mechanisms of Action |
|---|---|
| Immunoglobulin | ↓ p-tau (mitochondrial stress and apoptosis) ↓ caveolin (oxidative stress) |
| Cell-Based | ↓ oxidative stress ↓ inflammatory cell migration |
| MSC-Exosomes | ↓ oxidative stress ↓ ER stress |
| CCR5 Antagonists | ↓ Inflammatory cell migration |
| Extra-synaptic NMDA Receptor Inhibitors | ↓ mitochondrial stress |
| Selective Ca2+ Channel Inhibitors | ↓ mitochondrial and ER stress |
| eIF2α Phosphorylation | ↓ unfolded protein production and ER stress |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aghili-Mehrizi, S.; Williams, E.; Yan, S.; Willman, M.; Willman, J.; Lucke-Wold, B. Secondary Mechanisms of Neurotrauma: A Closer Look at the Evidence. Diseases 2022, 10, 30. https://doi.org/10.3390/diseases10020030
Aghili-Mehrizi S, Williams E, Yan S, Willman M, Willman J, Lucke-Wold B. Secondary Mechanisms of Neurotrauma: A Closer Look at the Evidence. Diseases. 2022; 10(2):30. https://doi.org/10.3390/diseases10020030
Chicago/Turabian StyleAghili-Mehrizi, Sina, Eric Williams, Sandra Yan, Matthew Willman, Jonathan Willman, and Brandon Lucke-Wold. 2022. "Secondary Mechanisms of Neurotrauma: A Closer Look at the Evidence" Diseases 10, no. 2: 30. https://doi.org/10.3390/diseases10020030
APA StyleAghili-Mehrizi, S., Williams, E., Yan, S., Willman, M., Willman, J., & Lucke-Wold, B. (2022). Secondary Mechanisms of Neurotrauma: A Closer Look at the Evidence. Diseases, 10(2), 30. https://doi.org/10.3390/diseases10020030