
Proficient Novel Biomarkers Guide Early Detection of Acute Kidney Injury: A Review



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
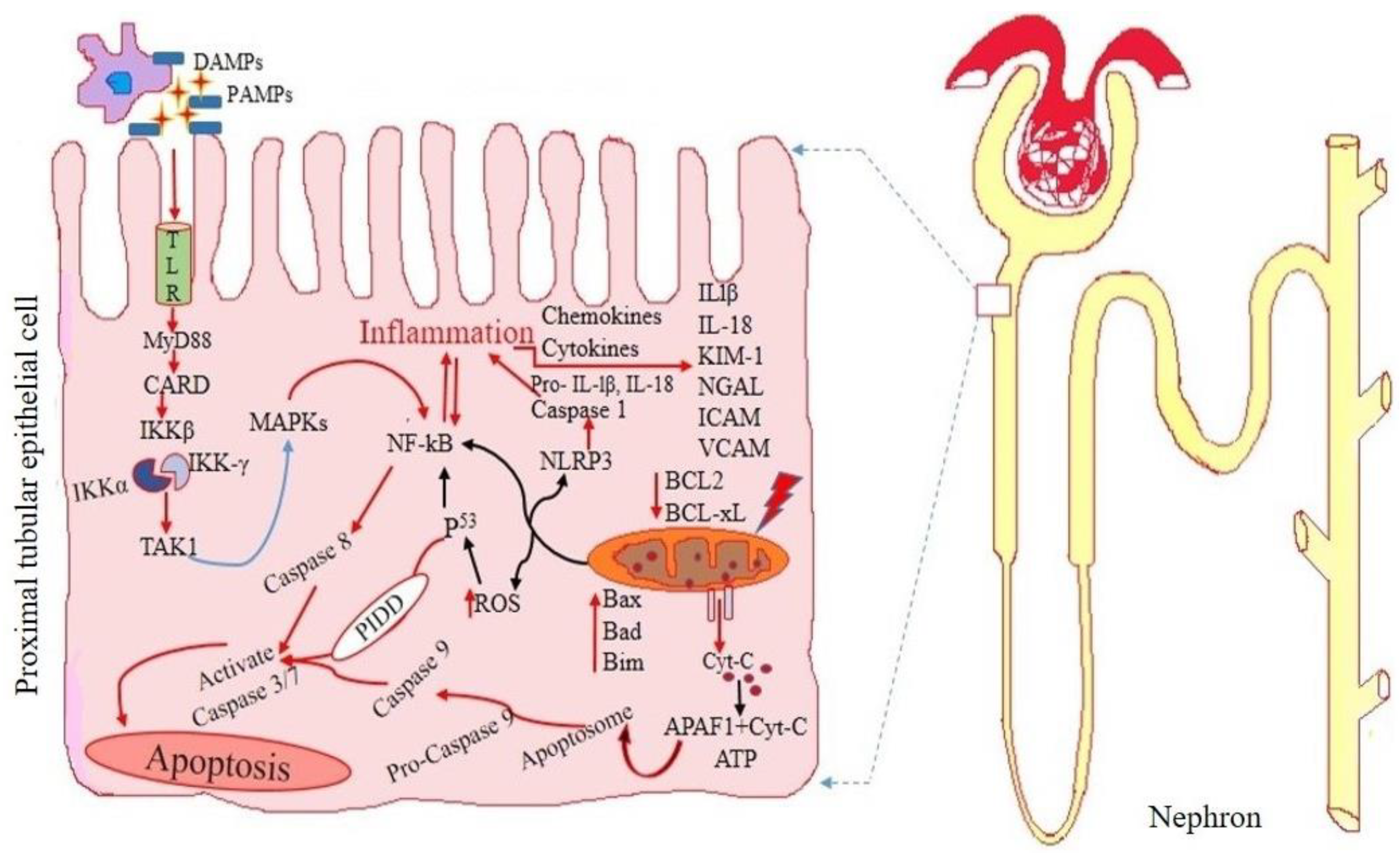
2. Emerging Novel Biomarkers for Detecting Acute Kidney Injury
2.1. Biomarkers for Glomerular Injury
2.1.1. Immunoglobulin G
2.1.2. Nephrin
2.1.3. Podocalyxin
2.1.4. Podocin
2.1.5. Transferrin
2.1.6. Netrin-1
2.1.7. Pyruvate Kinase M2
2.2. Tubular Injury Diagnostic Markers
2.2.1. Kidney Injury Molecule-1
2.2.2. Interleukin 18
2.2.3. Neutrophil Gelatinase-Associated Lipocalin (NGAL)
2.2.4. Uromodulin
2.2.5. Calprotectin
2.2.6. Vanin 1
2.2.7. Galectin-3
2.2.8. Platelet Derived Growth Factor (PDGF)
2.2.9. Urinary Na+/H+ Exchanger Isoform 3 (NHE3)
2.2.10. Retinol Binding Protein
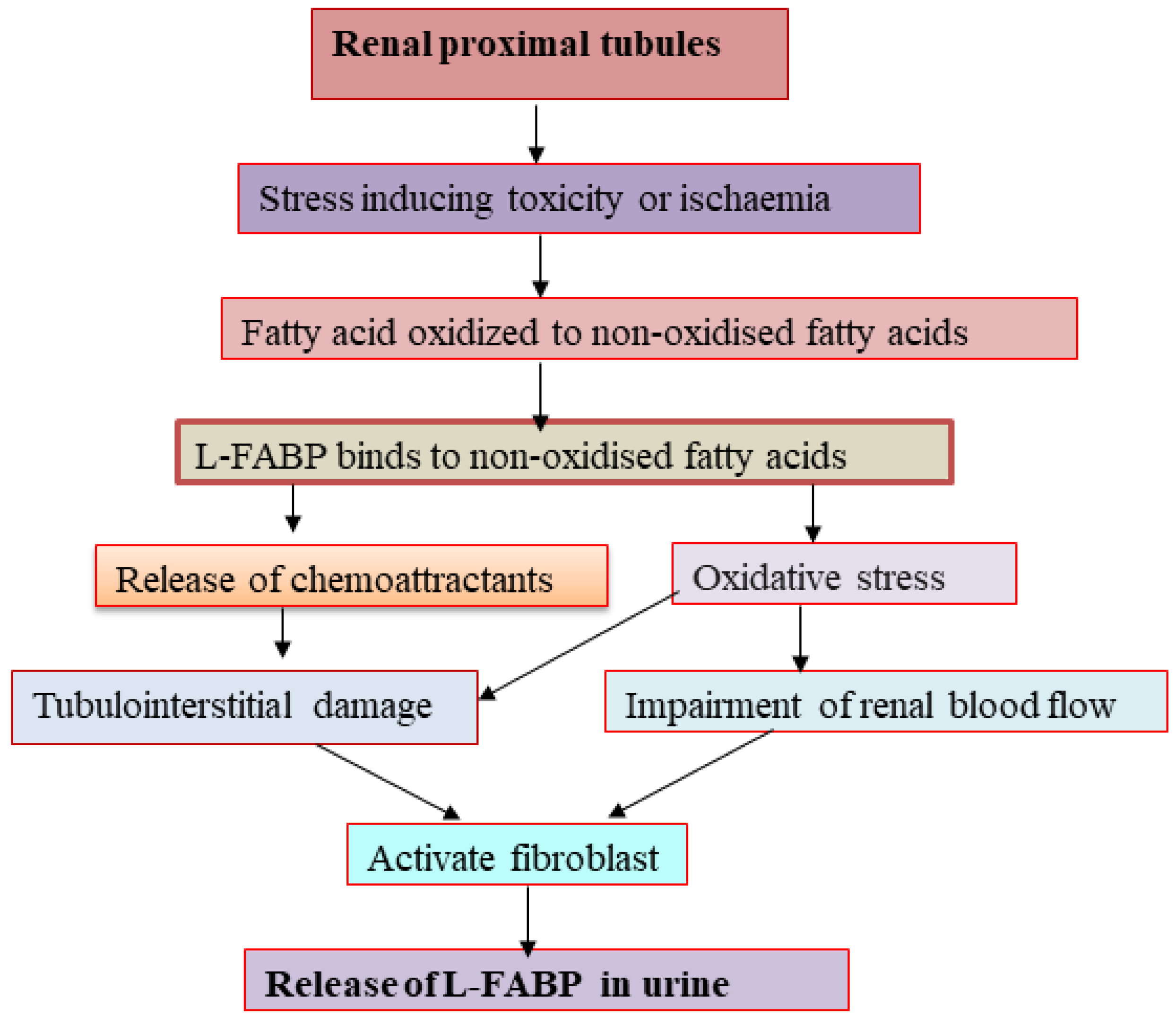
2.2.11. Liver-Type Fatty Acid Binding Protein (L-FABP)
2.2.12. β2-Microglobulin (B2M)
2.2.13. Cysteine-Rich Protein 61 (CYR61)
2.2.14. Transforming Growth Factor Beta (TGF-β)
2.2.15. N-Acetyl-β-Glucosaminidase (NAG)
2.2.16. Osteopontin
2.2.17. Clusterin
2.2.18. IL-6
2.2.19. E-Cadherin
2.2.20. Calbindin
2.2.21. Cystatin C
2.2.22. TIMP-2 × IGFBP7
3. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kellum, J.A.; Lameire, N.; Aspelin, P.; Barsoum, R.S.; Burdmann, E.A.; Goldstein, S.L. KDIGO AKI guidelines. Kidney Int. 2012, 2, 1–38. [Google Scholar]
- Lewington, A.J.; Cerdá, J.; Mehta, R.L. Raising awareness of acute kidney injury: A global perspective of a silent killer. Kidney Int. 2013, 84, 457–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Susantitaphong, P.; Cruz, D.N.; Cerda, J.; Abulfaraj, M.; Alqahtani, F.; Koulouridis, I.; Jaber, B.L. World incidence of AKI: A meta-analysis. Clin. J. Am. Soc. Nephrol. 2013, 8, 1482–1493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xue, J.L.; Daniels, F.; Star, R.A.; Kimmel, P.L.; Eggers, P.W.; Molitoris, B.A.; Himmelfarb, J.; Collins, A.J. Incidence and Mortality of Acute Renal Failure in Medicare Beneficiaries, 1992 to 2001. J. Am. Soc. Nephrol. 2006, 17, 1135–1142. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.R.; Thiessen-Philbrook, H.; Garg, A.X.; Kadiyala, D.; Shlipak, M.G.; Koyner, J.L.; Edelstein, C.L.; Devarajan, P.; Patel, U.D.; Zappitelli, M.; et al. Performance of Kidney Injury Molecule-1 and Liver Fatty Acid-Binding Protein and Combined Biomarkers of AKI after Cardiac Surgery. Clin. J. Am. Soc. Nephrol. 2013, 8, 1079–1088. [Google Scholar] [CrossRef] [Green Version]
- Malyszko, J. Biomarkers of Acute Kidney Injury in Different Clinical Settings: A Time to Change the Paradigm? Kidney Blood Press. Res. 2010, 33, 368–382. [Google Scholar] [CrossRef]
- Zhang, W.R.; Parikh, C.R. Biomarkers of Acute and Chronic Kidney Disease. Annu. Rev. Physiol. 2019, 81, 309–333. [Google Scholar] [CrossRef]
- Hou, J.; Cheng, Y.; Hou, Y.; Wu, H. Lower Serum and Higher Urine Immunoglobulin G Are Associated with an Increased Severity of Idiopathic Membranous Nephropathy. Ann. Clin. Lab. Sci. 2019, 49, 777–784. [Google Scholar]
- Hu, Q.; Wu, K.; Pan, W.; Zeng, Y.; Hu, K.; Chen, D.; Huang, X.; Zhang, Q. Intestinal flora alterations in patients with early chronic kidney disease: A case-control study among the Han population in southwestern China. J. Int. Med Res. 2020, 48, 0300060520926033. [Google Scholar] [CrossRef]
- Abdou, A.E.; Anani, H.A.; Ibrahim, H.F.; Ebrahem, E.E.; Seliem, N.; Youssef, E.M.; Ghoraba, N.M.; Hassan, A.S.; Ramadan, M.A.A.; Mahmoud, E.; et al. Urinary IgG, serum CX3CL1 and miRNA-152-3p: As predictors of nephropathy in Egyptian type 2 diabetic patients. Tissue Barriers 2021, 10, 1994823. [Google Scholar] [CrossRef]
- Singh, S.S.; Heijmans, R.; Meulen, C.K.; Lieverse, A.G.; Gornik, O.; Sijbrands, E.J.; Lauc, G.; van Hoek, M. Association of the IgG N-glycome with the course of kidney function in type 2 diabetes. BMJ Open Diabetes Res. Care 2020, 8, e001026. [Google Scholar] [CrossRef] [PubMed]
- Cravedi, P.; Remuzzi, G. Pathophysiology of proteinuria and its value as an outcome measure in chronic kidney disease. Br. J. Clin. Pharmacol. 2013, 76, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Bazzi, C.; Rizza, V.; Casellato, D.; Tofik, R.; Berg, A.-L.; Gallieni, M.; D’Amico, G.; Bakoush, O. Fractional excretion of IgG in idiopathic membranous nephropathy with nephrotic syndrome: A predictive marker of risk and drug responsiveness. BMC Nephrol. 2014, 15, 74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandasamy, Y.; Smith, R.; Lumbers, E.R.; Rudd, D. Nephrin–a biomarker of early glomerular injury. Biomark. Res. 2014, 2, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostovska, I.; Tosheska-Trajkovska, K.; Topuzovska, S.; Cekovska, S.; Spasovski, G.; Kostovski, O.; Labudovic, D. Urinary nephrin is earlier, more sensitive and specific marker of diabetic nephropathy than microalbuminuria. J. Med. Biochem. 2019, 39, 83–90. [Google Scholar] [CrossRef]
- Silambanan, S.; Kondapi, K.; Kumar, N.L.; Moorthy, S. A study of association of urinary nephrin with albuminuria in patients with diabetic nephropathy. Indian J. Nephrol. 2021, 31, 142–148. [Google Scholar] [CrossRef]
- Kostovska, I.; Trajkovska, K.T.; Cekovska, S.; Topuzovska, S.; Kavrakova, J.B.; Spasovski, G.; Kostovski, O.; Labudovic, D. Role of urinary podocalyxin in early diagnosis of diabetic nephropathy. Rom. J. Intern. Med. 2020, 58, 233–241. [Google Scholar] [CrossRef]
- Akankwasa, G.; Jianhua, L.; Guixue, C.; Changjuan, A.; Xiaosong, Q. Urine markers of podocyte dysfunction: A review of podocalyxin and nephrin in selected glomerular diseases. Biomark. Med. 2018, 12, 927–935. [Google Scholar] [CrossRef]
- Asao, R.; Asanuma, K.; Kodama, F.; Akiba-Takagi, M.; Nagai-Hosoe, Y.; Seki, T.; Takeda, Y.; Ohsawa, I.; Mano, S.; Matsuoka, K.; et al. Relationships between Levels of Urinary Podocalyxin, Number of Urinary Podocytes, and Histologic Injury in Adult Patients with IgA Nephropathy. Clin. J. Am. Soc. Nephrol. 2012, 7, 1385–1393. [Google Scholar] [CrossRef] [Green Version]
- Zeng, J.; Zhang, X.; Yu, R.; Tang, Y.; Luo, W.-J.; Chen, C.; Wu, Y.-J. Research on the Combined Detection of Urine UmAlb and Urinary Nephrin, Podocalyxin in Podocyte of MKR Mice with Diabetic Nephropathy. Sichuan Da Xue Xue Bao. Yi Xue Ban = J. Sichuan Univ. Med. Sci. Ed. 2015, 46, 722–725. [Google Scholar]
- Huber, T.B.; Simons, M.; Hartleben, B.; Sernetz, L.; Schmidts, M.; Gundlach, E.; Saleem, M.A.; Walz, G.; Benzing, T. Molecular basis of the functional podocin–nephrin complex: Mutations in the NPHS2 gene disrupt nephrin targeting to lipid raft microdomains. Hum. Mol. Genet. 2003, 12, 3397–3405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollet, G.; Ratelade, J.; Boyer, O.; Muda, A.O.; Morisset, L.; Lavin, T.A.; Kitzis, D.; Dallman, M.; Bugeon, L.; Hubner, N.; et al. Podocin Inactivation in Mature Kidneys Causes Focal Segmental Glomerulosclerosis and Nephrotic Syndrome. J. Am. Soc. Nephrol. 2009, 20, 2181–2189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elshaarawy, A.; Behairy, M.A.; Bawady, S.A.; Abdelsattar, H.A.; Shadad, E. Urinary podocin level as a predictor of diabetic kidney disease. J. Nephropathol. 2018, 8, 26. [Google Scholar] [CrossRef]
- Abdel Rahman, H.S.; Hadhoud, K.; Bakr, H.G.; Youssef, K.M. Assessment of urinary podocin level as an early indicator in diabetic nephropathy. Zagazig Univ. Med. J. 2019, 25, 682–688. [Google Scholar] [CrossRef]
- Zhang, D.; Meyron-Holtz, E.; Rouault, T.A. Renal Iron Metabolism: Transferrin Iron Delivery and the Role of Iron Regulatory Proteins. J. Am. Soc. Nephrol. 2007, 18, 401–406. [Google Scholar] [CrossRef]
- Vicente-Vicente, L.; Ferreira, L.; González-Buitrago, J.M.; López-Hernández, F.J.; López-Novoa, J.M.; Morales, A.I. Increased urinary excretion of albumin, hemopexin, transferrin and VDBP correlates with chronic sensitization to gentamicin nephrotoxicity in rats. Toxicology 2013, 304, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Brian Reeves, W.; Kwon, O.; Ramesh, G. Netrin-1 and kidney injury. II. Netrin-1 is an early biomarker of acute kidney injury. Am. J. Physiol.-Ren. Physiol. 2008, 294, F731–F738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, Y.; Wang, H.; Sun, R.; Ni, Y.; Ma, L.; Xv, F.; Hu, X.; Jiang, L.; Wu, A.; Chen, X.; et al. Urinary netrin-1 and KIM-1 as early biomarkers for septic acute kidney injury. Ren. Fail. 2014, 36, 1559–1563. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Rong, S.; Zhou, J.; Yuan, W. The role and mechanism of PKM2 in the development of LPS-induced acute kidney injury. Histol. Histopathol. 2021, 36, 845–852. [Google Scholar]
- Alquraishi, M.; Chahed, S.; Alani, D.; Puckett, D.L.; Dowker, P.D.; Hubbard, K.; Zhao, Y.; Kim, J.Y.; Nodit, L.; Fatima, H.; et al. Podocyte specific deletion of PKM2 ameliorates LPS-induced podocyte injury through beta-catenin. Cell Commun. Signal. 2022, 20, 76. [Google Scholar] [CrossRef]
- Geng, J.; Qiu, Y.; Qin, Z.; Su, B. The value of kidney injury molecule 1 in predicting acute kidney injury in adult patients: A systematic review and Bayesian meta-analysis. J. Transl. Med. 2021, 19, 105. [Google Scholar] [CrossRef] [PubMed]
- Medić, B.; Rovčanin, B.; Jovanović, G.B.; Radojević-Škodrić, S.; Prostran, M. Kidney Injury Molecule-1 and Cardiovascular Diseases: From Basic Science to Clinical Practice. BioMed Res. Int. 2015, 2015, 854070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Estrela, G.R.; Wasinski, F.; Felizardo, R.J.F.; Souza, L.L.; Câmara, N.O.S.; Bader, M.; Araujo, R.C. MATE-1 modulation by kinin B1 receptor enhances cisplatin efflux from renal cells. Mol. Cell. Biochem. 2017, 428, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Ma, H.; Shao, J.; Wu, J.; Zhou, L.; Zhang, Z.; Wang, Y.; Huang, Z.; Ren, J.; Liu, S.; et al. A Role for Tubular Necroptosis in Cisplatin-Induced AKI. J. Am. Soc. Nephrol. 2015, 26, 2647–2658. [Google Scholar] [CrossRef]
- Qi, Z.; Li, Z.; Li, W.; Liu, Y.; Wang, C.; Lin, H.; Liu, J.; Li, P. Pseudoginsengenin DQ exhibits therapeutic effects in cisplatin-induced acute kidney injury via Sirt1/NF-κB and caspase signaling pathway without compromising its antitumor activity in mice. Molecules 2018, 23, 3038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirooka, Y.; Nozaki, Y. Interleukin-18 in Inflammatory Kidney Disease. Front. Med. 2021, 8, 639103. [Google Scholar] [CrossRef] [PubMed]
- Parikh, C.R.; Devarajan, P. New biomarkers of acute kidney injury. Crit. Care Med. 2008, 36, S159–S165. [Google Scholar] [CrossRef]
- Araki, S.; Haneda, M.; Koya, D.; Sugimoto, T.; Isshiki, K.; Chin-Kanasaki, M.; Uzu, T.; Kashiwagi, A. Predictive impact of elevated serum level of IL-18 for early renal dysfunction in type 2 diabetes: An observational follow-up study. Diabetologia 2007, 50, 867–873. [Google Scholar] [CrossRef] [Green Version]
- Guo, L.; Zhu, B.; Yuan, H.; Zhao, W. Evaluation of serum neutrophil gelatinase-associated lipocalin in older patients with chronic kidney disease. Aging Med. 2020, 3, 35–42. [Google Scholar] [CrossRef] [Green Version]
- McMahon, B.A.; Galligan, M.; Redahan, L.; Martin, T.; Meaney, E.; Cotter, E.J.; Murphy, N.; Hannon, C.; Doran, P.; Marsh, B.; et al. Biomarker Predictors of Adverse Acute Kidney Injury Outcomes in Critically Ill Patients: The Dublin Acute Biomarker Group Evaluation Study. Am. J. Nephrol. 2019, 50, 19–28. [Google Scholar] [CrossRef]
- Soto, K.; Papoila, A.L.; Coelho, S.; Bennett, M.; Ma, Q.; Rodrigues, B.; Fidalgo, P.; Frade, F.; Devarajan, P. Plasma NGAL for the Diagnosis of AKI in Patients Admitted from the Emergency Department Setting. Clin. J. Am. Soc. Nephrol. 2013, 8, 2053–2063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rysz, J.; Gluba-Brzózka, A.; Franczyk, B.; Jabłonowski, Z.; Ciałkowska-Rysz, A. Novel Biomarkers in the Diagnosis of Chronic Kidney Disease and the Prediction of Its Outcome. Int. J. Mol. Sci. 2017, 18, 1702. [Google Scholar] [CrossRef] [PubMed]
- Li, A.; Yi, B.; Liu, Y.; Wang, J.; Dai, Q.; Huang, Y.; Li, Y.C.; Zhang, H. Urinary NGAL and RBP Are Biomarkers of Normoalbuminuric Renal Insufficiency in Type 2 Diabetes Mellitus. J. Immunol. Res. 2019, 2019, 5063089. [Google Scholar] [CrossRef] [PubMed]
- Lhotta, K. Uromodulin and Chronic Kidney Disease. Kidney Blood Press. Res. 2010, 33, 393–398. [Google Scholar] [CrossRef] [PubMed]
- El-Achkar, T.M.; McCracken, R.; Liu, Y.; Heitmeier, M.R.; Bourgeois, S.; Ryerse, J.; Wu, X.-R. Tamm-Horsfall protein translocates to the basolateral domain of thick ascending limbs, interstitium, and circulation during recovery from acute kidney injury. Am. J. Physiol. Physiol. 2013, 304, F1066–F1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stríz, I.; Trebichavský, I. Calprotectin—A pleiotropic molecule in acute and chronic inflammation. Physiol. Res. 2004, 53, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Moghtaderi, M.; Vakili, M.; Fahimi, D.; Esfahani, S.-T.; Sharifzadeh, M. Comparative analysis between urinary calprotectin and serum creatinine for early detection of intrinsic acute kidney injury. Indian J. Nephrol. 2021, 31, 353. [Google Scholar] [CrossRef] [PubMed]
- Schrezenmeier, E.V.; Barasch, J.; Budde, K.; Westhoff, T.; Schmidt-Ott, K.M. Biomarkers in acute kidney injury–pathophysiological basis and clinical performance. Acta Physiol. 2017, 219, 556–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heller, F.; Frischmann, S.; Grufcnbaum, M.; Zidek, W.; Westhoff, T.H. Urinary Calprotectin and the Distinction between Prerenal and Intrinsic Acute Kidney Injury. Clin. J. Am. Soc. Nephrol. 2011, 6, 2347–2355. [Google Scholar] [CrossRef] [Green Version]
- Groschet, A.; Morton, A.J.; Polyak, M.M.R.; Matyjaszek, S.; Freeman, D.E. Detection of calprotectin and its correlation to the accumulation of neutrophils within equine large colon during ischaemia and reperfusion. Equine Veter-J. 2008, 40, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Schrier, R.W. Early intervention in acute kidney injury. Nat. Rev. Nephrol. 2010, 6, 56–59. [Google Scholar] [CrossRef] [PubMed]
- Ebbing, J.; Mathia, S.; Seibert, F.S.; Pagonas, N.; Bauer, F.; Erber, B.; Günzel, K.; Kilic, E.; Kempkensteffen, C.; Miller, K.; et al. Urinary calprotectin: A new diagnostic marker in urothelial carcinoma of the bladder. World J. Urol. 2013, 32, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- Seibert, F.S.; Rosenberger, C.; Mathia, S.; Arndt, R.; Arns, W.; Andrea, H.; Pagonas, N.; Bauer, F.; Zidek, W.; Westhoff, T.H. Urinary Calprotectin Differentiates Between Prerenal and Intrinsic Acute Renal Allograft Failure. Transplantation 2017, 101, 387–394. [Google Scholar] [CrossRef]
- Hosohata, K.; Matsuoka, H.; Iwanaga, K.; Kumagai, E. Urinary vanin-1 associated with chronic kidney disease in hypertensive patients: A pilot study. J. Clin. Hypertens. 2020, 22, 1458–1465. [Google Scholar] [CrossRef] [PubMed]
- Bartucci, R.; Salvati, A.; Olinga, P.; Boersma, Y.L. Vanin 1: Its Physiological Function and Role in Diseases. Int. J. Mol. Sci. 2019, 20, 3891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosohata, K.; Ando, H.; Fujimura, A. Urinary Vanin-1 As a Novel Biomarker for Early Detection of Drug-Induced Acute Kidney Injury. J. Pharmacol. Exp. Ther. 2012, 341, 656–662. [Google Scholar] [CrossRef]
- Hosohata, K.; Matsuoka, H.; Kumagai, E. Association of urinary vanin-1 with kidney function decline in hypertensive patients. J. Clin. Hypertens. 2021, 23, 1316–1321. [Google Scholar] [CrossRef]
- Su, Y.; Lu, J.; Gong, P.; Chen, X.; Liang, C.; Zhang, J. Rapamycin induces autophagy to alleviate acute kidney injury following cerebral ischemia and reperfusion via the mTORC1/ATG13/ULK1 signaling pathway. Mol. Med. Rep. 2018, 18, 5445–5454. [Google Scholar] [CrossRef] [Green Version]
- von Mach, T.; Carlsson, M.C.; Straube, T.; Nilsson, U.; Leffler, H.; Jacob, R. Ligand binding and complex formation of galectin-3 is modulated by pH variations. Biochem. J. 2014, 457, 107–115. [Google Scholar] [CrossRef]
- O’Seaghdha, C.M.; Hwang, S.-J.; Ho, J.E.; Vasan, R.S.; Levy, D.; Fox, C.S. Elevated Galectin-3 Precedes the Development of CKD. J. Am. Soc. Nephrol. 2013, 24, 1470–1477. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.C.; Kuo, P.L. The role of galectin-3 in the kidneys. Int. J. Mol. Sci. 2016, 17, 565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hussain, M.; Habib, A.; Najmi, A. Potential predictive biomarkers for early detection of diabetic kidney disease. Int. J. Infect. Dis. 2020, 101, 355. [Google Scholar] [CrossRef]
- Fredriksson, L.; Li, H.; Eriksson, U. The PDGF family: Four gene products form five dimeric isoforms. Cytokine Growth Factor Rev. 2004, 15, 197–204. [Google Scholar] [CrossRef]
- Boor, P.; Ostendorf, T.; Floege, J. PDGF and the progression of renal disease. Nephrol. Dial. Transplant. 2014, 29, i45–i54. [Google Scholar] [CrossRef] [PubMed]
- Kok, H.M.; Falke, L.L.; Goldschmeding, R.; Nguyen, T.Q. Targeting CTGF, EGF and PDGF pathways to prevent progression of kidney disease. Nat. Rev. Nephrol. 2014, 10, 700–711. [Google Scholar] [CrossRef] [PubMed]
- Floege, J.; Eitner, F.; Alpers, C.E. A New Look at Platelet-Derived Growth Factor in Renal Disease. J. Am. Soc. Nephrol. 2007, 19, 12–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Zhong, X.; Jin, J.; Li, J.; Meng, X.-M. Potential targeted therapy and diagnosis based on novel insight into growth factors, receptors, and downstream effectors in acute kidney injury and acute kidney injury-chronic kidney disease progression. Signal Transduct. Target. Ther. 2020, 5, 9. [Google Scholar] [CrossRef] [Green Version]
- du Cheyron, D.; Daubin, C.; Poggioli, J.; Ramakers, M.; Houillier, P.; Charbonneau, P.; Paillard, M. Urinary measurement of Na+/H+ exchanger isoform 3 (NHE3) protein as new marker of tubule injury in critically ill patients with ARF. Am. J. Kidney Dis. 2003, 42, 497–506. [Google Scholar] [CrossRef]
- Li, X.C.; Zhu, D.; Chen, X.; Zheng, X.; Zhao, C.; Zhang, J.; Soleimani, M.; Rubera, I.; Tauc, M.; Zhou, X.; et al. Proximal tubule-specific deletion of the NHE3 (Na+/H+ exchanger 3) in the kidney attenuates Ang II (angiotensin II)-induced hypertension in mice. Hypertension 2019, 74, 526–535. [Google Scholar] [CrossRef]
- Ratajczyk, K.; Konieczny, A.; Czekaj, A.; Piotrów, P.; Fiutowski, M.; Krakowska, K.; Kowal, P.; Witkiewicz, W.; Marek-Bukowiec, K. The Clinical Significance of Urinary Retinol-Binding Protein 4: A Review. Int. J. Environ. Res. Public Health 2022, 19, 9878. [Google Scholar] [CrossRef]
- Garcon, G.; Leleu, B.; Zerimech, F.; Marez, T.; Haguenoer, J.-M.; Furon, D.; Shirali, P. Biologic Markers of Oxidative Stress and Nephrotoxicity as Studied in Biomonitoring of Adverse Effects of Occupational Exposure to Lead and Cadmium. J. Occup. Environ. Med. 2004, 46, 1180–1186. [Google Scholar] [CrossRef] [PubMed]
- D’Amico, G.; Bazzi, C. Urinary protein and enzyme excretion as markers of tubular damage. Curr. Opin. Nephrol. Hypertens. 2003, 12, 639–643. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-S.; Singh, R.J. Utilities of traditional and novel biomarkers in the management of acute kidney injury. Crit. Rev. Clin. Lab. Sci. 2019, 57, 215–226. [Google Scholar] [CrossRef]
- Matsui, K.; Kamijo-Ikemorif, A.; Sugaya, T.; Yasuda, T.; Kimura, K. Renal Liver-Type Fatty Acid Binding Protein (L-FABP) Attenuates Acute Kidney Injury in Aristolochic Acid Nephrotoxicity. Am. J. Pathol. 2011, 178, 1021–1032. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, A.; Kimura, K.; Sugaya, T.; Yamanouchi, M.; Hikawa, A.; Hirano, N.; Hirata, Y.; Goto, A.; Omata, M. Urinary fatty acid–binding protein as a new clinical marker of the progression of chronic renal disease. J. Lab. Clin. Med. 2004, 143, 23–30. [Google Scholar] [CrossRef]
- Manabe, K.; Kamihata, H.; Motohiro, M.; Senoo, T.; Yoshida, S.; Iwasaka, T. Urinary liver-type fatty acid-binding protein level as a predictive biomarker of contrast-induced acute kidney injury. Eur. J. Clin. Investig. 2011, 42, 557–563. [Google Scholar] [CrossRef] [PubMed]
- Kamijo, A.; Kimura, K.; Sugaya, T.; Yamanouchi, M.; Hase, H.; Kaneko, T.; Hirata, Y.; Goto, A.; Fujita, T.; Omata, M. Urinary free fatty acids bound to albumin aggravate tubulointerstitial damage. Kidney Int. 2002, 62, 1628–1637. [Google Scholar] [CrossRef] [Green Version]
- Kamijo-Ikemori, A.; Sugaya, T.; Kimura, K. Urinary fatty acid binding protein in renal disease. Clin. Chim. Acta 2006, 374, 1–7. [Google Scholar] [CrossRef]
- Assounga, A.G. Beta 2 microglobulin in kidney failure: A review and an algorithm for renal replacement therapy. Saudi J. Kidney Dis. Transplant. 2021, 32, 1214. [Google Scholar]
- Puthiyottil, D.; Priyamvada, P.; Kumar, M.N.; Chellappan, A.; Zachariah, B.; Parameswaran, S. Role of Urinary Beta 2 Microglobulin and Kidney Injury Molecule-1 in Predicting Kidney Function at One Year Following Acute Kidney Injury. Int. J. Nephrol. Renov. Dis. 2021, 14, 225–234. [Google Scholar] [CrossRef]
- Perbal, B. CCN proteins: Multifunctional signalling regulators. Lancet 2004, 363, 62–64. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-F.; Wang, J.-J.; Tu, Y.-C.; Hsu, C.-Y.; Wu, H.-Y.; Fang, C.-C.; Chen, Y.-M.; Wu, M.-S.; Tsai, T.-J. Associations between urinary cysteine-rich protein 61 excretion and kidney function decline in outpatients with chronic kidney disease: A prospective cohort study in Taiwan. BMJ Open 2021, 11, e051165. [Google Scholar] [CrossRef] [PubMed]
- Muramatsu, Y.; Tsujie, M.; Kohda, Y.; Pham, B.; Perantoni, A.O.; Zhao, H.; Jo, S.-K.; Yuen, P.S.; Craig, L.; Hu, X.; et al. Early detection of cysteine rich protein 61 (CYR61, CCN1) in urine following renal ischemic reperfusion injury. Kidney Int. 2002, 62, 1601–1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lai, C.-F.; Lin, S.-L.; Chiang, W.-C.; Chen, Y.-M.; Wu, V.-C.; Young, G.-H.; Ko, W.-J.; Kuo, M.-L.; Tsai, T.-J.; Wu, K.-D. Blockade of cysteine-rich protein 61 attenuates renal inflammation and fibrosis after ischemic kidney injury. Am. J. Physiol. Physiol. 2014, 307, F581–F592. [Google Scholar] [CrossRef] [Green Version]
- Schnaper Schnaper, H.W.; Jandeska, S.; Runyan, C.E.; Hubchak, S.C.; Basu, R.K.; Curley, J.F.; Smith, R.D.; Hayashida, T. TGF-beta signal transduction in chronic kidney disease. Front. Biosci. (Landmark Ed.) 2009, 14, 2448. [Google Scholar] [CrossRef]
- Harskamp, L.R.; Gansevoort, R.T.; Van Goor, H.; Meijer, E. The epidermal growth factor receptor pathway in chronic kidney diseases. Nat. Rev. Nephrol. 2016, 12, 496–506. [Google Scholar] [CrossRef]
- Samarakoon, R.; Dobberfuhl, A.D.; Cooley, C.; Overstreet, J.M.; Patel, S.; Goldschmeding, R.; Meldrum, K.K.; Higgins, P.J. Induction of renal fibrotic genes by TGF-β1 requires EGFR activation, p53 and reactive oxygen species. Cell. Signal. 2013, 25, 2198–2209. [Google Scholar] [CrossRef]
- Kilari, S.; Yang, B.; Sharma, A.; McCall, D.L.; Misra, S. Increased transforming growth factor beta (TGF-β) and pSMAD3 signaling in a Murine Model for Contrast Induced Kidney Injury. Sci. Rep. 2018, 8, 6630. [Google Scholar] [CrossRef]
- Lai, J.Y.; Luo, J.; O’Connor, C.; Jing, X.; Nair, V.; Ju, W.; Randolph, A.; Ben-Dov, I.Z.; Matar, R.N.; Briskin, D.; et al. MicroRNA-21 in glomerular injury. J. Am. Soc. Nephrol. 2015, 26, 805–816. [Google Scholar] [CrossRef] [Green Version]
- Roberts, A.B. Molecular and cell biology of TGF-β. Miner. Electrolyte Metab. 1998, 24, 111–119. [Google Scholar] [CrossRef]
- Chung, A.C.K.; Lan, H.Y. Molecular Mechanisms of TGF-β Signaling in Renal Fibrosis. Curr. Pathobiol. Rep. 2013, 1, 291–299. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-N.; Yang, Q.; Yang, C.; Cai, Y.-T.; Xing, T.; Gao, L.; Wang, F.; Chen, X.; Liu, X.-Q.; He, X.-Y.; et al. Smad3 promotes AKI sensitivity in diabetic mice via interaction with p53 and induction of NOX4-dependent ROS production. Redox Biol. 2020, 32, 101479. [Google Scholar] [CrossRef] [PubMed]
- Morita, A.; Numata, Y.; Kosugi, Y.; Noto, A.; Takeuchi, N.; Uchida, K. Stabilities of N-acetyl-β-d-glucosaminidase (NAG) isoenzymes in urine: Advantage of NAG isoenzyme B measurement in clinical applications. Clin. Chim. Acta 1998, 278, 35–43. [Google Scholar] [CrossRef]
- Hong, J.D.; Lim, I.S. Correlation between glomerular filtration rate and urinary N acetyl-beta-D glucosaminidase in children with persistent proteinuria in chronic glomerular disease. Korean J. Pediatr. 2012, 55, 136–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondiou, M.T.; Bourbouze, R.; Bernard, M.; Percheron, F.; Perez-Gonzalez, N.; Cabezas, J.A. Inhibition of A and B N-acetyl-β-d-glucosaminidase urinary isoenzymes by urea. Clin. Chim. Acta 1985, 149, 67–73. [Google Scholar] [CrossRef]
- Kim, S.R.; Lee, Y.H.; Lee, S.G.; Kang, E.S.; Cha, B.S.; Kim, J.H.; Lee, B.W. Urinary N-acetyl-β-D-glucosaminidase, an early marker of diabetic kidney disease, might reflect glucose excursion in patients with type 2 diabetes. Medicine 2016, 95, e4114. [Google Scholar] [CrossRef]
- Kadokura, T.; Saito, M.; Utsuno, A.; Kazuta, K.; Yoshida, S.; Kawasaki, S.; Nagase, I.; Kageyama, S. Ipragliflozin (ASP1941), a selective sodium-dependent glucose cotransporter 2 inhibitor, safely stimulates urinary glucose excretion without inducing hypoglycemia in healthy Japanese subjects. Diabetol. Int. 2011, 2, 172–182. [Google Scholar] [CrossRef]
- Hart, S.G. Assessment of renal injury in vivo. J. Pharmacol. Toxicol. Methods 2005, 52, 30–45. [Google Scholar] [CrossRef]
- Hartner, A.; Porst, M.; Gauer, S.; Pröls, F.; Veelken, R.; Hilgers, K.F. Glomerular osteopontin expression and macrophage infiltration in glomerulosclerosis of DOCA–salt rats. Am. J. Kidney Dis. 2001, 38, 153–164. [Google Scholar] [CrossRef]
- Varalakshmi, B.; Kiranmyai, V.S.; Aparna, B.; Ram, R.; Rao, P.V.L.N.S.; Kumar, V.S. Plasma osteopontin levels in patients with acute kidney injury requiring dialysis: A study in a tertiary care institute in South India. Int. Urol. Nephrol. 2020, 52, 917–921. [Google Scholar] [CrossRef]
- Magistroni, R.; D’Agati, V.D.; Appel, G.B.; Kiryluk, K. New developments in the genetics, pathogenesis, and therapy of IgA nephropathy. Kidney Int. 2015, 88, 974–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cattran, D.C.; Brenchley, P.E. Membranous nephropathy: Integrating basic science into improved clinical management. Kidney Int. 2017, 91, 566–574. [Google Scholar] [CrossRef] [PubMed]
- Mezzano, S.A.; Barría, M.; Droguett, M.A.; Burgos, M.E.; Ardiles, L.G.; Flores, C.; Egido, J. Tubular NF-κB and AP-1 activation in human proteinuric renal disease. Kidney Int. 2001, 60, 1366–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perkins, N.D. Integrating cell-signalling pathways with NF-κB and IKK function. Nat. Rev. Mol. Cell Biol. 2007, 8, 49–62. [Google Scholar] [CrossRef]
- Yan, X.; Sano, M.; Lu, L.; Wang, W.; Zhang, Q.; Zhang, R.; Wang, L.; Chen, Q.; Fukuda, K.; Shen, W. Plasma concentrations of osteopontin, but not thrombin-cleaved osteopontin, are associated with the presence and severity of nephropathy and coronary artery disease in patients with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2010, 9, 70. [Google Scholar] [CrossRef]
- Zhou, J.; Chen, H.; Fan, Y. Systematic analysis of the expression profile of non-coding RNAs involved in ischemia/reperfusion-induced acute kidney injury in mice using RNA sequencing. Oncotarget 2017, 8, 100196–100215. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Guan, Q.; Liu, X.; Wang, H.; Gleave, M.E.; Nguan, C.Y.C.; Du, C. Relationship of clusterin with renal inflammation and fibrosis after the recovery phase of ischemia-reperfusion injury. BMC Nephrol. 2016, 17, 133. [Google Scholar] [CrossRef] [Green Version]
- Davis, J.W., II; Goodsaid, F.M.; Bral, C.M.; Obert, L.A.; Mandakas, G.; Garner, I.I.C.E.; Collins, N.D.; Smith, R.J.; Rosenblum, I.Y. Quantitative gene expression analysis in a nonhuman primate model of antibiotic-induced nephrotoxicity. Toxicol. Appl. Pharmacol. 2004, 200, 16–26. [Google Scholar] [CrossRef]
- Park, S.; Mathis, K.W.; Lee, I.K. The physiological roles of apolipoprotein J/clusterin in metabolic and cardiovascular diseases. Rev. Endocr. Metab. Disord. 2013, 15, 45–53. [Google Scholar] [CrossRef]
- Wadey, R.M.; Pinches, M.G.; Jones, H.B.; Riccardi, D.; Price, S. Tissue Expression and Correlation of a Panel of Urinary Biomarkers Following Cisplatin-induced Kidney Injury. Toxicol. Pathol. 2013, 42, 591–602. [Google Scholar] [CrossRef] [Green Version]
- Jones, S.A.; Horiuchi, S.; Topley, N.; Yamamoto, N.; Fuller, G.M. The soluble interleukin 6 receptor: Mechanisms of production and implications in disease. FASEB J. 2000, 15, 43–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalaby, M.; Waage, A.; Espevik, T. Cytokine regulation of interleukin 6 production by human endothelial cells. Cell. Immunol. 1989, 121, 372–382. [Google Scholar] [CrossRef] [PubMed]
- Nechemia-Arbely, Y.; Barkan, D.; Pizov, G.; Shriki, A.; Rose-John, S.; Galun, E.; Axelrod, J.H. IL-6/IL-6R axis plays a critical role in acute kidney injury. J. Am. Soc. Nephrol. 2008, 19, 1106–1115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taddei, M.L.; Giannoni, E.; Fiaschi, T.; Chiarugi, P. Anoikis: An emerging hallmark in health and diseases. J. Pathol. 2011, 226, 380–393. [Google Scholar] [CrossRef]
- Meng, X.-M.; Nikolic-Paterson, D.J.; Lan, H.Y. TGF-β: The master regulator of fibrosis. Nat. Rev. Nephrol. 2016, 12, 325–338. [Google Scholar] [CrossRef]
- Devaux, C.A.; Mezouar, S.; Mege, J.-L. The E-Cadherin Cleavage Associated to Pathogenic Bacteria Infections Can Favor Bacterial Invasion and Transmigration, Dysregulation of the Immune Response and Cancer Induction in Humans. Front. Microbiol. 2019, 10, 2598. [Google Scholar] [CrossRef]
- Koziolek, M.; Mueller, G.A.; Dihazi, G.H.; Jung, K.; Altubar, C.; Wallbach, M.; Markovic, I.; Raddatz, D.; Jahn, O.; Karaköse, H.; et al. Urine E-cadherin: A Marker for Early Detection of Kidney Injury in Diabetic Patients. J. Clin. Med. 2020, 9, 639. [Google Scholar] [CrossRef] [Green Version]
- Parmentier, M.; Ghysens, M.; Rypens, F.; Lawson, D.; Pasteels, J.; Pochet, R. Calbindin in vertebrate classes: Immunohistochemical localization and Western blot analysis. Gen. Comp. Endocrinol. 1987, 65, 399–407. [Google Scholar] [CrossRef]
- George, B.; Szilagyi, J.T.; Joy, M.S.; Aleksunes, L.M. Regulation of renal calbindin expression during cisplatin-induced kidney injury. J. Biochem. Mol. Toxicol. 2022, 36, e23068. [Google Scholar] [CrossRef]
- Eltounali, S.A.; Moodley, J.; Naicker, T. Role of kidney biomarkers [Kidney injury molecule-1, Calbindin, Interleukin-18 and Monocyte chemoattractant protein-1] in HIV associated pre-eclampsia. Hypertens. Pregnancy 2017, 36, 288–294. [Google Scholar] [CrossRef]
- Maiwall, R.; Kumar, A.; Bhardwaj, A.; Kumar, G.; Bhadoria, A.S.; Sarin, S.K. Cystatin C predicts acute kidney injury and mortality in cirrhotics: A prospective cohort study. Liver Int. 2017, 38, 654–664. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Lu, B.; Sheng, X.; Jin, N. Cystatin C in Prediction of Acute Kidney Injury: A Systemic Review and Meta-analysis. Am. J. Kidney Dis. 2011, 58, 356–365. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Wang, L.; Hou, Y.; Ma, J.; Chi, R.; Ye, H.; Zhai, Y.; Zhang, D.; Gao, L.; Hu, L.; et al. The influence of glycemic status on the performance of cystatin C for acute kidney injury detection in the critically ill. Ren. Fail. 2019, 41, 139–149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Q.-H.; Liu, D.-W.; Long, Y.; Liu, H.-Z.; Chai, W.-Z.; Wang, X.-T. Acute renal failure during sepsis: Potential role of cell cycle regulation. J. Infect. 2009, 58, 459–464. [Google Scholar] [CrossRef] [PubMed]
- Godi, I.; De Rosa, S.; Martino, F.; Bazzano, S.; Martin, M.; Boni, E.; Carta, M.R.; Diaz, C.T.; Mari, G.; Lorenzin, A.; et al. Urinary [TIMP-2] × [IGFBP7] and serum procalcitonin to predict and assess the risk for short-term outcomes in septic and non-septic critically ill patients. Ann. Intensiv. Care 2020, 10, 46. [Google Scholar] [CrossRef]
- Seo, D.-W.; Li, H.; Qu, C.-K.; Oh, J.; Kim, Y.-S.; Diaz, T.; Wei, B.; Han, J.-W.; Stetler-Stevenson, W.G. Shp-1 Mediates the Antiproliferative Activity of Tissue Inhibitor of Metalloproteinase-2 in Human Microvascular Endothelial Cells. J. Biol. Chem. 2006, 281, 3711–3721. [Google Scholar] [CrossRef] [Green Version]
- Gunnerson, K.J.; Shaw, A.D.; Chawla, L.; Bihorac, A.; Al-Khafaji, A.; Kashani, K.; Lissauer, M.; Shi, J.; Walker, M.G.; Kellum, J.A. TIMP2•IGFBP7 biomarker panel accurately predicts acute kidney injury in high-risk surgical patients. J. Trauma Inj. Infect. Crit. Care 2016, 80, 243–249. [Google Scholar] [CrossRef]
- Maizel, J.; Daubin, D.; Van Vong, L.; Titeca-Beauport, D.; Wetzstein, M.; Kontar, L.; Slama, M.; Klouche, K.; Vinsonneau, C. Urinary TIMP2 and IGFBP7 Identifies High Risk Patients of Short-Term Progression from Mild and Moderate to Severe Acute Kidney Injury during Septic Shock: A Prospective Cohort Study. Dis. Markers 2019, 2019, 3471215. [Google Scholar] [CrossRef]



Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jana, S.; Mitra, P.; Roy, S. Proficient Novel Biomarkers Guide Early Detection of Acute Kidney Injury: A Review. Diseases 2023, 11, 8. https://doi.org/10.3390/diseases11010008
Jana S, Mitra P, Roy S. Proficient Novel Biomarkers Guide Early Detection of Acute Kidney Injury: A Review. Diseases. 2023; 11(1):8. https://doi.org/10.3390/diseases11010008
Chicago/Turabian StyleJana, Sahadeb, Palash Mitra, and Suchismita Roy. 2023. "Proficient Novel Biomarkers Guide Early Detection of Acute Kidney Injury: A Review" Diseases 11, no. 1: 8. https://doi.org/10.3390/diseases11010008
APA StyleJana, S., Mitra, P., & Roy, S. (2023). Proficient Novel Biomarkers Guide Early Detection of Acute Kidney Injury: A Review. Diseases, 11(1), 8. https://doi.org/10.3390/diseases11010008