
Lipid Rafts Interaction of the ARID3A Transcription Factor with EZRIN and G-Actin Regulates B-Cell Receptor Signaling


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
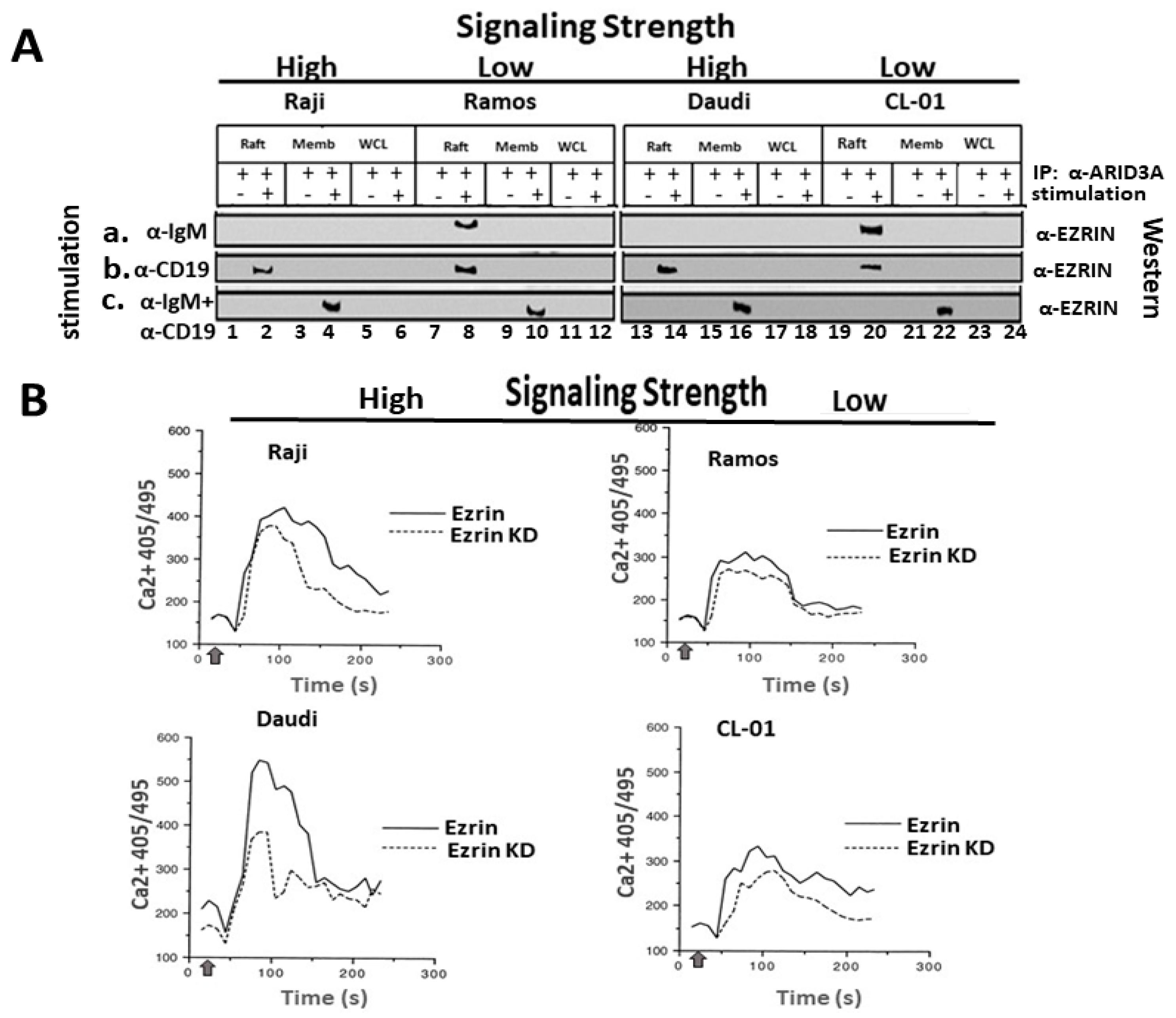
2.1. Variable Levels of ARID3A within Lipid Rafts in Unstimulated and Stimulated B-Cell Lymphomas
2.2. BCR Stimulation Induces a Rafts-Restricted, Transient Interaction of ARID3A with EZRIN Prior to Their Co-Discharge
2.3. EZRIN Knockdown Results in Reduced BCR Signaling Strength
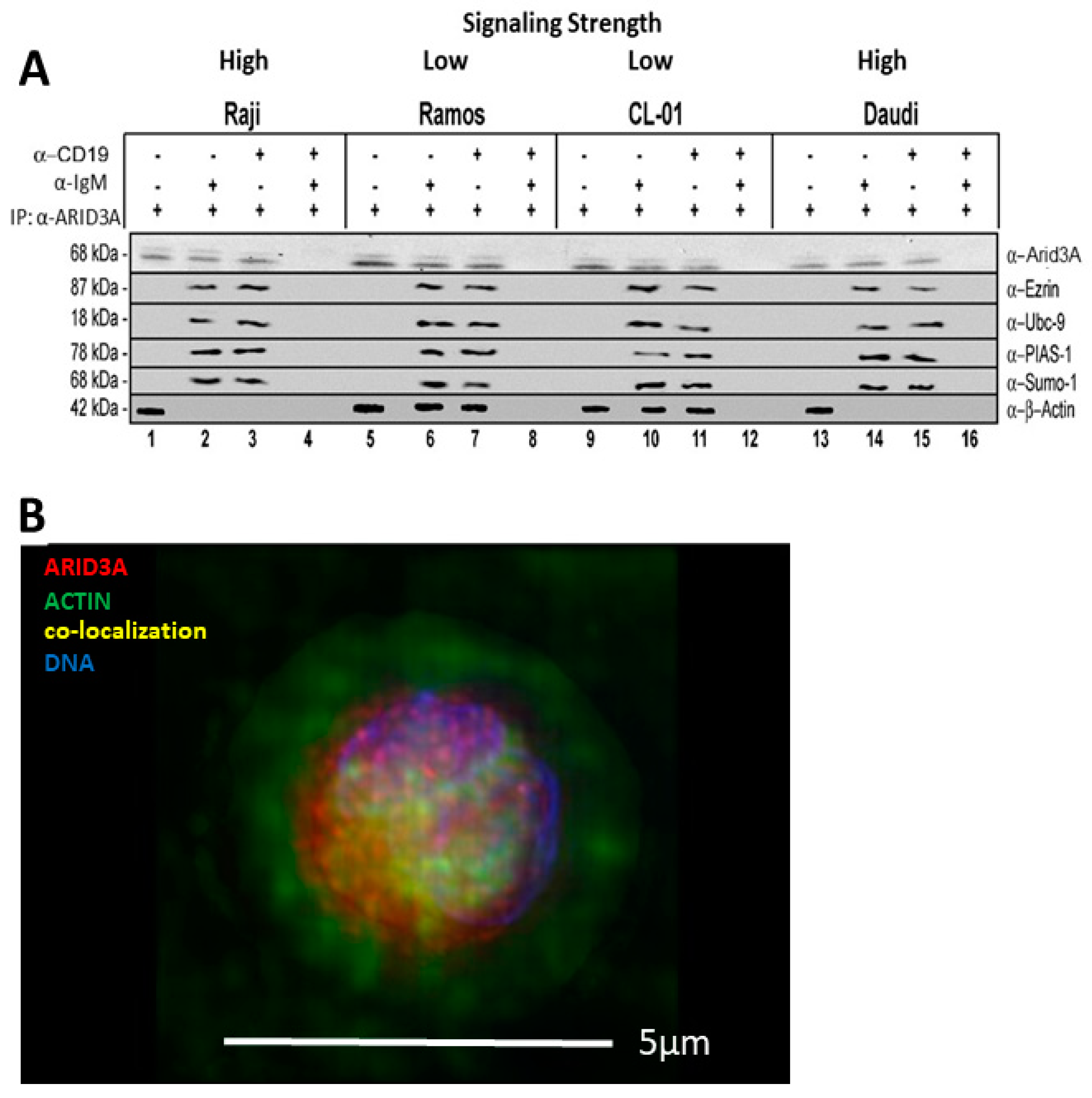
2.4. ARID3A Interacts via EZRIN with Actin in Leukemic Cell Lines and in Normal Murine B Cells
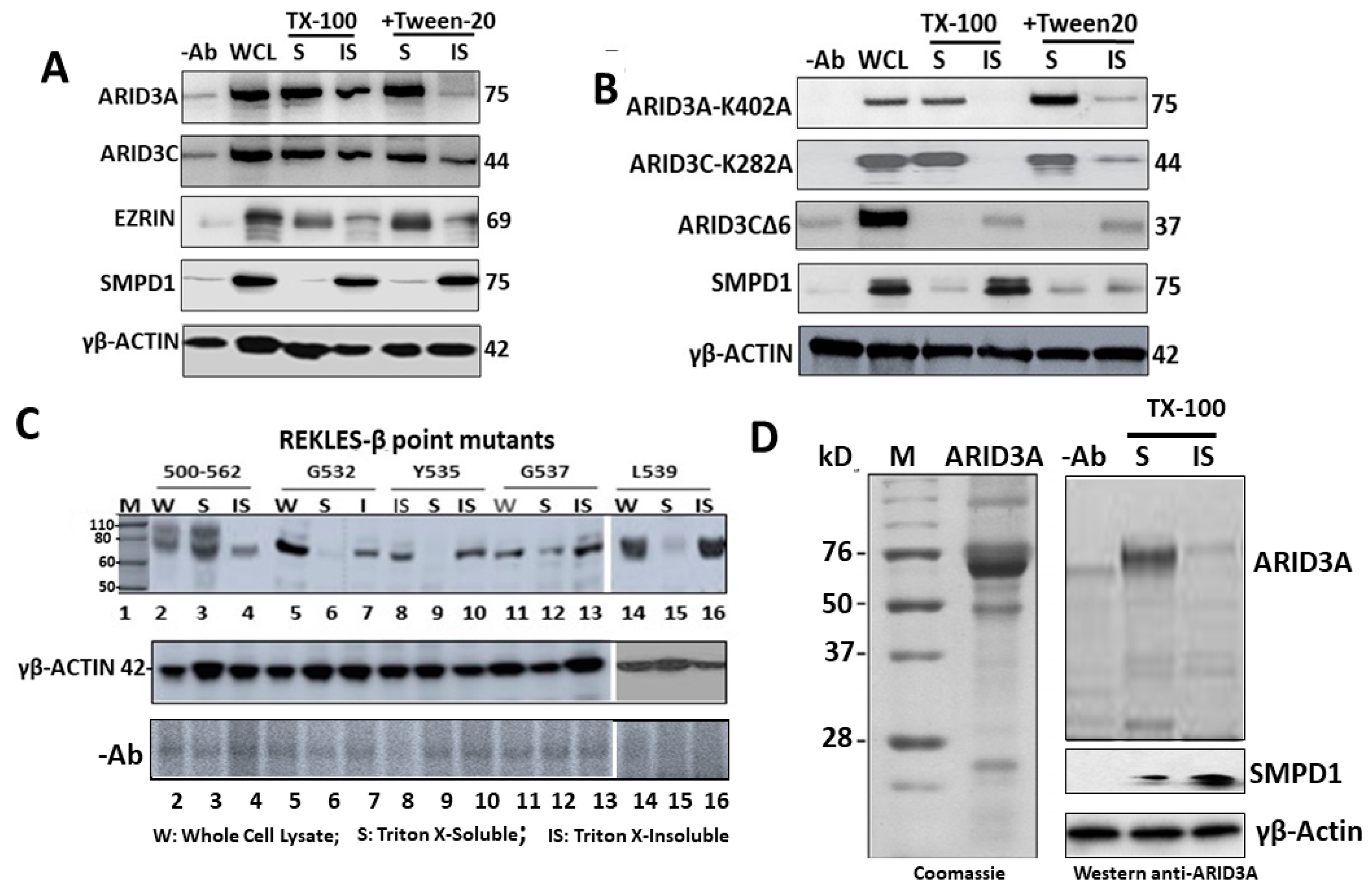
2.5. ARID3A Localizes within Monomeric G-Actin
2.6. SUMO Modification Does Not Control ARID3 Partitioning within G-Actin
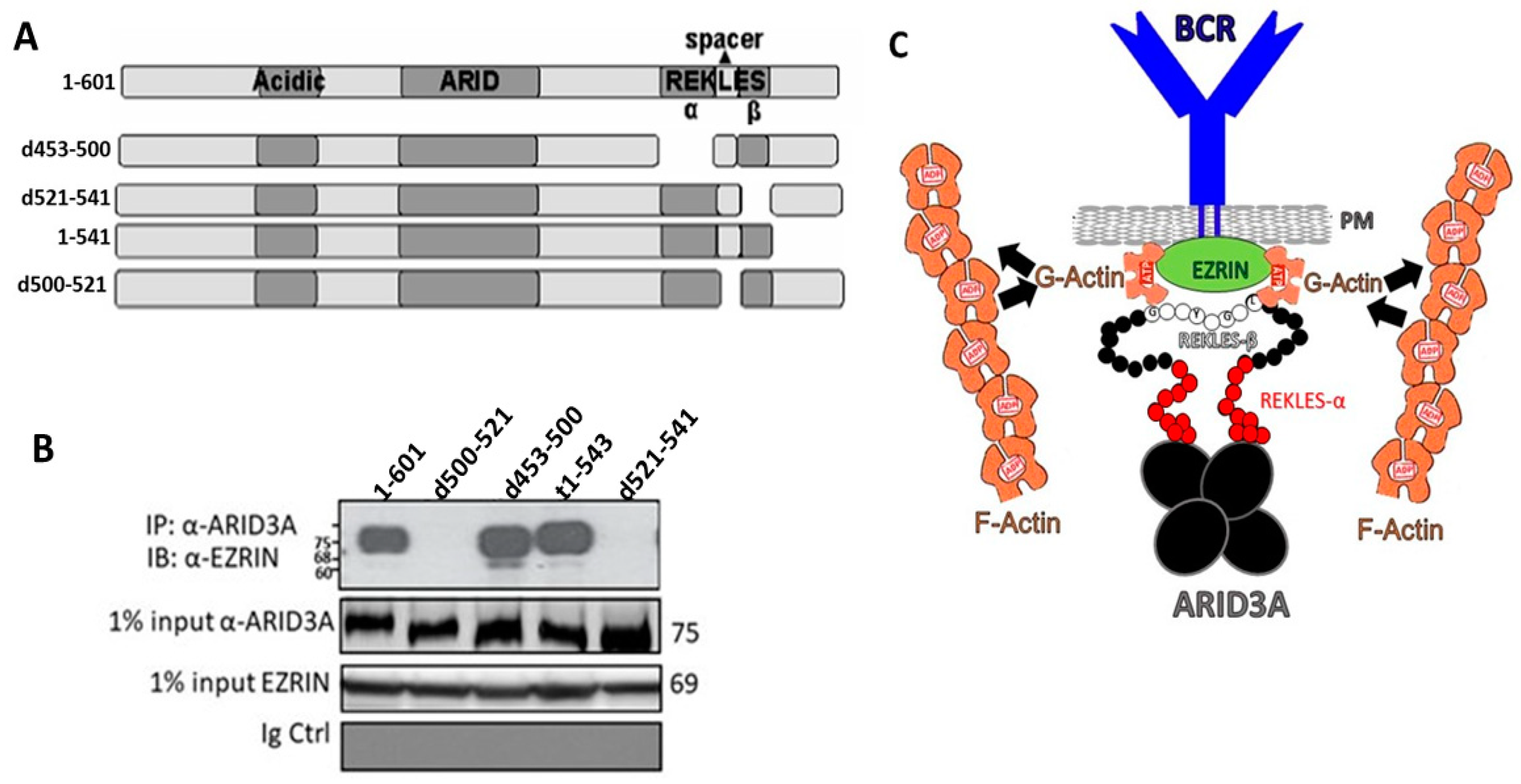
2.7. The REKLES-β Domain Is Essential for EZRIN–ARID3A Interaction
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. Constructs
4.3. Preparation of Stable sh-RNA Retrovirally Transduced B-Cell Lines
4.4. B-Cell Stimulation
4.5. Preparation of Lipid Rafts
4.6. Immunoprecipitation/Western Blot Analyses
4.7. Accumulation of Cytosolic Calcium
4.8. Calcium Calibration
4.9. Purification of Actin
4.10. Immunofluorescence Staining of Murine B Cells
4.11. Quantification of Blot Intensities with ImageJ
4.12. Modulated Polarization Microscopy (MPM)
4.13. Purification of ARID3A in Bacteria
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vedula, P.; Kashina, A. The makings of the ‘actin code’: Regulation of actin’s biological function at the amino acid and nucleotide level. J. Cell Sci. 2018, 131, jcs215509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lodish, H.; Berk, A.; Zipursky, S.L.; Matsudaira, P.; Baltimore, D.; Darnell, J. The actin cytoskeleton. In Molecular Cell Biology, 4th ed.; W. H. Freeman: New York, NY, USA, 2000. [Google Scholar]
- Salbreux, G.; Charras, G.; Paluch, E. Actin cortex mechanics and cellular morphogenesis. Trends Cell Biol. 2012, 22, 536–545. [Google Scholar] [CrossRef]
- Chugh, P.; Paluch, E.K. The actin cortex at a glance. J. Cell Sci. 2018, 131, 186254. [Google Scholar] [CrossRef] [Green Version]
- Winder, S.J. Actin-binding proteins. J. Cell Sci. 2005, 118, 651–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, S.A.; Vega, A.; Riedl, M.; Collins, R.F.; Ostrowski, P.P.; Woods, E.C.; Bertozzi, C.R.; Tammi, M.I.; Lidke, D.S.; Johnson, P.; et al. Transmembrane Pickets Connect Cyto- and Pericellular Skeletons Forming Barriers to Receptor Engagement. Cell 2018, 172, 305–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Yin, W.; Jing, Y.; Kang, D.; Yang, L.; Cheng, J.; Yu, Z.; Peng, Z.; Li, X.; Wen, Y.; et al. The Coordination Between B Cell Receptor Signaling and the Actin Cytoskeleton During B Cell Activation. Front. Immunol. 2019, 9, 3096. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Ma, X.; Zhang, M. Ezrin Mediates Invasion and Metastasis in Tumorigenesis: A Review. Front. Cell Dev. Biol. 2020, 8, 588801. [Google Scholar] [CrossRef]
- Ponuwei, G.A. A glimpse of the ERM proteins. J. Biomed. Sci. 2016, 23, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michie, K.A.; Bermeister, A.; Robertson, N.O.; Goodchild, S.C.; Curmi, P.M.G. Two Sides of the Coin: Ezrin/Radixin/Moesin and Merlin Control Membrane Structure and Contact Inhibition. Int. J. Mol. Sci. 2019, 20, 1996. [Google Scholar] [CrossRef] [Green Version]
- Treanor, B.; Depoil, D.; Bruckbauer, A.; Batista, F.D. Dynamic cortical actin remodeling by ERM proteins controls BCR microcluster organization and integrity. J. Exp. Med. 2011, 208, 1055–1068. [Google Scholar] [CrossRef] [Green Version]
- Hao, S.; August, A. Actin Depolymerization Transduces the Strength of B-Cell Receptor Stimulation. Mol. Biol. Cell 2005, 16, 2275–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, B.; Han, M.; Bernier, M.; Wen, J.-K. Nuclear actin and actin-binding proteins in the regulation of transcription and gene expression. FEBS J. 2009, 276, 2669–2685. [Google Scholar] [CrossRef] [PubMed]
- Miralles, F.; Posern, G.; Zaromytidou, A.-I.; Treisman, R. Actin Dynamics Control SRF Activity by Regulation of Its Coactivator MAL. Cell 2003, 113, 329–342. [Google Scholar] [CrossRef] [Green Version]
- Posern, G.; Miralles, F.; Guettler, S.; Treisman, R. Mutant actins that stabilise F-actin use distinct mechanisms to activate the SRF coactivator MAL. EMBO J. 2004, 23, 3973–3983. [Google Scholar] [CrossRef] [Green Version]
- Haller, K.; Rambaldi, I.; Daniels, E.; Featherstone, M. Subcellular Localization of Multiple PREP2 Isoforms Is Regulated by Actin, Tubulin, and Nuclear Export. J. Biol. Chem. 2004, 279, 49384–49394. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Sime, P.J.; Xu, H.; Williams, M.A.; LaRussa, L.; Georas, S.N.; Guo, J. Yin Yang 1 Is a Novel Regulator of Pulmonary Fibrosis. Am. J. Respir. Crit. Care Med. 2011, 183, 1689–1697. [Google Scholar] [CrossRef] [Green Version]
- Stewart, D.M.; Tian, L.; Nelson, D.L. Mutations that cause the Wiskott-Aldrich syndrome impair the interaction of Wiskott-Aldrich syndrome protein (WASP) with WASP interacting protein. J. Immunol. 1999, 162, 5019–5024. [Google Scholar]
- Pfajfer, L.; Seidel, M.G.; Houmadi, R.; Rey-Barroso, J.; Hirschmugl, T.; Salzer, E.; Antón, I.M.; Urban, C.; Schwinger, W.; Boztug, K.; et al. WIP deficiency severely affects human lymphocyte architecture during migration and synapse assembly. Blood 2017, 130, 1949–1953. [Google Scholar] [CrossRef]
- Pore, D.; Bodo, J.; Danda, A.; Yan, D.; Phillips, J.G.; Lindner, D.; Hill, B.T.; Smith, M.R.; Hsi, E.D.; Gupta, N. Identification of Ezrin-Radixin-Moesin proteins as novel regulators of pathogenic B-cell receptor signaling and tumor growth in diffuse large B-cell lymphoma. Leukemia 2015, 29, 1857–1867. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Luo, Z.; Yang, H. Peripheral B Cell Subsets in Autoimmune Diseases: Clinical Implications and Effects of B Cell-Targeted Therapies. J. Immunol. Res. 2020, 2020, 9518137. [Google Scholar] [CrossRef]
- Schmidt, C.; Kim, D.; Ippolito, G.C.; Naqvi, H.R.; Probst, L.; Mathur, S.; Rosas-Acosta, G.; Wilson, V.G.; Oldham, A.L.; Poenie, M.; et al. Signalling of the BCR is regulated by a lipid rafts-localised transcription factor, Bright. EMBO J. 2009, 28, 711–724. [Google Scholar] [CrossRef] [Green Version]
- Herrscher, R.F.; Kaplan, M.H.; Lelsz, D.L.; Das, C.; Scheuermann, R.; Tucker, P.W. The immunoglobulin heavy-chain matrix-associating regions are bound by Bright: A B cell-specific trans-activator that describes a new DNA-binding protein family. Genes Dev. 1995, 9, 3067–3082. [Google Scholar] [CrossRef] [Green Version]
- Ratliff, M.L.P.; Templeton, T.D.; Ward, J.M.; Webb, C.F.P. The Bright Side of Hematopoiesis: Regulatory Roles of ARID3a/Bright in Human and Mouse Hematopoiesis. Front. Immunol. 2014, 5, 113. [Google Scholar] [CrossRef]
- Saeki, K.; Miura, Y.; Akihiko, Y.; Kurosaki, T.; Yoshimura, A. The B cell-specific major raft protein, Raftlin, is necessary for the integrity of lipid raft and BCR signal transduction. EMBO J. 2003, 22, 3015–3026. [Google Scholar] [CrossRef]
- Sproul, T.W.; Malapati, S.; Kim, J.; Pierce, S.K. Cutting Edge: B Cell Antigen Receptor Signaling Occurs Outside Lipid Rafts in Immature B Cells. J. Immunol. 2000, 165, 6020–6023. [Google Scholar] [CrossRef] [Green Version]
- Putnam, M.A.; Moquin, A.E.; Merrihew, M.; Outcalt, C.; Sorge, E.; Caballero, A.; Gondré-Lewis, T.A.; Drake, J.R. Lipid Raft-Independent B Cell Receptor-Mediated Antigen Internalization and Intracellular Trafficking. J. Immunol. 2003, 170, 905–912. [Google Scholar] [CrossRef] [Green Version]
- Pollard, T.D. Actin and Actin-Binding Proteins. Cold Spring Harb. Perspect. Biol. 2016, 8, e018226. [Google Scholar] [CrossRef] [Green Version]
- Clarke, M.; Spudich, A.J. Nonmuscle Contractile Proteins: The Role of Actin and Myosin in Cell Motility and Shape Determination. Annu. Rev. Biochem. 1977, 46, 797–822. [Google Scholar] [CrossRef]
- Ng, S.Y.; Gunning, P.; Eddy, R.; Ponte, P.; Leavitt, J.; Shows, T.; Kedes, L. Evolution of the functional human beta-actin gene and its multi-pseudogene family: Conservation of noncoding regions and chromosomal dispersion of pseudogenes. Mol. Cell. Biol. 1985, 5, 2720–2732. [Google Scholar] [CrossRef]
- Gressin, L.; Guillotin, A.; Guérin, C.; Blanchoin, L.; Michelot, A. Architecture Dependence of Actin Filament Network Disassembly. Curr. Biol. 2015, 25, 1437–1447. [Google Scholar] [CrossRef] [Green Version]
- Keren, K.; Yam, P.T.; Kinkhabwala, A.; Mogilner, A.; Theriot, J.A. Intracellular fluid flow in rapidly moving cells. Nat. Cell Biol. 2009, 11, 1219–1224. [Google Scholar] [CrossRef]
- Tidwell, J.A.; Schmidt, C.; Heaton, P.; Wilson, V.; Tucker, P.W. Characterization of a new ARID family transcription factor (Brightlike/ARID3C) that co-activates Bright/ARID3A-mediated immunoglobulin gene transcription. Mol. Immunol. 2011, 49, 260–272. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Tucker, P.W. A Regulated Nucleocytoplasmic Shuttle Contributes to Bright’s Function as a Transcriptional Activator of Immunoglobulin Genes. Mol. Cell. Biol. 2006, 26, 2187–2201. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Probst, L.; Das, C.; Tucker, P.W. REKLES Is an ARID3-restricted Multifunctional Domain. J. Biol. Chem. 2007, 282, 15768–15777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, R.-T.; Das, C.; Tucker, P.W. Regulation of matrix attachment region-dependent, lymphocyte-restricted transcription through differential localization within promyelocytic leukemia nuclear bodies. EMBO J. 2000, 19, 4123–4133. [Google Scholar] [CrossRef] [PubMed]
- Sehgal, P.B.; Guo, G.G.; Shah, M.; Kumar, V.; Patel, K. Cytokine Signaling. J. Biol. Chem. 2002, 277, 12067–12074. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Siegel, R.; Roeder, R.G. Interaction of the B Cell-specific Transcriptional Coactivator OCA-B and Galectin-1 and a Possible Role in Regulating BCR-mediated B Cell Proliferation. J. Biol. Chem. 2006, 281, 15505–15516. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Wang, L.; Luo, Y.; Roeder, R.G. Identification and Characterization of a Novel OCA-B Isoform. Immunity 2001, 14, 157–167. [Google Scholar] [CrossRef] [Green Version]
- Caraveo, G.; Van Rossum, D.B.; Patterson, R.L.; Snyder, S.H.; Desiderio, S.; Nakamura, S.; Sakurada, S.; Salahuddin, S.; Osada, Y.; Tanaka, N.; et al. Action of TFII-I Outside the Nucleus as an Inhibitor of Agonist-Induced Calcium Entry. Science 2006, 314, 122–125. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Huang, Y.J.; Xiao, R.; Wang, D.; Acton, T.B.; Montelione, G.T. Solution NMR structure of the ARID domain of human AT-rich interactive domain-containing protein 3A: A human cancer protein interaction network target. Proteins Struct. Funct. Bioinform. 2010, 78, 2170–2175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Invernizzi, G.; Tiberti, M.; Lambrughi, M.; Lindorff-Larsen, K.; Papaleo, E. Communication Routes in ARID Domains between Distal Residues in Helix 5 and the DNA-Binding Loops. PLoS Comput. Biol. 2014, 10, e1003744. [Google Scholar] [CrossRef] [PubMed]
- Gupta, N.; Wollscheid, B.; Watts, J.D.; Scheer, B.; Aebersold, R.; DeFranco, A.L. Quantitative proteomic analysis of B cell lipid rafts reveals that ezrin regulates antigen receptor–mediated lipid raft dynamics. Nat. Immunol. 2006, 7, 625–633. [Google Scholar] [CrossRef] [PubMed]
- Koltzscher, M.; Neumann, C.; König, S.; Gerke, V. Ca2+-dependent Binding and Activation of Dormant Ezrin by Dimeric S100P. Mol. Biol. Cell 2003, 14, 2372–2384. [Google Scholar] [CrossRef] [Green Version]
- Uhlén, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A pathology atlas of the human cancer transcriptome. Science 2017, 357, 2507. [Google Scholar] [CrossRef] [Green Version]
- Dekker, J.D.; Park, D.; Shaffer, I.A.L.; Kohlhammer, H.; Deng, W.; Lee, B.-K.; Ippolito, G.C.; Georgiou, G.; Iyer, V.R.; Staudt, L.M.; et al. Subtype-specific addiction of the activated B-cell subset of diffuse large B-cell lymphoma to FOXP1. Proc. Natl. Acad. Sci. USA 2016, 113, 577–586. [Google Scholar] [CrossRef] [Green Version]
- Deng, W.; Dekker, J.D.; Tucker, H.O. FOXP1 and ARID3A collaborate to activate transcription of core embryonic stem cell factors in Activated B-Cell Diffuse Large B cell Lymphoma. J. Embryol. Stem Cell Res. 2018, 2, e000110. [Google Scholar]
- Wang, Y.; Cao, J.; Liu, W.; Zhang, J.; Wang, Z.; Zhang, Y.; Hou, L.; Chen, S.; Hao, P.; Zhang, L.; et al. Protein tyrosine phosphatase receptor type R (PTPRR) antagonizes the Wnt signaling pathway in ovarian cancer by dephosphorylating and inactivating β-catenin. J. Biol. Chem. 2019, 294, 18306–18323. [Google Scholar] [CrossRef]
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Chan, M.M.; Iritani, B.M. Hem-1: Putting the “WAVE” into actin polymerization during an immune response. FEBS Lett. 2010, 584, 4923–4932. [Google Scholar] [CrossRef] [Green Version]
- Lebensohn, A.M.; Kirschner, M.W. Activation of the WAVE complex by coincident signals controls actin assembly. Mol Cell. 2009, 36, 512–524. [Google Scholar] [CrossRef] [Green Version]
- Suetsugu, S.; Kurisu, S.; Oikawa, T.; Yamazaki, D.; Oda, A.; Takenawa, T. Optimization of WAVE2 complex–induced actin polymerization by membrane-bound IRSp53, PIP3, and Rac. J. Cell Biol. 2006, 173, 571–585. [Google Scholar] [CrossRef] [PubMed]
- Pollitt, A.Y.; Insall, R.H. WASP and SCAR/WAVE proteins: The drivers of actin assembly. J. Cell Sci. 2009, 122, 2575–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takenawa, T.; Suetsugu, S. The WASP-WAVE protein network: Connecting the membrane to the cytoskeleton. Nat. Rev. Mol. Cell Biol. 2007, 8, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Bos, J.L. Linking Rap to cell adhesion. Curr. Opin. Cell Biol. 2005, 17, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Zou, L.; Ding, Z.; Roy, P. Profilin-1 overexpression inhibits proliferation of MDA-MB-231 breast cancer cells partly through p27kip1upregulation. J. Cell. Physiol. 2010, 223, 623–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McConnell, F.M.; Shears, S.B.; Lane, P.J.L.; Scheibel, M.S.; Clark, A.E. Relationships between the degree of cross-linking of surface immunoglobulin and the associated inositol 1,4,5-trisphosphate and Ca2+ signals in human B cells. Biochem. J. 1992, 284, 447–455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, F.; Gregory, C.; Sample, C.; Rowe, M.; Liebowitz, D.; Murray, R.; Rickinson, A.; Kieff, E. Epstein-Barr virus latent membrane protein (LMP1) and nuclear proteins 2 and 3C are effectors of phenotypic changes in B lymphocytes: EBNA-2 and LMP1 cooperatively induce CD23. J. Virol. 1990, 64, 2309–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cerimele, F.; Battle, T.; Lynch, R.; Frank, D.A.; Murad, E.; Cohen, C.; Macaron, N.; Sixbey, J.; Smith, K.; Watnick, R.S.; et al. Reactive oxygen signaling and MAPK activation distinguish Epstein-Barr Virus (EBV)-positive versus EBV-negative Burkitt’s lymphoma. Proc. Natl. Acad. Sci. USA 2004, 102, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Laskov, R.; Berger, N.; Scharff, M.D.; Horwitz, M.S. Tumor necrosis factor-α and CD40L modulate cell surface morphology and induce aggregation in Ramos Burkitt’s lymphoma cells. Leuk. Lymphoma 2006, 47, 507–519. [Google Scholar] [CrossRef]
- Kuhn, J.R.; Poenie, M. Dynamic Polarization of the Microtubule Cytoskeleton during CTL-Mediated Killing. Immunity 2002, 16, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Riddell, A.; Gardner, R.; Perez-Gonzalez, A.; Lopes, T.; Martinez, L. Rmax: A systematic approach to evaluate instrument sort performance using center stream catch. Methods 2015, 82, 64–73. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.; Ippolito, G.C.; Zong, R.-T.; Bryant, J.; Koslovsky, J.; Tucker, P. Bright/ARID3A contributes to chromatin accessibility of the immunoglobulin heavy chain enhancer. Mol. Cancer 2007, 6, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schmidt, C.; Christian, L.; Smith, T.A.; Tidwell, J.; Kim, D.; Tucker, H.O. Lipid Rafts Interaction of the ARID3A Transcription Factor with EZRIN and G-Actin Regulates B-Cell Receptor Signaling. Diseases 2021, 9, 22. https://doi.org/10.3390/diseases9010022
Schmidt C, Christian L, Smith TA, Tidwell J, Kim D, Tucker HO. Lipid Rafts Interaction of the ARID3A Transcription Factor with EZRIN and G-Actin Regulates B-Cell Receptor Signaling. Diseases. 2021; 9(1):22. https://doi.org/10.3390/diseases9010022
Chicago/Turabian StyleSchmidt, Christian, Laura Christian, Tyler A. Smith, Josephine Tidwell, Dongkyoon Kim, and Haley O. Tucker. 2021. "Lipid Rafts Interaction of the ARID3A Transcription Factor with EZRIN and G-Actin Regulates B-Cell Receptor Signaling" Diseases 9, no. 1: 22. https://doi.org/10.3390/diseases9010022
APA StyleSchmidt, C., Christian, L., Smith, T. A., Tidwell, J., Kim, D., & Tucker, H. O. (2021). Lipid Rafts Interaction of the ARID3A Transcription Factor with EZRIN and G-Actin Regulates B-Cell Receptor Signaling. Diseases, 9(1), 22. https://doi.org/10.3390/diseases9010022