Generation of a Collision Cross Section Library for Multi-Dimensional Plant Metabolomics Using UHPLC-Trapped Ion Mobility-MS/MS



Abstract
:1. Introduction
2. Results and Discussion
2.1. CCS Library
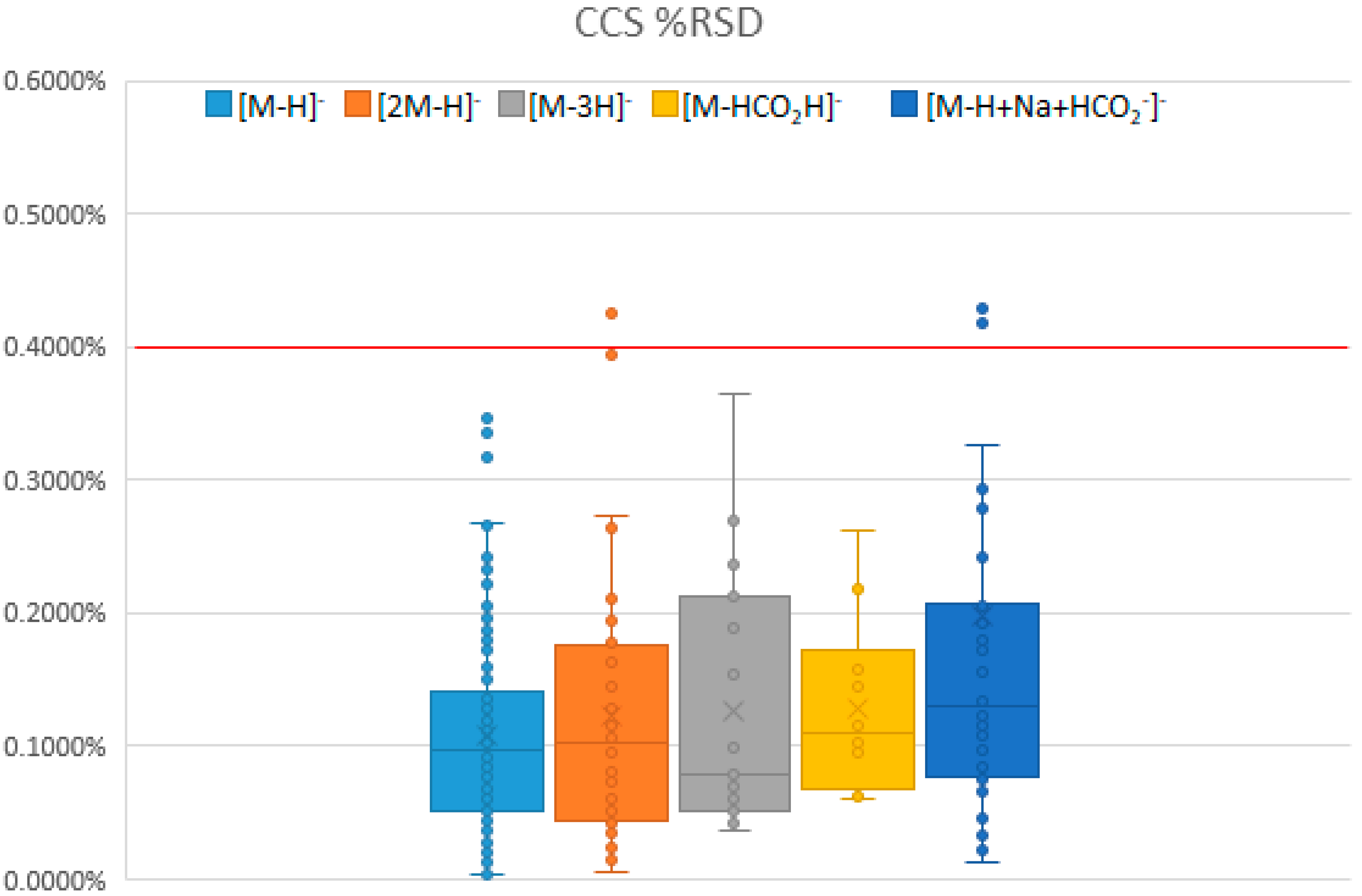
2.1.1. Statistics and Reproducibility
2.1.2. Comparison of CCS Values with Published Data
2.1.3. Isomer Comparisons
2.2. Ion Mobility Spectra Interpretations
2.3. In-TIMS Ion Manipulation
2.4. CCS Matching with Authentic Standards
2.5. CCS Value Matching with Plant Extracts
3. Materials and Methods
3.1. Chemicals
3.2. UHPLC-ESI-TIMS-QTOF-MS
3.2.1. UHPLC
3.2.2. ESI-TIMS-QTOF-MS
3.2.3. Calibration
3.3. Plant Extract Preparation
3.4. Data Processing and CCS Determination
3.5. TIMS Δ6 Adduct Investigations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sumner, L.W.; Lei, Z.; Nikolau, B.J.; Saito, K. Modern plant metabolomics: Advanced natural product gene discoveries, improved technologies, and future prospects. Nat. Prod. Rep. 2015, 32, 212–229. [Google Scholar] [CrossRef] [PubMed]
- Saito, K.; Matsuda, F. Metabolomics for Functional Genomics, Systems Biology, and Biotechnology. Annu. Rev. Plant Biol. 2010, 61, 463–489. [Google Scholar] [CrossRef] [PubMed]
- Sumner, L.W.; Mendes, P.; Dixon, R.A. Plant metabolomics: Large-scale phytochemistry in the functional genomics era. Phytochemistry 2003, 62, 817–836. [Google Scholar] [CrossRef] [Green Version]
- Guy, C.; Kaplan, F.; Kopka, J.; Selbig, J.; Hincha, D.K. Metabolomics of temperature stress. Physiol. Plant. 2008, 132, 220–235. [Google Scholar] [CrossRef]
- Hall, R.D. Plant metabolomics: From holistic hope, to hype, to hot topic. New Phytol. 2006, 169, 453–468. [Google Scholar] [CrossRef]
- Shulaev, V.; Cortes, D.; Miller, G.; Mittler, R. Metabolomics for plant stress response. Physiol. Plant. 2008, 132, 199–208. [Google Scholar] [CrossRef]
- Sumner, L.W.; Styczynski, M.; McLean, J.; Fiehn, O.; Jander, G.; Liao, J.; Sumner, S.; Britz-McKibbin, P.; Welti, R.; Jones, A.D.; et al. Introducing the USA Plant, Algae and Microbial Metabolomics Research Coordination Network (PAMM-NET). Metabolomics 2015, 11, 3–5. [Google Scholar] [CrossRef] [Green Version]
- Neue, U.D. Theory of peak capacity in gradient elution. J. Chromatogr. A 2005, 1079, 153–161. [Google Scholar] [CrossRef]
- Plumb, R.; Castro-Perez, J.; Granger, J.; Beattie, I.; Joncour, K.; Wright, A. Ultra-performance liquid chromatography coupled to quadrupole-orthogonal time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2004, 18, 2331–2337. [Google Scholar] [CrossRef]
- Adahchour, M.; Beens, J.; Brinkman, U.A.T. Recent developments in the application of comprehensive two-dimensional gas chromatography. J. Chromatogr. A 2008, 1186, 67–108. [Google Scholar] [CrossRef]
- Stoll, D.R.; Li, X.; Wang, X.; Carr, P.W.; Porter, S.E.G.; Rutan, S.C. Fast, comprehensive two-dimensional liquid chromatography. J. Chromatogr. A 2007, 1168, 3–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoll, D.R.; Cohen, J.D.; Carr, P.W. Fast, comprehensive online two-dimensional high performance liquid chromatography through the use of high temperature ultra-fast gradient elution reversed-phase liquid chromatography. J. Chromatogr. A 2006, 1122, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Eiceman, G.A.; Karpas, Z.; Hill, H., Jr. Ion Mobility Spectrometry, 3rd ed.; CRC Press: Boca Raton, FL, USA, 2013; ISBN 1439859973. [Google Scholar]
- May, J.C.; McLean, J.A. Ion Mobility-Mass Spectrometry: Time-Dispersive Instrumentation. Anal. Chem. 2015, 87, 1422–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelmann, K.; Silveira, J.A.; Ridgeway, M.E.; Park, M.A. Fundamentals of trapped ion mobility spectrometry. J. Am. Soc. Mass Spectrom. 2015, 26, 14–24. [Google Scholar] [CrossRef]
- Silveira, J.A.; Michelmann, K.; Ridgeway, M.E.; Park, M.A. Fundamentals of Trapped Ion Mobility Spectrometry Part II: Fluid Dynamics. J. Am. Soc. Mass Spectrom. 2016, 27, 585–595. [Google Scholar] [CrossRef]
- Dixon, R.A.; Pasinetti, G.M. Flavonoids and Isoflavonoids: From Plant Biology to Agriculture and Neuroscience. Plant Physiol. 2010, 154, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Dixon, R.A.; Sumner, L.W. Legume Natural Products: Understanding and Manipulating Complex Pathways for Human and Animal Health. Plant Physiol. 2003, 131, 878–885. [Google Scholar] [CrossRef] [Green Version]
- Cseke, L.; Kirakosyan, A.; Kaufman, P.; Warber, S.; Duke, J.; Brielmann, H. Natural Products from Plants, 2nd ed.; CRC Press Taylor & Francis Group: Boca Raton, FL, USA, 2006; ISBN 9780849329760. [Google Scholar]
- Fiehn, O.; Robertson, D.; Griffin, J.; van der Werf, M.; Nikolau, B.; Morrison, N.; Sumner, L.W.; Goodacre, R.; Hardy, N.W.; Taylor, C.; et al. The metabolomics standards initiative (MSI). Metabolomics 2007, 3, 175–178. [Google Scholar] [CrossRef]
- Sumner, L.W.; Amberg, A.; Barrett, D.; Beale, M.H.; Beger, R.; Daykin, C.A.; Fan, T.W.-M.; Fiehn, O.; Goodacre, R.; Griffin, J.L.; et al. Proposed minimum reporting standards for chemical analysis. Metabolomics 2007, 3, 211–221. [Google Scholar] [CrossRef] [Green Version]
- Creek, D.J.; Dunn, W.B.; Fiehn, O.; Griffin, J.L.; Hall, R.D.; Lei, Z.; Mistrik, R.; Neumann, S.; Schymanski, E.L.; Sumner, L.W.; et al. Metabolite identification: Are you sure? And how do your peers gauge your confidence? Metabolomics 2014, 10, 350–353. [Google Scholar] [CrossRef]
- Lei, Z.; Jing, L.; Qiu, F.; Zhang, H.; Huhman, D.; Zhou, Z.; Sumner, L.W. Construction of an Ultrahigh Pressure Liquid Chromatography-Tandem Mass Spectral Library of Plant Natural Products and Comparative Spectral Analyses. Anal. Chem. 2015, 87, 7373–7381. [Google Scholar] [CrossRef]
- Fine, D.; Wherritt, D.; Barsch, A.; Sumner, L.W. Bruker-Sumner Metabobase Plant Libraries 1.0. 2015. Available online: https://www.bruker.com/service/support-upgrades/software-downloads/mass-spectrometry.html (accessed on 15 July 2016).
- Kanu, A.B.; Dwivedi, P.; Tam, M.; Matz, L.; Hill, H.H. Ion mobility-mass spectrometry. J. Mass Spectrom. 2008, 43, 1–22. [Google Scholar] [CrossRef]
- May, J.C.; Morris, C.B.; McLean, J.A. Ion Mobility Collision Cross Section Compendium. Anal. Chem. 2017, 89, 1032–1044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabelica, V.; Shvartsburg, A.A.; Afonso, C.; Barran, P.; Benesch, J.L.P.; Bleiholder, C.; Bowers, M.T.; Bilbao, A.; Bush, M.F.; Campbell, J.L.; et al. Recommendations for reporting ion mobility Mass Spectrometry measurements. Mass Spectrom. Rev. 2019, 38, 291–320. [Google Scholar] [CrossRef] [Green Version]
- Silveira, J.A.; Ridgeway, M.E.; Park, M.A. High Resolution Trapped Ion Mobility Spectrometery of Peptides. Anal. Chem. 2014, 86, 5624–5627. [Google Scholar] [CrossRef]
- Stow, S.M.; Causon, T.J.; Zheng, X.; Kurulugama, R.T.; Mairinger, T.; May, J.C.; Rennie, E.E.; Baker, E.S.; Smith, R.D.; McLean, J.A.; et al. An Interlaboratory Evaluation of Drift Tube Ion Mobility–Mass Spectrometry Collision Cross Section Measurements. Anal. Chem. 2017, 89, 9048–9055. [Google Scholar] [CrossRef] [Green Version]
- Zheng, X.; Aly, N.A.; Zhou, Y.; Dupuis, K.T.; Bilbao, A.; Paurus, V.L.; Orton, D.J.; Wilson, R.; Payne, S.H.; Smith, R.D.; et al. A structural examination and collision cross section database for over 500 metabolites and xenobiotics using drift tube ion mobility spectrometry. Chem. Sci. 2017, 8, 7724–7736. [Google Scholar] [CrossRef] [Green Version]
- Dodds, J.N.; May, J.C.; Mclean, J.A. Correlating Resolving Power, Resolution and Collision Cross Section: Unifying Cross Platform Assessment of Separation Efficiency in Ion Mobility Spectrometry Graphical abstract HHS Public Access. Anal. Chem. 2017, 89, 12176–12184. [Google Scholar] [CrossRef] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UHPLC-TIMS-MS Library CCS 1 | Direct Infusion Mixture CCS | CCS % Difference | |
|---|---|---|---|
| Rutin | 231.05 | 232.37 | 0.56679% |
| Naringin | 215.92 | 215.75 | 0.07876% |
| Naringenin | 163.01 | 162.99 | 0.00818% |
| Chrysin | 156.14 | 156.15 | 0.00854% |
| 6-hydroxyflavone | 155.91 | 155.61 | 0.19688% |
| Average | - | - | 0.16557% |
| Compounds | CCS without Matrix | CCS with Matrix | CCS % Difference |
|---|---|---|---|
| Rutin | 232.56 | 232.64 | 0.03583% |
| Naringin | 215.66 | 215.90 | 0.11431% |
| Naringenin | 162.91 | 162.95 | 0.02455% |
| Chrysin | 156.14 | 156.25 | 0.07042% |
| 6-hydroxyflavone | 156.05 | 155.94 | 0.07265% |
| Average | - | - | 0.06355% |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schroeder, M.; Meyer, S.W.; Heyman, H.M.; Barsch, A.; Sumner, L.W. Generation of a Collision Cross Section Library for Multi-Dimensional Plant Metabolomics Using UHPLC-Trapped Ion Mobility-MS/MS. Metabolites 2020, 10, 13. https://doi.org/10.3390/metabo10010013
Schroeder M, Meyer SW, Heyman HM, Barsch A, Sumner LW. Generation of a Collision Cross Section Library for Multi-Dimensional Plant Metabolomics Using UHPLC-Trapped Ion Mobility-MS/MS. Metabolites. 2020; 10(1):13. https://doi.org/10.3390/metabo10010013
Chicago/Turabian StyleSchroeder, Mark, Sven W. Meyer, Heino M. Heyman, Aiko Barsch, and Lloyd W. Sumner. 2020. "Generation of a Collision Cross Section Library for Multi-Dimensional Plant Metabolomics Using UHPLC-Trapped Ion Mobility-MS/MS" Metabolites 10, no. 1: 13. https://doi.org/10.3390/metabo10010013
APA StyleSchroeder, M., Meyer, S. W., Heyman, H. M., Barsch, A., & Sumner, L. W. (2020). Generation of a Collision Cross Section Library for Multi-Dimensional Plant Metabolomics Using UHPLC-Trapped Ion Mobility-MS/MS. Metabolites, 10(1), 13. https://doi.org/10.3390/metabo10010013