Cervicovaginal Microbiome and Urine Metabolome Paired Analysis Reveals Niche Partitioning of the Microbiota in Patients with Human Papilloma Virus Infections


Abstract
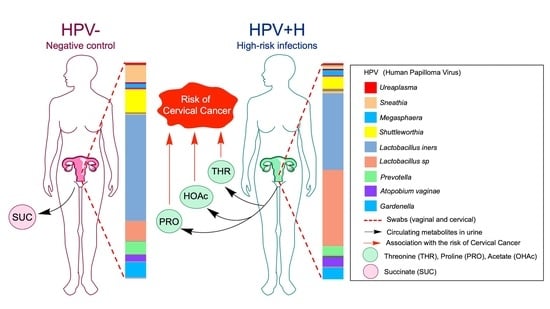
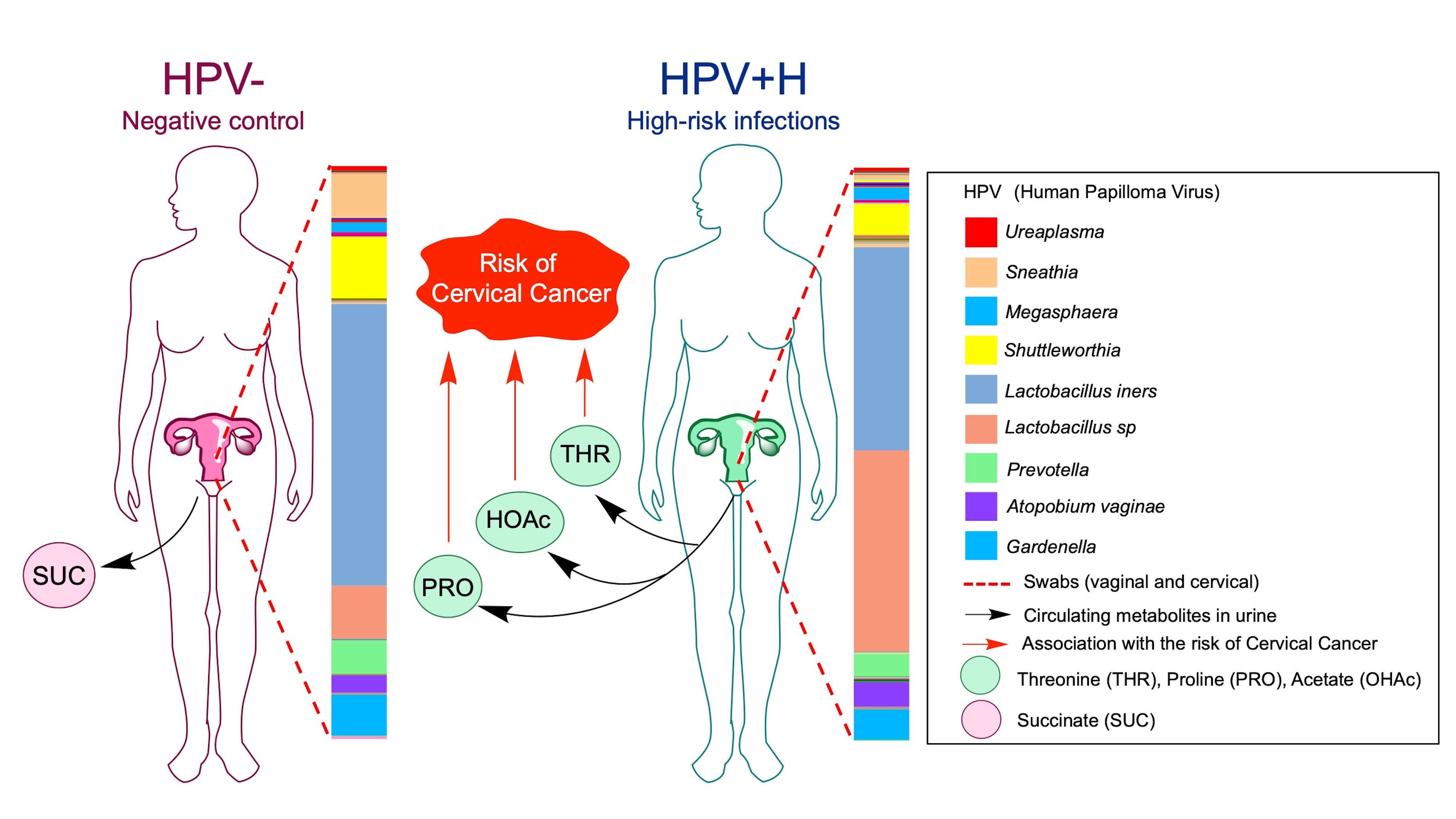
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
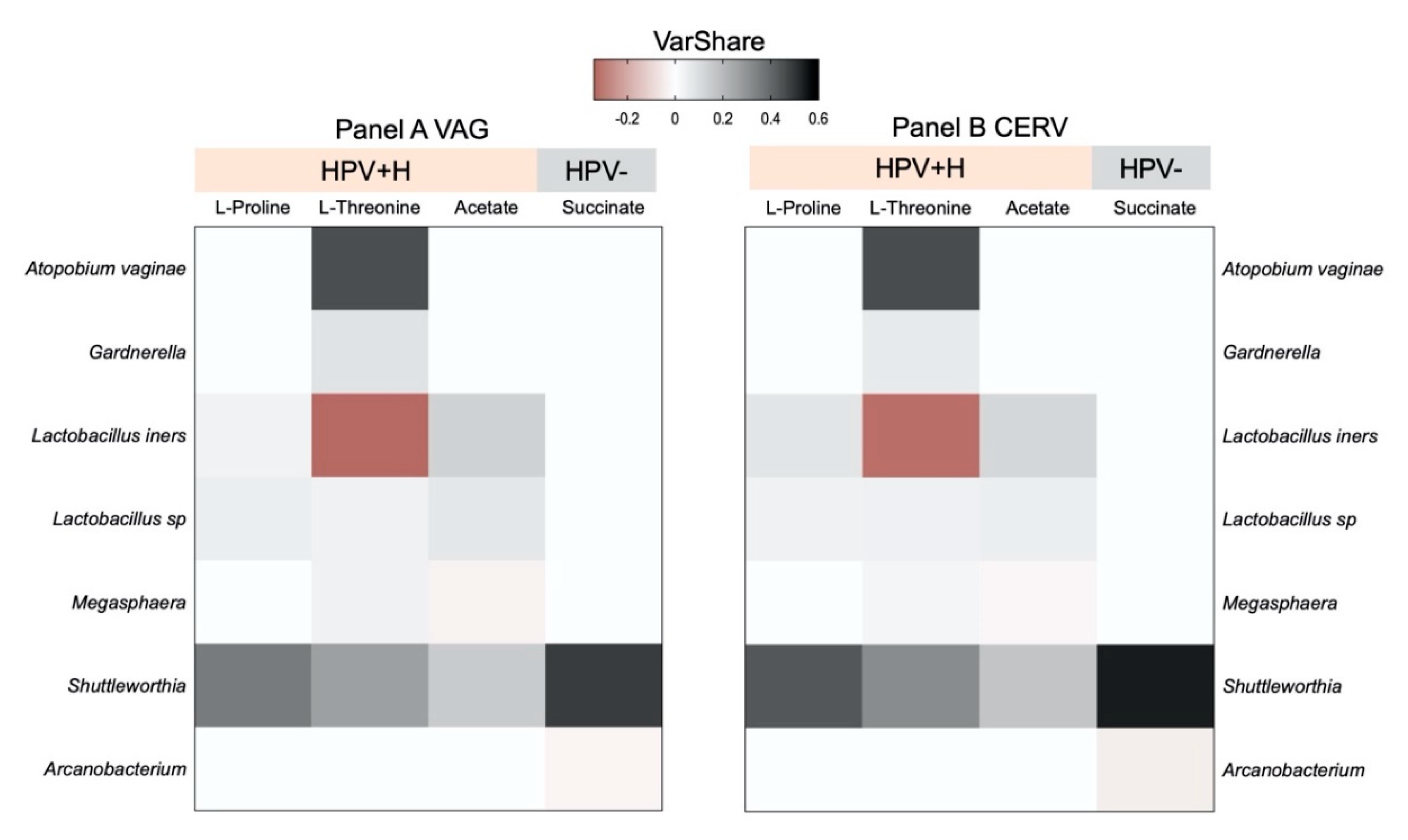{kind=link}
1. Introduction
2. Results
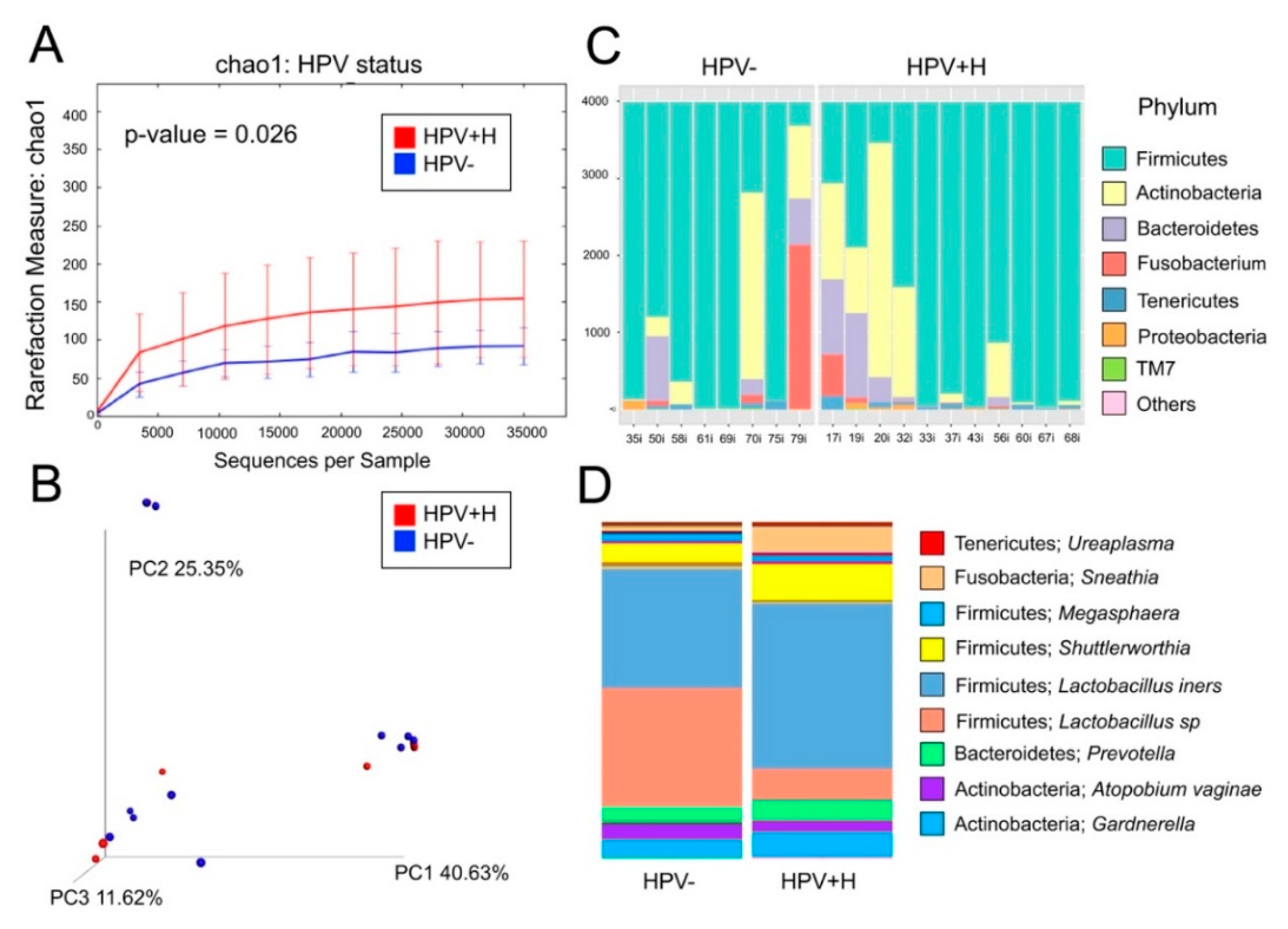
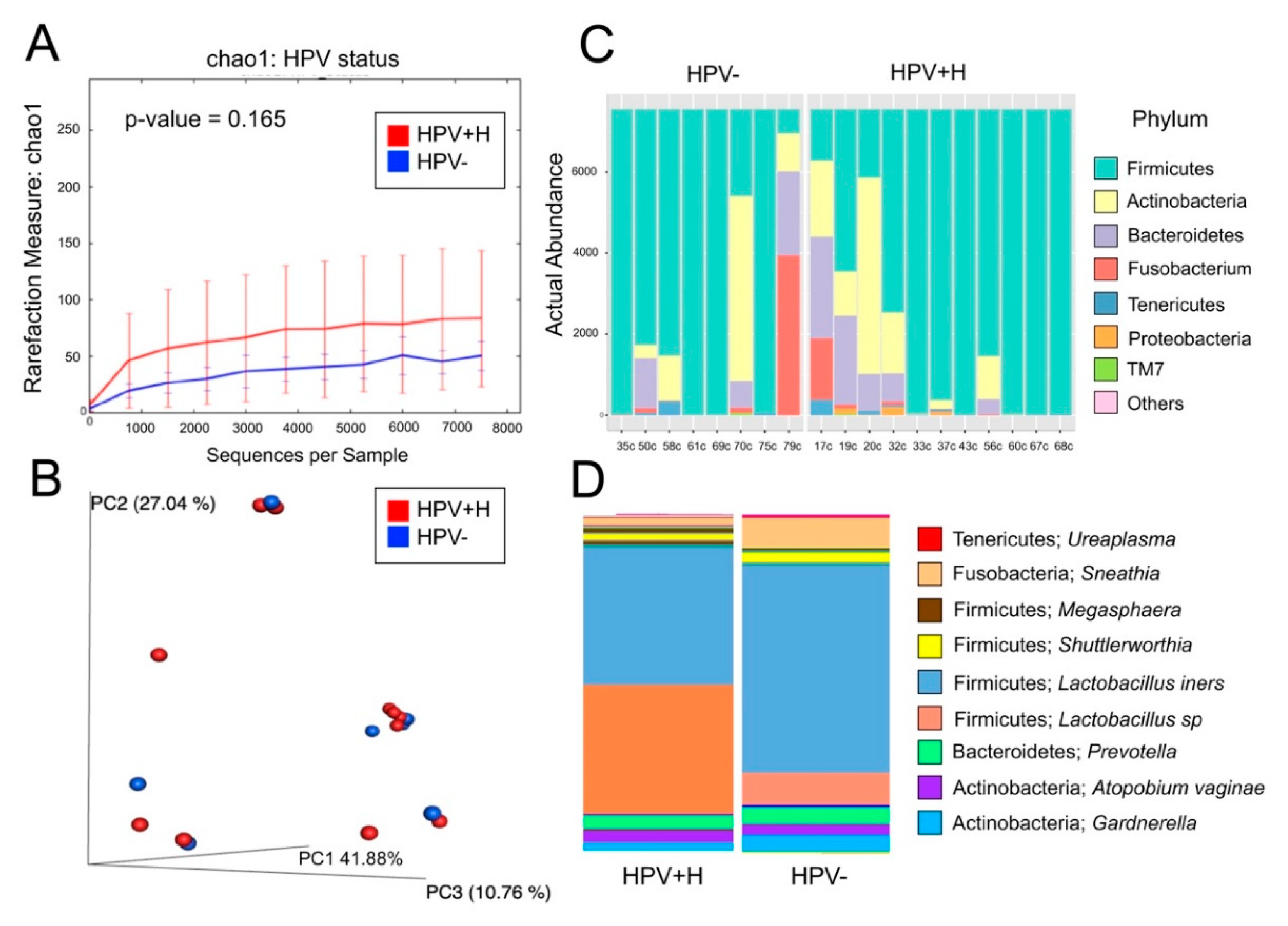
2.1. Bacterial Taxa Associated with Vaginal Samples
2.2. Bacterial Taxa Associated with Cervical Samples
2.3. Association of Urine Metabolites with the Shift in Cervicovaginal Microbiome Induced by HPV+H Infections
2.4. Integration of 16S Community Profiling with Urine Metabolome Identified Taxa Associated with Enzymatic Reactions in HPV+H and HPV−
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Patient Recruitment, Sampling Acquisition, and Human Papilloma Virus Genotyping
5.2. Microbiota Analyses
5.3. Metabolomics Analysis
5.4. Bioinformatic Analysis using Multi-Omics Integration of Metabolome and Microbiome
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mitra, A.; MacIntyre, D.A.; Lee, Y.S.; Smith, A.; Marchesi, J.R.; Lehne, B.; Bhatia, R.; Lyons, D.; Paraskevaidis, E.; Li, J.V.; et al. Cervical intraepithelial neoplasia disease progression is associated with increased vaginal microbiome diversity. Sci. Rep. 2015, 5, 16865. [Google Scholar] [CrossRef] [Green Version]
- Godoy-Vitorino, F.; Romaguera, J.; Zhao, C.; Vargas-Robles, D.; Ortiz-Morales, G.; Vazquez-Sanchez, F.; Sanchez-Vazquez, M.; De la Garza-Casillas, M.; Martinez-Ferrer, M.; White, J.R.; et al. Cervicovaginal Fungi and Bacteria Associated With Cervical Intraepithelial Neoplasia and High-Risk Human Papillomavirus Infections in a Hispanic Population. Front. Microbiol. 2018, 9, 2533. [Google Scholar] [CrossRef] [Green Version]
- Gao, W.; Weng, J.; Gao, Y.; Chen, X. Comparison of the vaginal microbiota diversity of women with and without human papillomavirus infection: A cross-sectional study. BMC Infect. Dis. 2013, 13, 271. [Google Scholar] [CrossRef] [Green Version]
- Abramowitz, L.; Lacau Saint Guily, J.; Moyal-Barracco, M.; Bergeron, C.; Borne, H.; Dahlab, A.; Bresse, X.; Uhart, M.; Cancalon, C.; Catella, L.; et al. Epidemiological and economic burden of potentially HPV-related cancers in France. PLoS ONE 2018, 13, e0202564. [Google Scholar] [CrossRef]
- Dube Mandishora, R.S.; Christiansen, I.K.; Chin’ombe, N.; Duri, K.; Ngara, B.; Rounge, T.B.; Meisal, R.; Ambur, O.H.; Palefsky, J.M.; Stray-Pedersen, B.; et al. Genotypic diversity of anogenital human papillomavirus in women attending cervical cancer screening in Harare, Zimbabwe. J. Med. Virol. 2017, 89, 1671–1677. [Google Scholar] [CrossRef]
- Vargas-Robles, D.; Magris, M.; Morales, N.; de Koning, M.N.C.; Rodríguez, I.; Nieves, T.; Godoy-Vitorino, F.; Sánchez, G.I.; Alcaraz, L.D.; Forney, L.J.; et al. High Rate of Infection by Only Oncogenic Human Papillomavirus in Amerindians. mSphere 2018, 3, e00176-18. [Google Scholar] [CrossRef] [Green Version]
- Garrett, W.S. The gut microbiota and colon cancer. Science 2019, 364, 1133–1135. [Google Scholar] [CrossRef]
- Elinav, E.; Garrett, W.S.; Trinchieri, G.; Wargo, J. The cancer microbiome. Nat. Rev. Cancer 2019, 19, 371–376. [Google Scholar] [CrossRef]
- Brennan, C.A.; Garrett, W.S. Gut Microbiota, Inflammation, and Colorectal Cancer. Annu. Rev. Microbiol. 2016, 70, 395–411. [Google Scholar] [CrossRef] [Green Version]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Brusselaers, N.; Shrestha, S.; Van de Wijgert, J.; Verstraelen, H. Vaginal dysbiosis and the risk of human papillomavirus and cervical cancer: Systematic review and meta-analysis. Am. J. Obstet. Gynecol. 2019, 221, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Ravel, J.; Gajer, P.; Abdo, Z.; Schneider, G.M.; Koenig, S.S.; McCulle, S.L.; Karlebach, S.; Gorle, R.; Russell, J.; Tacket, C.O.; et al. Vaginal microbiome of reproductive-age women. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4680–4687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łaniewski, P.; Barnes, D.; Goulder, A.; Cui, H.; Roe, D.J.; Chase, D.M.; Herbst-Kralovetz, M.M. Linking cervicovaginal immune signatures, HPV and microbiota composition in cervical carcinogenesis in non-Hispanic and Hispanic women. Sci. Rep. 2018, 8, 7593. [Google Scholar] [CrossRef] [PubMed]
- Audirac-Chalifour, A.; Torres-Poveda, K.; Bahena-Roman, M.; Tellez-Sosa, J.; Martinez-Barnetche, J.; Cortina-Ceballos, B.; Lopez-Estrada, G.; Delgado-Romero, K.; Burguete-Garcia, A.I.; Cantu, D.; et al. Cervical Microbiome and Cytokine Profile at Various Stages of Cervical Cancer: A Pilot Study. PLoS ONE 2016, 11, e0153274. [Google Scholar] [CrossRef] [PubMed]
- Onywera, H.; Williamson, A.-L.; Mbulawa, Z.Z.A.; Coetzee, D.; Meiring, T.L. Factors associated with the composition and diversity of the cervical microbiota of reproductive-age Black South African women: A retrospective cross-sectional study. PeerJ 2019, 7, e7488. [Google Scholar] [CrossRef] [PubMed]
- Wiik, J.; Sengpiel, V.; Kyrgiou, M.; Nilsson, S.; Mitra, A.; Tanbo, T.; Monceyron Jonassen, C.; Moller Tannaes, T.; Sjoborg, K. Cervical microbiota in women with cervical intra-epithelial neoplasia, prior to and after local excisional treatment, a Norwegian cohort study. BMC Women’s Health 2019, 19, 30. [Google Scholar] [CrossRef]
- Huang, X.; Li, C.; Li, F.; Zhao, J.; Wan, X.; Wang, K. Cervicovaginal microbiota composition correlates with the acquisition of high-risk human papillomavirus types. Int. J. Cancer 2018, 143, 621–634. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.; Broeck, D.V.; Rossi, R.; Ogbe, E.; Harmon, S.; Mabeya, H. Associations between Vaginal Infections and Potential High-risk and High-risk Human Papillomavirus Genotypes in Female Sex Workers in Western Kenya. Clin. Ther. 2016, 38, 2567–2577. [Google Scholar] [CrossRef]
- Zhang, X.; Li, L.; Butcher, J.; Stintzi, A.; Figeys, D. Advancing functional and translational microbiome research using meta-omics approaches. Microbiome 2019, 7, 154. [Google Scholar] [CrossRef]
- Lamichhane, S.; Sen, P.; Dickens, A.M.; Oresic, M.; Bertram, H.C. Gut metabolome meets microbiome: A methodological perspective to understand the relationship between host and microbe. Methods 2018, 149, 3–12. [Google Scholar] [CrossRef]
- Ilhan, Z.E.; Laniewski, P.; Thomas, N.; Roe, D.J.; Chase, D.M.; Herbst-Kralovetz, M.M. Deciphering the complex interplay between microbiota, HPV, inflammation and cancer through cervicovaginal metabolic profiling. EBioMedicine 2019, 44, 675–690. [Google Scholar] [CrossRef] [Green Version]
- Gardner, A.; Parkes, H.G.; So, P.-W.; Carpenter, G.H. Determining bacterial and host contributions to the human salivary metabolome. J. Oral Microbiol. 2019, 11, 1617014. [Google Scholar] [CrossRef] [Green Version]
- Kho, Z.Y.; Lal, S.K. The Human Gut Microbiome—A Potential Controller of Wellness and Disease. Front. Microbiol. 2018, 9, 1835. [Google Scholar] [CrossRef] [Green Version]
- Shaffer, M.; Armstrong, A.J.S.; Phelan, V.V.; Reisdorph, N.; Lozupone, C.A. Microbiome and metabolome data integration provides insight into health and disease. Transl. Res. 2017, 189, 51–64. [Google Scholar] [CrossRef]
- Liu, R.; Hong, J.; Xu, X.; Feng, Q.; Zhang, D.; Gu, Y.; Shi, J.; Zhao, S.; Liu, W.; Wang, X.; et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 2017, 23, 859–868. [Google Scholar] [CrossRef]
- Pedersen, H.K.; Gudmundsdottir, V.; Nielsen, H.B.; Hyotylainen, T.; Nielsen, T.; Jensen, B.A.; Forslund, K.; Hildebrand, F.; Prifti, E.; Falony, G.; et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 2016, 535, 376–381. [Google Scholar] [CrossRef]
- Kurilshikov, A.; Van den Munckhof, I.C.L.; Chen, L.; Bonder, M.J.; Schraa, K.; Rutten, J.H.W.; Riksen, N.P.; De Graaf, J.; Oosting, M.; Sanna, S.; et al. Gut Microbial Associations to Plasma Metabolites Linked to Cardiovascular Phenotypes and Risk. Circ. Res. 2019, 124, 1808–1820. [Google Scholar] [CrossRef]
- Meyer, T.W.; Hostetter, T.H. Uremic solutes from colon microbes. Kidney Int. 2012, 81, 949–954. [Google Scholar] [CrossRef] [Green Version]
- Jain, A.; Li, X.H.; Chen, W.N. An untargeted fecal and urine metabolomics analysis of the interplay between the gut microbiome, diet and human metabolism in Indian and Chinese adults. Sci. Rep. 2019, 9, 9191. [Google Scholar] [CrossRef] [Green Version]
- Tynkkynen, T.; Wang, Q.; Ekholm, J.; Anufrieva, O.; Ohukainen, P.; Vepsäläinen, J.; Männikkö, M.; Keinänen-Kiukaanniemi, S.; Holmes, M.V.; Goodwin, M.; et al. Proof of concept for quantitative urine NMR metabolomics pipeline for large-scale epidemiology and genetics. Int. J. Epidemiol. 2019, 48, 978–993. [Google Scholar] [CrossRef] [Green Version]
- Holmes, E.; MacIntyre, D.; Modi, N.; Marchesi, J.R. Chapter 8—Handing on Health to the Next Generation: Early Life Exposures. In Metabolic Phenotyping in Personalized and Public Healthcare; Holmes, E., Nicholson, J.K., Darzi, A.W., Lindon, J.C., Eds.; Academic Press: Boston, MA, USA, 2016; pp. 213–264. [Google Scholar]
- Godoy-Vitorino, F.; Ortiz-Morales, G.; Romaguera, J.; Sanchez, M.M.; Martinez-Ferrer, M.; Chorna, N. Discriminating high-risk cervical Human Papilloma Virus infections with urinary biomarkers via non-targeted GC-MS-based metabolomics. PLoS ONE 2018, 13, e0209936. [Google Scholar] [CrossRef] [PubMed]
- Noecker, C.; Eng, A.; Srinivasan, S.; Theriot, C.M.; Young, V.B.; Jansson, J.K.; Fredricks, D.N.; Borenstein, E. Metabolic Model-Based Integration of Microbiome Taxonomic and Metabolomic Profiles Elucidates Mechanistic Links between Ecological and Metabolic Variation. mSystems 2016, 1, e00013–e00015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godoy-Vitorino, F. Human microbial ecology and the rising new medicine. Ann. Transl. Med. 2019, 7, 342. [Google Scholar] [CrossRef] [PubMed]
- Harmon, J.P.; Moran, N.A.; Ives, A.R. Species response to environmental change: Impacts of food web interactions and evolution. Science 2009, 323, 1347–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diether, N.E.; Willing, B.P. Microbial Fermentation of Dietary Protein: An Important Factor in Diet(-)Microbe(-)Host Interaction. Microorganisms 2019, 7, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwasniewski, W.; Wolun-Cholewa, M.; Kotarski, J.; Warchol, W.; Kuzma, D.; Kwasniewska, A.; Gozdzicka-Jozefiak, A. Microbiota dysbiosis is associated with HPV-induced cervical carcinogenesis. Oncol. Lett. 2018, 16, 7035–7047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefani, M.; Taddei, N.; Ramponi, G. Insights into acylphosphatase structure and catalytic mechanism. Cell. Mol. Life Sci. 1997, 53, 141–151. [Google Scholar] [CrossRef]
- Martinez-Ramirez, I.; Carrillo-Garcia, A.; Contreras-Paredes, A.; Ortiz-Sanchez, E.; Cruz-Gregorio, A.; Lizano, M. Regulation of Cellular Metabolism by High-Risk Human Papillomaviruses. Int. J. Mol. Sci. 2018, 19, 1839. [Google Scholar] [CrossRef] [Green Version]
- Moore, H.E.; Davenport, E.L.; Smith, E.M.; Muralikrishnan, S.; Dunlop, A.S.; Walker, B.A.; Krige, D.; Drummond, A.H.; Hooftman, L.; Morgan, G.J.; et al. Aminopeptidase inhibition as a targeted treatment strategy in myeloma. Mol. Cancer 2009, 8, 762–770. [Google Scholar] [CrossRef] [Green Version]
- Polatti, F. Bacterial vaginosis, Atopobium vaginae and nifuratel. Curr. Clin. Pharm. 2012, 7, 36–40. [Google Scholar] [CrossRef] [Green Version]
- Taylor, B.D.; Totten, P.A.; Astete, S.G.; Ferris, M.J.; Martin, D.H.; Ness, R.B.; Haggerty, C.L. Toll-like receptor variants and cervical Atopobium vaginae infection in women with pelvic inflammatory disease. Am. J. Reprod. Immunol. 2018, 79, e12804. [Google Scholar] [CrossRef] [PubMed]
- Haggerty, C.L.; Totten, P.A.; Tang, G.; Astete, S.G.; Ferris, M.J.; Norori, J.; Bass, D.C.; Martin, D.H.; Taylor, B.D.; Ness, R.B. Identification of novel microbes associated with pelvic inflammatory disease and infertility. Sex. Transm. Infect. 2016, 92, 441–446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geissdorfer, W.; Bohmer, C.; Pelz, K.; Schoerner, C.; Frobenius, W.; Bogdan, C. Tuboovarian abscess caused by Atopobium vaginae following transvaginal oocyte recovery. J. Clin. Microbiol. 2003, 41, 2788–2790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Paola, M.; Sani, C.; Clemente, A.M.; Iossa, A.; Perissi, E.; Castronovo, G.; Tanturli, M.; Rivero, D.; Cozzolino, F.; Cavalieri, D.; et al. Characterization of cervico-vaginal microbiota in women developing persistent high-risk Human Papillomavirus infection. Sci. Rep. 2017, 7, 10200. [Google Scholar] [CrossRef]
- Francklyn, C.S.; Mullen, P. Progress and challenges in aminoacyl-tRNA synthetase-based therapeutics. J. Biol. Chem. 2019, 294, 5365–5385. [Google Scholar] [CrossRef] [Green Version]
- Dewan, V.; Reader, J.; Forsyth, K.M. Role of aminoacyl-tRNA synthetases in infectious diseases and targets for therapeutic development. Top. Curr. Chem. 2014, 344, 293–329. [Google Scholar]
- Pham, J.S.; Dawson, K.L.; Jackson, K.E.; Lim, E.E.; Pasaje, C.F.; Turner, K.E.; Ralph, S.A. Aminoacyl-tRNA synthetases as drug targets in eukaryotic parasites. Int. J. Parasitol. Drugs Drug Resist. 2014, 4, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Arita, T.; Morimoto, M.; Yamamoto, Y.; Miyashita, H.; Kitazawa, S.; Hirayama, T.; Sakamoto, S.; Miyamoto, K.; Adachi, R.; Iwatani, M.; et al. Prolyl-tRNA synthetase inhibition promotes cell death in SK-MEL-2 cells through GCN2-ATF4 pathway activation. Biochem. Biophys. Res. Commun. 2017, 488, 648–654. [Google Scholar] [CrossRef]
- Wellman, T.L.; Eckenstein, M.; Wong, C.; Rincon, M.; Ashikaga, T.; Mount, S.L.; Francklyn, C.S.; Lounsbury, K.M. Threonyl-tRNA synthetase overexpression correlates with angiogenic markers and progression of human ovarian cancer. BMC Cancer 2014, 14, 620. [Google Scholar] [CrossRef] [Green Version]
- Weiss, S.; Van Treuren, W.; Lozupone, C.; Faust, K.; Friedman, J.; Deng, Y.; Xia, L.C.; Xu, Z.Z.; Ursell, L.; Alm, E.J.; et al. Correlation detection strategies in microbial data sets vary widely in sensitivity and precision. ISME J. 2016, 10, 1669–1681. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Pena, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [Green Version]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chorna, N.; Romaguera, J.; Godoy-Vitorino, F. Cervicovaginal Microbiome and Urine Metabolome Paired Analysis Reveals Niche Partitioning of the Microbiota in Patients with Human Papilloma Virus Infections. Metabolites 2020, 10, 36. https://doi.org/10.3390/metabo10010036
Chorna N, Romaguera J, Godoy-Vitorino F. Cervicovaginal Microbiome and Urine Metabolome Paired Analysis Reveals Niche Partitioning of the Microbiota in Patients with Human Papilloma Virus Infections. Metabolites. 2020; 10(1):36. https://doi.org/10.3390/metabo10010036
Chicago/Turabian StyleChorna, Nataliya, Josefina Romaguera, and Filipa Godoy-Vitorino. 2020. "Cervicovaginal Microbiome and Urine Metabolome Paired Analysis Reveals Niche Partitioning of the Microbiota in Patients with Human Papilloma Virus Infections" Metabolites 10, no. 1: 36. https://doi.org/10.3390/metabo10010036
APA StyleChorna, N., Romaguera, J., & Godoy-Vitorino, F. (2020). Cervicovaginal Microbiome and Urine Metabolome Paired Analysis Reveals Niche Partitioning of the Microbiota in Patients with Human Papilloma Virus Infections. Metabolites, 10(1), 36. https://doi.org/10.3390/metabo10010036