The Role of Metabolic Enzymes in the Regulation of Inflammation


Abstract
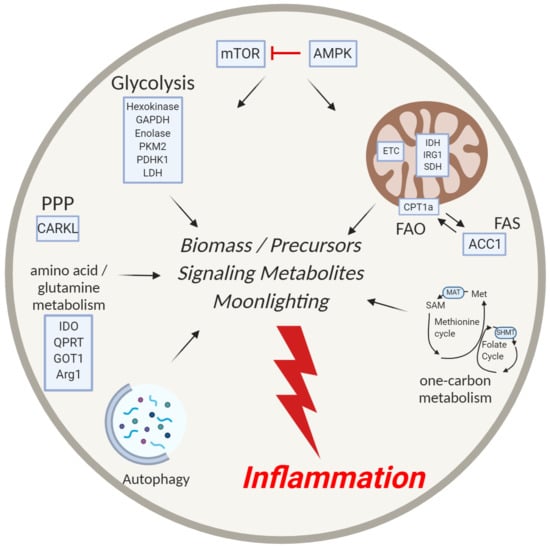
:
1. Introduction
2. mTOR and AMPK—The Master Regulators of Metabolism
3. Glycolysis
3.1. Hexokinase
3.2. Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH)
3.3. Enolase
3.4. Pyruvate Kinase M2 (PKM2)
3.5. Pyruvate Dehydrogenase Kinase 1 (PDHK1)
3.6. Lactate Dehydrogenase (LDH)
4. Mitochondrial Metabolism—TCA Cycle and Electron Transport Chain
4.1. TCA Cycle
4.1.1. Isocitrate Dehydrogenase (IDH)
4.1.2. Immune-Responsive Gene 1 Protein (IRG1)
4.1.3. Succinate Dehydrogenase (SDH)
4.2. Electron Transport Chain (ETC) and ROS
5. The Pentose Phosphate Pathway
CARKL
6. Fatty Acid Metabolism
7. One-Carbon Metabolism
8. Amino Acid Metabolism
9. Autophagy
10. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Patel, C.H.; Leone, R.D.; Horton, M.R.; Powell, J.D. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat. Rev. Drug Discov. 2019, 18, 669–688. [Google Scholar] [CrossRef] [PubMed]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waickman, A.T.; Powell, J.D. mTOR, metabolism, and the regulation of T-cell differentiation and function. Immunol. Rev. 2012, 249, 43–58. [Google Scholar] [CrossRef] [Green Version]
- Weichhart, T.; Hengstschläger, M.; Linke, M. Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 2015, 15, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Saunders, R.N.; Metcalfe, M.S.; Nicholson, M.L. Rapamycin in transplantation: A review of the evidence. Kidney Int. 2001, 59, 3–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Powell, J.D. The mTOR Kinase Differentially Regulates Effector and Regulatory T Cell Lineage Commitment. Immunity 2009, 30, 832–844. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–304. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.; Gudapati, P.; Dragovic, S.; Spencer, C.; Joyce, S.; Killeen, N.; Boothby, M. Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 2010, 32, 743–753. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Zhu, T.; Guan, K.L. TSC2 mediates cellular energy response to control cell growth and survival. Cell 2003, 115, 577–590. [Google Scholar] [CrossRef] [Green Version]
- Gwinn, D.M.; Shackelford, D.B.; Egan, D.F.; Mihaylova, M.M.; Mery, A.; Vasquez, D.S.; Turk, B.E.; Shaw, R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell 2008, 30, 214–226. [Google Scholar] [CrossRef] [Green Version]
- Hardie, D.G. AMP-activated protein kinase-an energy sensor that regulates all aspects of cell function. Genes Dev. 2011, 25, 1895–1908. [Google Scholar] [CrossRef] [Green Version]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef] [Green Version]
- Davies, S.P.; Sim, A.T.R.; Hardie, D.G. Location and function of three sites phosphorylated on rat acetyl-CoA carboxylase by the AMP-activated protein kinase. Eur. J. Biochem. 1990, 187, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Blagih, J.; Coulombe, F.; Vincent, E.E.; Dupuy, F.; Galicia-Vázquez, G.; Yurchenko, E.; Raissi, T.C.; vanderWindt, G.J.W.; Viollet, B.; Pearce, E.L.; et al. The energy sensor AMPK regulates T Cell metabolic adaptation and effector responses invivo. Immunity 2015, 42, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Mayer, A.; Denanglaire, S.; Viollet, B.; Leo, O.; Andris, F. AMP-activated protein kinase regulates lymphocyte responses to metabolic stress but is largely dispensable for immune cell development and function. Eur. J. Immunol. 2008, 38, 948–956. [Google Scholar] [CrossRef] [PubMed]
- Tamás, P.; Hawley, S.A.; Clarke, R.G.; Mustard, K.J.; Green, K.; Hardie, D.G.; Cantrell, D.A. Regulation of the energy sensor AMP-activated protein kinase by antigen receptor and Ca2+ in T lymphocytes. J. Exp. Med. 2006, 203, 1665–1670. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, E.H.; Poffenberger, M.C.; Wong, A.H.T.; Jones, R.G. The role of AMPK in T cell metabolism and function. Curr. Opin. Immunol. 2017, 46, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Rao, E.; Zhang, Y.; Zhu, G.; Hao, J.; Persson, X.M.T.; Egilmez, N.K.; Suttles, J.; Li, B. Deficiency of AMPK in CD8+ T cells suppresses their anti-tumor function by inducing protein phosphatase-mediated cell death. Oncotarget 2015, 6, 7944–7958. [Google Scholar] [CrossRef]
- Pearce, E.L.; Walsh, M.C.; Cejas, P.J.; Harms, G.M.; Shen, H.; Wang, L.S.; Jones, R.G.; Choi, Y. Enhancing CD8 T-cell memory by modulating fatty acid metabolism. Nature 2009, 460, 103–107. [Google Scholar] [CrossRef]
- Rolf, J.; Zarrouk, M.; Finlay, D.K.; Foretz, M.; Viollet, B.; Cantrell, D.A. AMPKα1: A glucose sensor that controls CD8 T-cell memory. Eur. J. Immunol. 2013, 43, 889–896. [Google Scholar] [CrossRef] [Green Version]
- Escobar, D.A.; Botero-Quintero, A.M.; Kautza, B.C.; Luciano, J.; Loughran, P.; Darwiche, S.; Rosengart, M.R.; Zuckerbraun, B.S.; Gomez, H. Adenosine monophosphate-activated protein kinase activation protects against sepsis-induced organ injury and inflammation. J. Surg. Res. 2015, 194, 262–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, A.; Ma, A.G.; Yong, M.; Weiss, C.R.; Ma, Y.; Guan, Q.; Bernstein, C.N.; Peng, Z. AMPK agonist downregulates innate and adaptive immune responses in TNBS-induced murine acute and relapsing colitis. Biochem. Pharmacol. 2010, 80, 1708–1717. [Google Scholar] [CrossRef] [PubMed]
- Paintlia, A.S.; Paintlia, M.K.; Singh, I.; Singh, A.K. Immunomodulatory effect of combination therapy with lovastatin and 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside alleviates neurodegeneration in experimental autoimmune encephalomyelitis. Am. J. Pathol. 2006, 169, 1012–1025. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, Y.; Choi, S.C.; Xu, Z.; Perry, D.J.; Seay, H.; Croker, B.P.; Morel, L. Normalization of CD4+ T cell metabolism reverses lupus. Sci. Transl. Med. 2015, 7, 274ra18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.F.; Lo, Y.C.; Cheng, C.H.; Furtmüller, G.J.; Oh, B.; Andrade-Oliveira, V.; Thomas, A.G.; Bowman, C.E.; Slusher, B.S.; Wolfgang, M.J.; et al. Preventing allograft rejection by targeting immune metabolism. Cell Rep. 2015, 13, 760–770. [Google Scholar] [CrossRef] [Green Version]
- Frauwirth, K.A.; Riley, J.L.; Harris, M.H.; Parry, R.V.; Rathmell, J.C.; Plas, D.R.; Elstrom, R.L.; June, C.H.; Thompson, C.B. The CD28 signaling pathway regulates glucose metabolism. Immunity 2002, 16, 769–777. [Google Scholar] [CrossRef] [Green Version]
- Cham, C.M.; Driessens, G.; O’Keefe, J.P.; Gajewski, T.F. Glucose deprivation inhibits multiple key gene expression events and effector functions in CD8+ T cells. Eur. J. Immunol. 2008, 38, 2438–2450. [Google Scholar] [CrossRef] [Green Version]
- Cham, C.M.; Gajewski, T.F. Glucose Availability regulates IFN-γ production and p70S6 Kinase activation in CD8 + effector T Cells. J. Immunol. 2005, 174, 4670–4677. [Google Scholar] [CrossRef] [Green Version]
- Kornberg, M.D. The immunologic Warburg effect: Evidence and therapeutic opportunities in autoimmunity. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, 12, e1486. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Dillon, C.P.; Shi, L.Z.; Milasta, S.; Carter, R.; Finkelstein, D.; McCormick, L.L.; Fitzgerald, P.; Chi, H.; Munger, J.; et al. The transcription factor myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 2011, 35, 871–882. [Google Scholar] [CrossRef] [Green Version]
- Shi, L.Z.; Wang, R.; Huang, G.; Vogel, P.; Neale, G.; Green, D.R.; Chi, H. HIF1α-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 2011, 208, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Byles, V.; Covarrubias, A.J.; Ben-Sahra, I.; Lamming, D.W.; Sabatini, D.M.; Manning, B.D.; Horng, T. The TSC-mTOR pathway regulates macrophage polarization. Nat. Commun. 2013, 4, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saeed, S.; Quintin, J.; Kerstens, H.H.D.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Stunnenberg, H.G. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345. [Google Scholar] [CrossRef] [Green Version]
- Gerriets, V.A.; Kishton, R.J.; Johnson, M.O.; Cohen, S.; Siska, P.J.; Nichols, A.G.; Rathmell, J.C. Foxp3 and toll-like receptor signaling balance T reg cell anabolic metabolism for suppression. Nat. Immunol. 2016, 17, 1459–1466. [Google Scholar] [CrossRef]
- de Kivit, S.; Mensink, M.; Hoekstra, A.T. Stable human regulatory T cells switch to glycolysis following TNF receptor 2 costimulation. Nat. Metab. 2020, 2, 1046–1061. [Google Scholar] [CrossRef]
- Vats, D.; Mukundan, L.; Odegaard, J.I.; Zhang, L.; Smith, K.L.; Morel, C.R.; Chawla, A. Oxidative metabolism and PGC-1β attenuate macrophage-mediated inflammation. Cell Metab. 2006, 4, 13–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Covarrubias, A.J.; Aksoylar, H.I.; Yu, J. Akt-mTORC1 signaling regulates Acly to integrate metabolic input to control of macrophage activation. eLife 2016, 5, e11612. [Google Scholar] [CrossRef]
- Huang, S.C.C.; Smith, A.M.; Everts, B. Metabolic reprogramming mediated by the mTORC2-IRF4 signaling axis is essential for macrophage alternative activation. Immunity 2016, 45, 817–830. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Chu, Z.; Zhu, L. 2-Deoxy-d-glucose treatment decreases anti-inflammatory M2 macrophage polarization in mice with tumor and allergic airway inflammation. Front. Immunol. 2017, 8, 637. [Google Scholar] [CrossRef]
- Wang, F.; Zhang, S.; Vuckovic, I. Glycolytic Stimulation Is Not a Requirement for M2 Macrophage Differentiation. Cell Metab. 2018, 28, 463–475. [Google Scholar] [CrossRef] [Green Version]
- Gnanaprakasam, J.N.R.; Wang, R. MYC in regulating immunity: Metabolism and beyond. Genes 2017, 8, 88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, S.; Kaminiski, R.; Deshmane, S.; Langford, D.; Khalili, K.; Amini, S.; Datta, P.K. Role of hexokinase-1 in the survival of hiv-1-infected macrophages. Cell Cycle 2015, 14, 980–989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumar, M.; Liu, J.; Ji, Y.; Subramanian, M.; Crompton, J.G.; Yu, Z.; Roychoudhuri, R.; Palmer, D.C.; Muranski, P.; Karoly, E.D.; et al. Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J. Clin. Investig. 2013, 123, 4479–4488. [Google Scholar] [CrossRef] [PubMed]
- Everts, B.; Pearce, E.J. Metabolic control of dendritic cell activation and function: Recent advances and clinical implications. Front. Immunol. 2014, 5, 203. [Google Scholar] [PubMed] [Green Version]
- Everts, B.; Amiel, E.; van der Windt, G.J.W.; Freitas, T.C.; Chott, R.; Yarasheski, K.E.; Pearce, E.L.; Pearce, E.J. Commitment to glycolysis sustains survival of NO-producing inflammatory dendritic cells. Blood 2012, 120, 1422–1431. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, C.M.; Holowka, T.; Sun, J.; Blagih, J.; Amiel, E.; DeBerardinis, R.J.; Cross, J.R.; Jung, E.; Thompson, C.B.; Jones, R.G.; et al. Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 2010, 115, 4742–4749. [Google Scholar] [CrossRef] [Green Version]
- Tannahill, G. Succinate is an inflammatory signal that induces IL-1beta through HIF-1alpha. Nature 2013, 496, 238–242. [Google Scholar] [CrossRef]
- Abboud, G.; Choi, S.C.; Kanda, N.; Zeumer-Spataro, L.; Roopenian, D.C.; Morel, L. Inhibition of glycolysis reduces disease severity in an autoimmune model of rheumatoid arthritis. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef]
- Okano, T.; Saegusa, J.; Nishimura, K.; Takahashi, S.; Sendo, S.; Ueda, Y.; Morinobu, A. 3-bromopyruvate ameliorate autoimmune arthritis by modulating Th17/Treg cell differentiation and suppressing dendritic cell activation. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Seki, S.M.; Stevenson, M.; Rosen, A.M.; Arandjelovic, S.; Gemta, L.; Bullock, T.N.J.; Gaultier, A. Lineage-specific metabolic properties and vulnerabilities of T Cells in the demyelinating central nervous system. J. Immunol. 2017, 198, 4607–4617. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, G.; Xu, Z.G.; Tu, H.; Hu, F.; Dai, J.; Lin, H.K. Lactate is a natural suppressor of RLR signaling by targeting MAVS. Cell 2019, 178, 176–189.e15. [Google Scholar] [CrossRef] [PubMed]
- Wolf, A.J.; Reyes, C.N.; Liang, W.; Becker, C.; Shimada, K.; Wheeler, M.L.; Cho, H.C.; Popescu, N.I.; Coggeshall, K.M.; Arditi, M.; et al. Hexokinase is an innate immune receptor for the detection of bacterial peptidoglycan. Cell 2016, 166, 624–636. [Google Scholar] [CrossRef] [PubMed]
- Liberti, M.V.; Dai, Z.; Wardell, S.E.; Baccile, J.A.; Liu, X.; Gao, X.; Baldi, R.; Mehrmohamadi, M.; Johnson, M.O.; Madhukar, N.S.; et al. A predictive model for selective targeting of the Warburg effect through GAPDH inhibition with a natural product. Cell Metab. 2017, 26, 648–659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shestov, A.A.; Liu, X.; Ser, Z.; Cluntun, A.A.; Hung, Y.P.; Huang, L.; Kim, D.; Le, A.; Yellen, G.; Albeck, J.G.; et al. Quantitative determinants of aerobic glycolysis identify flux through the enzyme GAPDH as a limiting step. eLife 2014, 3, 1–18. [Google Scholar] [CrossRef]
- Nagy, E.; Rigby, W.F.C. Glyceraldehyde-3-phosphate dehydrogenase selectively binds AU-rich RNA in the NAD+-binding region (Rossmann fold). J. Biol. Chem. 1995, 270, 2755–2763. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Curtis, J.D.; Maggi, L.B.; Faubert, B.; Villarino, A.V.; O’Sullivan, D.; Huang, S.C.C.; van der Windt, G.J.W.; Blagih, J.; Qiu, J.; et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 2013, 153, 1239. [Google Scholar] [CrossRef] [Green Version]
- Millet, P.; Vachharajani, V.; McPhail, L.; Yoza, B.; McCall, C.E. GAPDH binding to TNF-alpha mRNA contributes to posttranscriptional repression in monocytes: A novel mechanism of communication between inflammation and metabolism. J. Immunol. 2016, 196, 2541–2551. [Google Scholar] [CrossRef]
- Galván-Peña, S.; Carroll, R.G.; Newman, C.; Hinchy, E.C.; Palsson-McDermott, E.; Robinson, E.K.; Covarrubias, S.; Nadin, A.; James, A.M.; Haneklaus, M.; et al. Malonylation of GAPDH is an inflammatory signal in macrophages. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Yun, J.; Mullarky, E.; Lu, C.; Bosch, K.N.; Kavalier, A.; Rivera, K.; Roper, J.; Chio, I.I.C.; Giannopoulou, E.G.; Rago, C.; et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science 2015, 350, 1391–1396. [Google Scholar] [CrossRef] [Green Version]
- Kornberg, M.D.; Bhargava, P.; Kim, P.M.; Putluri, V.; Snowman, A.M.; Putluri, N.; Calabresi, P.A.; Snyder, S.H. Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 2018, 360, 449–453. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.T.; Han, C.; Xu, D.Q.; Fu, X.W.; Wang, J.S.; Kong, L.Y. 4-Octyl itaconate inhibits aerobic glycolysis by targeting GAPDH to exert anti-inflammatory effects. Nat. Commun. 2019, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollong, M.J.; Lee, G.; Coukos, J.S.; Yun, H.; Zambaldo, C.; Chang, J.W.; Chin, E.N.; Ahmad, I.; Chatterjee, A.K.; Lairson, L.L.; et al. A metabolite-derived protein modification integrates glycolysis with KEAP1–NRF2 signalling. Nature 2018, 562, 600–604. [Google Scholar] [CrossRef]
- Eberhard, T.; Kronvall, G.; Ullberg, M. Surface bound plasmin promotes migration of Streptococcus pneumoniae through reconstituted basement membranes. Microb. Pathog. 1999, 26, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Bergmann, S.; Wild, D.; Diekmann, O.; Frank, R.; Bracht, D.; Chhatwal, G.S.; Hammerschmidt, S. Identification of a novel plasmin(ogen)-binding motif in surface displayed α-enolase of Streptococcus pneumoniae. Mol. Microbiol. 2003, 49, 411–423. [Google Scholar] [CrossRef] [PubMed]
- Sha, J.; Erova, T.E.; Alyea, R.A.; Wang, S.; Olano, J.P.; Pancholi, V.; Chopra, A.K. Surface-expressed enolase contributes to the pathogenesis of clinical isolate ssu of Aeromonas hydrophilaa. J. Bacteriol. 2009, 191, 3095–3107. [Google Scholar] [CrossRef] [Green Version]
- Wygrecka, M.; Marsh, L.M.; Morty, R.E.; Henneke, I.; Guenther, A.; Lohmeyer, J.; Markart, P.; Preissner, K.T. Enolase-1 promotes plasminogen-mediated recruitment of monocytes to the acutely inflamed lung. Blood 2009, 113, 5588–5598. [Google Scholar] [CrossRef] [Green Version]
- De Rosa, V.; Galgani, M.; Porcellini, A.; Colamatteo, A.; Santopaolo, M.; Zuchegna, C.; Matarese, G. Glycolysis controls the induction of human regulatory T cells by modulating the expression of FOXP3 exon 2 splicing variants. Nat. Immunol. 2015, 16, 1174–1184. [Google Scholar] [CrossRef] [Green Version]
- Tamada, M.; Suematsu, M.; Saya, H. Pyruvate kinase M2: Multiple faces for conferring benefits on cancer cells. Clin. Cancer Res. 2012, 18, 5554–5561. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.; Rathmell, J.C.; Macintyre, A.N. Metabolic reprogramming towards aerobic glycolysis correlates with greater proliferative ability and resistance to metabolic inhibition in CD8 versus CD4 T cells. PLoS ONE 2014, 9, e104104. [Google Scholar] [CrossRef] [Green Version]
- Palsson-Mcdermott, E.M.; Curtis, A.M.; Goel, G.; Lauterbach, M.A.R.; Sheedy, F.J.; Gleeson, L.E.; van den Bosch, M.W.M.; Quinn, S.R.; Domingo-Fernandez, R.; Johnson, D.G.W.; et al. Pyruvate kinase M2 regulates hif-1α activity and il-1β induction and is a critical determinant of the Warburg effect in LPS-activated macrophages. Cell Metab. 2015, 21, 65–80. [Google Scholar] [CrossRef] [Green Version]
- He, C.L.; Bian, Y.Y.; Xue, Y.; Liu, Z.X.; Zhou, K.Q.; Yao, C.F.; Lin, Y.; Zou, H.F.; Luo, F.X.; Qu, Y.Y.; et al. Pyruvate Kinase M2 Activates mTORC1 by Phosphorylating AKT1S1. Sci. Rep. 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Mancuso, A.; Tong, X.; Ward, P.S.; Fan, J.; Rabinowitz, J.D.; Thompson, C.B. Pyruvate kinase M2 promotes de novo serine synthesis to sustain mTORC1 activity and cell proliferation. Proc. Natl. Acad. Sci. USA 2012, 109, 6904–6909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, W.; Hu, H.; Chang, R.; Zhong, J.; Knabel, M.; O’Meally, R.; Cole, R.N.; Pandey, A.; Semenza, G.L. Pyruvate kinase M2 is a PHD3-stimulated coactivator for hypoxia-inducible factor 1. Cell 2011, 145, 732–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angiari, S.; Runtsch, M.C.; Sutton, C.E.; Pearce, E.L.; Mills, K.H.G.; O’neill, L.A.J. Pharmacological activation of pyruvate kinase M2 inhibits T Cell pathogenicity and suppresses autoimmunity. Cell Metab. 2020, 31, 391–405. [Google Scholar] [CrossRef] [Green Version]
- Kono, M.; Maeda, K.; Stocton-Gavanescu, I.; Pan, W.; Umeda, M.; Katsuyama, E.; Burbano, C.; Orite, S.Y.K.; Vukelic, M.; Tsokos, M.G.; et al. Pyruvate kinase M2 is requisite for Th1 and Th17 differentiation. JCI Insight 2019, 4. [Google Scholar] [CrossRef]
- Damasceno, L.E.A.; Prado, D.S.; Veras, F.P.; Fonseca, M.M.; Toller-Kawahisa, J.E.; Rosa, M.H.; Alves-Filho, J.C. PKM2 promotes Th17 cell differentiation and autoimmune inflammation by fine-tuning STAT3 activation. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Pucino, V.; Certo, M.; Bombardieri, M.; Pitzalis, C. Lactate buildup at the site of chronic inflammation promotes disease by inducing CD4+ T Cell Metabolic Rewiring. Cell Metab. 2019, 30, 1055–1074. [Google Scholar] [CrossRef] [Green Version]
- Xie, M.; Yu, Y.; Kang, R.; Zhu, S.; Yang, L.; Zeng, L.; Sun, X.; Yang, M.; Billiar, T.R.; Wang, H.; et al. PKM2-Dependent glycolysis promotes NLRP3 and AIM2 inflammasome activation. Nat. Commun. 2016, 7, 1–13. [Google Scholar] [CrossRef]
- Walls, J.F.; Subleski, J.J.; Palmieri, E.M.; Gonzalez Cotto, M.; Gardiner, C.M.; McVicar, D.W.; Finlay, D.K. Metabolic but not transcriptional regulation by PKM2 is important for Natural Killer cell responses. eLife 2020, 9. [Google Scholar] [CrossRef]
- Menk, A.; Scharping, N.E.; Moreci, R.S.; Zeng, X.; Guy, C.; Salvatore, S.; Bae, H.; Xie, J.; Young, H.A.; Wendell, S.G.; et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. 2018, 22, 1509–1521. [Google Scholar] [CrossRef] [Green Version]
- Gerriets, V.A.; Kishton, R.J.; Nichols, A.G.; MacIntyre, A.N.; Inoue, M.; Ilkayeva, O.; Winter, P.S.; Liu, X.; Priyadharshini, B.; Slawinska, M.E.; et al. Metabolic programming and PDHK1 control CD4+ T cell subsets and inflammation. J. Clin. Investig. 2015, 125, 194–207. [Google Scholar] [CrossRef] [PubMed]
- Bian, L.; Josefsson, E.; Jonsson, I.M.; Verdrengh, M.; Ohlsson, C.; Bokarewa, M.; Tarkowski, A.; Magnusson, M. Dichloroacetate alleviates development of collagen II-induced arthritis in female DBA/1 mice. Arthritis Res. 2009, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostroukhova, M.; Goplen, N.; Karim, M.Z.; Michalec, L.; Guo, L.; Liang, Q.; Alam, R. The role of low-level lactate production in airway inflammation in asthma. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 302. [Google Scholar] [CrossRef] [Green Version]
- Peng, M.; Yin, N.; Chhangawala, S.; Xu, K.; Leslie, C.S.; Li, M.O. Aerobic glycolysis promotes T helper 1 cell differentiation through an epigenetic mechanism. Science 2016, 354, 481–484. [Google Scholar] [CrossRef]
- Kaushik, D.K.; Bhattacharya, A.; Mirzaei, R.; Rawji, K.S.; Ahn, Y.; Rho, J.M.; Yong, V.W. Enhanced glycolytic metabolism supports transmigration of brain-infiltrating macrophages in multiple sclerosis. J. Clin. Investig. 2019, 129, 3277–3292. [Google Scholar] [CrossRef] [PubMed]
- Pioli, P.A.; Jonell Hamilton, B.; Connolly, J.E.; Brewer, G.; Rigby, W.F.C. Lactate dehydrogenase is an AU-rich element-binding protein that directly interacts with AUF1. J. Biol. Chem. 2002, 277, 35738–35745. [Google Scholar] [CrossRef] [Green Version]
- Calcinotto, A.; Filipazzi, P.; Grioni, M.; Iero, M.; De Milito, A.; Ricupito, A.; Rivoltini, L. Modulation of microenvironment acidity reverses anergy in human and murine tumor-infiltrating T lymphocytes. Cancer Res. 2012, 72. [Google Scholar] [CrossRef] [Green Version]
- Fischer, K.; Hoffmann, P.; Voelkl, S.; Meidenbauer, N.; Ammer, J.; Edinger, M.; Kreutz, M. Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 2007, 109, 3812–3819. [Google Scholar] [CrossRef]
- Angelin, A.; Gil-de-Gómez, L.; Dahiya, S.; Jiao, J.; Guo, L.; Levine, M.H.; Beier, U.H. Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab. 2017, 25, 1282–1293. [Google Scholar] [CrossRef] [Green Version]
- Comito, G.; Iscaro, A.; Bacci, M.; Morandi, A.; Ippolito, L.; Parri, M.; Chiarugi, P. Lactate modulates CD4 + T-cell polarization and induces an immunosuppressive environment, which sustains prostate carcinoma progression via TLR8/miR21 axis. Oncogene 2019, 38, 3681–3695. [Google Scholar] [CrossRef]
- Colegio, O.R.; Chu, N.Q.; Szabo, A.L.; Chu, T.; Rhebergen, A.M.; Jairam, V.; Medzhitov, R. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014, 513, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Luo, J.; Kuang, D.; Xu, S.; Duan, Y.; Xia, Y.; Yang, X.P. Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2α–mediated tumor progression. J. Clin. Investig. 2019, 129, 631–646. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohn, T.; Rapp, S.; Luther, N.; Klein, M.; Bruehl, T.J.; Kojima, N.; Bopp, T. Tumor immunoevasion via acidosis-dependent induction of regulatory tumor-associated macrophages. Nat. Immunol. 2018, 56, KV-092. [Google Scholar] [CrossRef]
- Zhang, D.; Tang, Z.; Huang, H.; Zhou, G.; Cui, C.; Weng, Y.; Liu, W.; Kim, S.; Lee, S.; Perez-Neut, M.; et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019, 574, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Mehta, M.M.; Weinberg, S.E.; Chandel, N.S. Mitochondrial control of immunity: Beyond ATP. Nat. Rev. Immunol. 2017, 17, 608–620. [Google Scholar] [CrossRef]
- Bailis, W.; Shyer, J.A.; Zhao, J.; Canaveras, J.C.G.; al Khazal, F.J.; Qu, R.; Steach, H.R.; Bielecki, P.; Khan, O.; Jackson, R.; et al. Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 2019, 571, 403–407. [Google Scholar] [CrossRef]
- Ryan, D.G.; O’Neill, L.A.J. Krebs cycle reborn in macrophage immunometabolism. Annu. Rev. Immunol. 2020, 38, 289–313. [Google Scholar] [CrossRef] [Green Version]
- Yamada, K.J.; Heim, C.E.; Xi, X.; Attri, K.S.; Wang, D.; Zhang, W.; Singh, P.K.; Bronich, T.K.; Kielian, T. Monocyte metabolic reprogramming promotes pro-inflammatory activity and Staphylococcus aureus biofilm clearance. PLoS Pathog. 2020, 16, e1008354. [Google Scholar] [CrossRef] [PubMed]
- Bailey, J.D.; Diotallevi, M.; Nicol, T.; McNeill, E.; Shaw, A.; Chuaiphichai, S.; Hale, A.; Starr, A.; Nandi, M.; Stylianou, E.; et al. Nitric Oxide Modulates Metabolic Remodeling in Inflammatory Macrophages through TCA Cycle Regulation and Itaconate Accumulation. Cell Rep. 2019, 28, 218–230. [Google Scholar] [CrossRef] [Green Version]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 1–17. [Google Scholar] [CrossRef]
- Infantino, V.; Convertini, P.; Cucci, L.; Panaro, M.A.; di Noia, M.A.; Calvello, R.; Palmieri, F.; Iacobazzi, V. The mitochondrial citrate carrier: A new player in inflammation. Biochem. J. 2011, 438, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meiser, J.; Krämer, L.; Sapcariu, S.C.; Battello, N.; Ghelfi, J.; D’Herouel, A.F.; Skupin, A.; Hiller, K. Pro-inflammatory macrophages sustain pyruvate oxidation through pyruvate dehydrogenase for the synthesis of itaconate and to enable cytokine expression. J. Biol. Chem. 2016, 291, 3932–3946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witkowski, A.; Thweatt, J.; Smith, S. Mammalian ACSF3 protein is a malonyl-CoA synthetase that supplies the chain extender units for mitochondrial fatty acid synthesis. J. Biol. Chem. 2011, 286, 33729–33736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019, 19, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Tallam, A.; Perumal, T.M.; Antony, P.M.; Jäger, C.; Fritz, J.V.; Vallar, L.; Balling, R.; del Sol, A.; Michelucci, A. Gene regulatory network inference of immunoresponsive gene 1 (IRG1) identifies interferon regulatory factor 1 (IRF1) as its transcriptional regulator in mammalian macrophages. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, R.; Zhang, P.; Wang, Y.; Tao, K. Itaconate: A metabolite regulates inflammation response and oxidative stress. Oxidative Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.C.; Griss, T.; et al. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef]
- Mills, E.L.; Kelly, B.; Logan, A.; Costa, A.S.H.; Varma, M.; Bryant, C.E.; Tourlomousis, P.; Däbritz, J.H.M.; Gottlieb, E.; Latorre, I.; et al. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 2016, 167, 457–470. [Google Scholar] [CrossRef] [Green Version]
- Tarasenko, T.N.; Pacheco, S.E.; Koenig, M.K.; Gomez-Rodriguez, J.; Kapnick, S.M.; Diaz, F.; Zerfas, P.M.; Barca, E.; Sudderth, J.; DeBerardinis, R.J.; et al. Cytochrome c oxidase activity is a metabolic checkpoint that regulates cell fate decisions during T cell activation and differentiation. Cell Metab. 2017, 25, 1254–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific t cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garaude, J.; Acín-Pérez, R.; Martínez-Cano, S.; Enamorado, M.; Ugolini, M.; Nistal-Villán, E.; Hervás-Stubbs, S.; Pelegrín, P.; Sander, L.E.; Enríquez, J.A.; et al. Mitochondrial respiratory-chain adaptations in macrophages contribute to antibacterial host defense. Nat. Immunol. 2016, 17, 1037–1045. [Google Scholar] [CrossRef] [Green Version]
- Clementi, E.; Brown, G.C.; Feelisch, M.; Moncada, S. Persistent inhibition of cell respiration by nitric oxide: Crucial role of S-nitrosylation of mitochondrial complex I and protective action of glutathione. Proc. Natl. Acad. Sci. USA 1998, 95, 7631–7636. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- NCT02762500. Available online: www.clinicaltrials.gov (accessed on 6 July 2020).
- Puleston, D.J.; Buck, M.D.; Klein, R.I.; Pearce, E.J.; Balabanov, S.; Pearce, E.L. Polyamines and eIF5A hypusination modulate mitochondrial respiration and macrophage activation. Cell Metab. 2019, 30, 352–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.H.; Wolff, E.C. Hypusine, a polyamine-derived amino acid critical for eukaryotic translation. J. Biol. Chem. 2018, 293, 18710–18718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, R.Z.; Jiang, S.; Zhang, L.; Yu, Z. Mitochondrial electron transport chain, ROS generation and uncoupling (Review). Int. J. Mol. Med. 2019, 44, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Cameron, A.M.; Castoldi, A.; Sanin, D.E.; Flachsmann, L.J.; Field, C.S.; Puleston, D.J.; Kyle, R.L.; Patterson, A.E.; Hässler, F.; Buescher, J.M.; et al. Inflammatory macrophage dependence on NAD + salvage is a consequence of reactive oxygen species–mediated DNA damage. Nat. Immunol. 2019, 20, 420–432. [Google Scholar] [CrossRef]
- Zhang, D.; Jin, W.; Wu, R.; Li, J.; Park, S.A.; Tu, E.; Zanvit, P.; Xu, J.; Liu, O.; Cain, A.; et al. High Glucose Intake Exacerbates Autoimmunity through Reactive-Oxygen-Species-Mediated TGF-β Cytokine Activation. Immunity. 2019 51, 671–681. [CrossRef]
- Park, M.; Lee, S.; Moon, S. Metformin attenuates graft-versus-host disease via restricting mammalian target of rapamycin/signal transducer and activator of transcription 3 and promoting adenosine monophosphate-activated protein kinase-autophagy for the balance between T helper 17 and Tregs. Transl. Res. 2016, 173, 115–130. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Kishton, R.J.; Rathmell, J. A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 2016, 16, 553–565. [Google Scholar] [CrossRef] [Green Version]
- Nagy, C.; Haschemi, A. Time and demand are two critical dimensions of immunometabolism: The process of macrophage activation and the pentose phosphate pathway. Front. Immunol. 2015, 6. [Google Scholar] [CrossRef] [Green Version]
- Gaber, T.; Strehl, C.; Buttgereit, F. Metabolic regulation of inflammation. Nat. Rev. Rheumatol. 2017, 13, 267–279. [Google Scholar] [CrossRef] [PubMed]
- Haschemi, A.; Kosma, P.; Gille, L.; Evans, C.R.; Burant, C.F.; Starkl, P.; Knapp, B.; Haas, R.; Schmid, J.A.; Jandl, C.; et al. The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 2012, 15, 813–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oronsky, B.; Scicinski, J.; Ning, S.; Peehl, D.; Oronsky, A.; Cabrales, P.; Bednarski, M.; Knox, S. RRx-001, A novel dinitroazetidine radiosensitizer. Investig. New Drugs 2016, 34, 371–377. [Google Scholar] [CrossRef] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4 + T Cell Subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- van der Windt, G.J.W.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of cd8 + t cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Raud, B.; Roy, D.G.; Divakaruni, A.S. Etomoxir actions on regulatory and memory T cells are independent of Cpt1a-mediated fatty acid oxidation. Cell Metab. 2018, 28, 504–515. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, D.; vanderWindt, G.W.J.; Huang, S.C.C.; Curtis, J.D.; Chang, C.H.; Buck, M.D.L.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M.; et al. Memory CD8 + T Cells use cell-intrinsic lipolysis to support the metabolic programming necessary for development. Immunity 2014, 41, 75–88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malandrino, M.I.; Fucho, R.; Weber, M.; Calderon-Dominguez, M.; Mir, J.F.; Valcarcel, L.; Escoté, X.; Gómez-Serrano, M.; Peral, B.; Salvadó, L.; et al. Enhanced fatty acid oxidation in adipocytes and macrophages reduces lipid-induced triglyceride accumulation and inflammation. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E756–E769. [Google Scholar] [CrossRef] [Green Version]
- Shriver, L.P.; Manchester, M. Inhibition of fatty acid metabolism ameliorates disease activity in an animal model of multiple sclerosis. Sci. Rep. 2011, 1. [Google Scholar] [CrossRef]
- Byersdorfer, C.A.; Tkachev, V.; Opipari, A.W.; Goodell, S.; Swanson, J.; Sandquist, S.; Glick, G.D.; Ferrara, J.L.M. Effector T cells require fatty acid metabolism during murine graft-versus-host disease. Blood 2013, 122, 3230–3237. [Google Scholar] [CrossRef] [Green Version]
- Everts, B.; Amiel, E.; Huang, S.C.C.; Smith, A.M.; Chang, C.H.; Lam, W.Y.; Redmann, V.; Freitas, T.C.; Blagih, J.; van der Windt, G.J.W.; et al. TLR-driven early glycolytic reprogramming via the kinases TBK1-IKKε supports the anabolic demands of dendritic cell activation. Nat. Immunol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angela, M.; Endo, Y.; Asou, H.K.; Yamamoto, T.; Tumes, D.J.; Tokuyama, H.; Yokote, K.; Nakayama, T. Fatty acid metabolic reprogramming via mTOR-mediated inductions of PPARγ directs early activation of T cells. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Bai, Y.; Xiong, Y.; Zhang, J.; Chen, S.; Zheng, X.; Meng, X.; Li, L.; Wang, J.; Xu, C.; et al. Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature 2016, 531, 651–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Walsh, M.C.; Hoehn, K.L.; James, D.E.; Wherry, E.J.; Choi, Y. Regulator of fatty acid metabolism, acetyl coenzyme a carboxylase 1, controls T cell immunity. J. Immunol. 2014, 192, 3190–3199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berod, L.; Friedrich, C.; Nandan, A.; Freitag, J.; Hagemann, S.; Harmrolfs, K.; Sandouk, A.; Hesse, C.; Castro, C.N.; BäHre, H.; et al. De novo fatty acid synthesis controls the fate between regulatory T and T helper 17 cells. Nat. Med. 2014, 20, 1327–1333. [Google Scholar] [CrossRef]
- Fox, J.T.; Stover, P.J. Chapter 1 Folate-Mediated One-Carbon Metabolism. Vitam. Horm. 2008, 79, 1–44. [Google Scholar]
- Ducker, G.S.; Rabinowitz, J.D. One-carbon metabolism in health and disease. Cell Metab. 2017, 25, 27–42. [Google Scholar] [CrossRef] [Green Version]
- Ron-Harel, N.; Santos, D.; Ghergurovich, J.M.; Sage, P.T.; Reddy, A.; Lovitch, S.B.; Dephoure, N.; Satterstrom, F.K.; Sheffer, M.; Spinelli, J.B.; et al. Mitochondrial biogenesis and proteome remodeling promote one-carbon metabolism for T cell activation. Cell Metab. 2016, 24, 104–117. [Google Scholar] [CrossRef] [Green Version]
- Ma, E.H.; Bantug, G.; Griss, T.; Condotta, S.; Johnson, R.M.; Samborska, B.; Mainolfi, N.; Suri, V.; Guak, H.; Balmer, M.L.; et al. Serine is an essential metabolite for effector t cell expansion. Cell Metab. 2017, 25, 345–357. [Google Scholar] [CrossRef]
- Ron-Harel, N.; Notarangelo, G.; Ghergurovich, J.M.; Paulo, J.A.; Sage, P.T.; Santos, D.; Kyle Satterstrom, F.; Gygi, S.P.; Rabinowitz, J.D.; Sharpe, A.H.; et al. Defective respiration and one-carbon metabolism contribute to impaired naïve T cell activation in aged mice. Proc. Natl. Acad. Sci. USA 2018, 115, 13347–13352. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Wang, Z.; Zhang, X.; Wu, Y.; Correspondence, D.W. One-carbon metabolism supports S-adenosylmethionine and histone methylation to drive inflammatory macrophages. Mol. Cell 2019, 75, 1147–1160. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, A.E.; Ducker, G.S.; Billingham, L.K.; Martinez, C.A.; Mainolfi, N.; Suri, V.; Friedman, A.; Manfredi, M.G.; Weinberg, S.E.; Rabinowitz, J.D.; et al. Serine metabolism supports macrophage IL-1β production. Cell Metab. 2019, 29, 1003–1011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducker, G.S.; Chen, L.; Morscher, R.J.; Ghergurovich, J.M.; Esposito, M.; Teng, X.; Kang, Y.; Rabinowitz, J.D. Reversal of cytosolic one-carbon flux compensates for loss of the mitochondrial folate pathway. Cell Metab. 2016, 23, 1140–1153. [Google Scholar] [CrossRef] [Green Version]
- Pålsson-McDermott, E.M.; O’Neill, L.A.J. Targeting immunometabolism as an anti-inflammatory strategy. Cell Res. 2020, 30, 300–314. [Google Scholar] [CrossRef] [Green Version]
- Sinclair, L.V.; Rolf, J.; Emslie, E.; Shi, Y.B.; Taylor, P.M.; Cantrell, D.A. Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 2013, 14, 500–508. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakaya, M.; Xiao, Y.; Zhou, X.; Chang, J.H.; Chang, M.; Cheng, X.; Blonska, M.; Lin, X.; Sun, S.C. Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 2014, 40, 692–705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, M.K.; Sinha, P.; Clements, V.K.; Rodriguez, P.; Ostrand-Rosenberg, S. Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res. 2010, 70, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Munn, D.H.; Zhou, M.; Attwood, J.T.; Bondarev, I.; Conway, S.J.; Marshall, B.; Brown, C.; Mellor, A.L. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science 1998, 281, 1191–1193. [Google Scholar] [CrossRef] [PubMed]
- Sedlmayr, P.; Blaschitz, A.; Stocker, R. The role of placental tryptophan catabolism. Front. Immunol. 2014, 5, 230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Triplett, T.A.; Garrison, K.C.; Marshall, N.; Donkor, M.; Blazeck, J.; Lamb, C.; Qerqez, A.; Dekker, J.D.; Tanno, Y.; Lu, W.C.; et al. Reversal of indoleamine 2,3-dioxygenase–Mediated cancer immune suppression by systemic kynurenine depletion with a therapeutic enzyme. Nat. Biotechnol. 2018, 36, 758. [Google Scholar] [CrossRef] [PubMed]
- Minhas, P.S.; Liu, L.; Moon, P.K.; Joshi, A.U.; Dove, C.; Mhatre, S.; Contrepois, K.; Wang, Q.; Lee, B.A.; Coronado, M.; et al. Macrophage de novo NAD+ synthesis specifies immune function in aging and inflammation. Nat. Immunol. 2019, 20, 50–63. [Google Scholar] [CrossRef]
- Albina, J.E.; Mills, C.D.; Barbul, A.; Thirkill, C.E.; Henry, W.L.; Mastrofrancesco, B.; Caldwell, M.D. Arginine metabolism in wounds. Am. J. Physiol. Endocrinol. Metab. 1988, 254. [Google Scholar] [CrossRef]
- El-Gayar, S.; Thüring-Nahler, H.; Pfeilschifter, J.; Röllinghoff, M.; Bogdan, C. Translational control of inducible nitric oxide synthase by il-13 and arginine availability in inflammatory macrophages. J. Immunol. 2003, 171, 4561–4568. [Google Scholar] [CrossRef] [Green Version]
- Duque-Correa, M.A.; Kühl, A.A.; Rodriguez, P.C.; Zedler, U.; Schommer-Leitner, S.; Rao, M.; Weiner, J.; Hurwitz, R.; Qualls, J.E.; Kosmiadi, G.A.; et al. Macrophage arginase-1 controls bacterial growth and pathology in hypoxic tuberculosis granulomas. Proc. Natl. Acad. Sci. USA 2014, 111, E4024–E4032. [Google Scholar] [CrossRef] [Green Version]
- Vigeland, C.L.; Beggs, H.S.; Collins, S.L.; Chan-Li, Y.; Powell, J.D.; Doerschuk, C.M.; Horton, M.R. Inhibition of glutamine metabolism accelerates resolution of acute lung injury. Physiol. Rep. 2019, 7. [Google Scholar] [CrossRef] [Green Version]
- Leone, R.D.; Zhao, L.; Englert, J.M.; Sun, I.M.; Oh, M.H.; Sun, I.H.; Arwood, M.L.; Bettencourt, I.A.; Patel, C.H.; Wen, J.; et al. Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 2019, 366, 1013–1021. [Google Scholar] [CrossRef]
- Altman, B.J.; Jacobs, S.R.; Mason, E.F.; Michalek, R.D.; MacIntyre, A.N.; Coloff, J.L.; Ilkayeva, O.; Jia, W.; He, Y.W.; Rathmell, J.C. Autophagy is essential to suppress cell stress and to allow BCR-Abl-mediated leukemogenesis. Oncogene 2011, 30, 1855–1867. [Google Scholar] [CrossRef] [Green Version]
- Mcleod, I.X.; Jia, W.; He, Y.W. The contribution of autophagy to lymphocyte survival and homeostasis. Immunol. Rev. 2012, 249, 195–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy is involved in T cell death after binding of HIV-1 envelope proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Riffelmacher, T.; Richter, F.C.; Simon, A.K. Autophagy dictates metabolism and differentiation of inflammatory immune cells. Autophagy 2018, 14, 199–206. [Google Scholar] [CrossRef] [Green Version]
- Kang, Y.H.; Cho, M.H.; Kim, J.Y.; Kwon, M.S.; Peak, J.J.; Kang, S.W.; Yoon, S.Y.; Song, Y. Impaired macrophage autophagy induces systemic insulin resistance in obesity. Oncotarget 2016, 7, 35577–35591. [Google Scholar] [CrossRef] [Green Version]
- Mouttie, L.E.; Vu, T.; Lineburg, K.E.; Kuns, R.D.; Bagger, F.O.; Teal, B.E.; Lor, M.; Boyle, G.M.; Bruedigam, C.; Mintern, J.D.; et al. Autophagy is required for stem cell mobilization by G-CSF. Blood 2015, 125, 2933–2936. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
| Enzymatic Target | Therapeutic Compound | Disease | Trial Status |
|---|---|---|---|
| mTOR [2,3,4,5] | Rapamycin | Organ Transplant | FDA-approved drug |
| Hexokinase [24,48,49,50] | 3-bromopyruvate | Rheumatoid Arthritis, Multiple Sclerosis | Pre-clinical |
| GADPH [60] | Dimethyl fumarate | Multiple Sclerosis | FDA-approved drug |
| Pyruvate kinase M2 [70,74,75] | TEPP-46 | Multiple Sclerosis | Pre-clinical |
| Pyruvate dehydrogenase kinase 1 [81,82,83] | Dichloroacetate | Rheumatoid Arthritis, Inflammatory Bowel Disease, MS, Asthma | Pre-clinical |
| Lactate dehydrogenase [85] | FX11 | Research ongoing | Pre-clinical |
| Complex I of the electron transport chain (ETC) | Metformin | Diabetes, others ongoing | FDA-approved drug |
| F1F0 ATP synthase [116] | LYC-30937-ec | Ulcerative Colitis | Clinical trials |
| ACC1 [139] | Soraphen A | Multiple Sclerosis | Pre-clinical |
| Dihydrofolate reductase [141] | Methotrexate | Rheumatoid Arthritis | FDA-approved drug |
| Glutamine metabolism [25,159,160] | 6-diazo-5-oxo-L-norleucine (DON) and prodrug JHU083 | Acute Lung Injury, Allograft Rejection, Anti-Tumor Response | Pre-clinical |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Godfrey, W.H.; Kornberg, M.D. The Role of Metabolic Enzymes in the Regulation of Inflammation. Metabolites 2020, 10, 426. https://doi.org/10.3390/metabo10110426
Godfrey WH, Kornberg MD. The Role of Metabolic Enzymes in the Regulation of Inflammation. Metabolites. 2020; 10(11):426. https://doi.org/10.3390/metabo10110426
Chicago/Turabian StyleGodfrey, Wesley H., and Michael D. Kornberg. 2020. "The Role of Metabolic Enzymes in the Regulation of Inflammation" Metabolites 10, no. 11: 426. https://doi.org/10.3390/metabo10110426
APA StyleGodfrey, W. H., & Kornberg, M. D. (2020). The Role of Metabolic Enzymes in the Regulation of Inflammation. Metabolites, 10(11), 426. https://doi.org/10.3390/metabo10110426