Biological Filtering and Substrate Promiscuity Prediction for Annotating Untargeted Metabolomics


 and
and 
Abstract
:1. Introduction
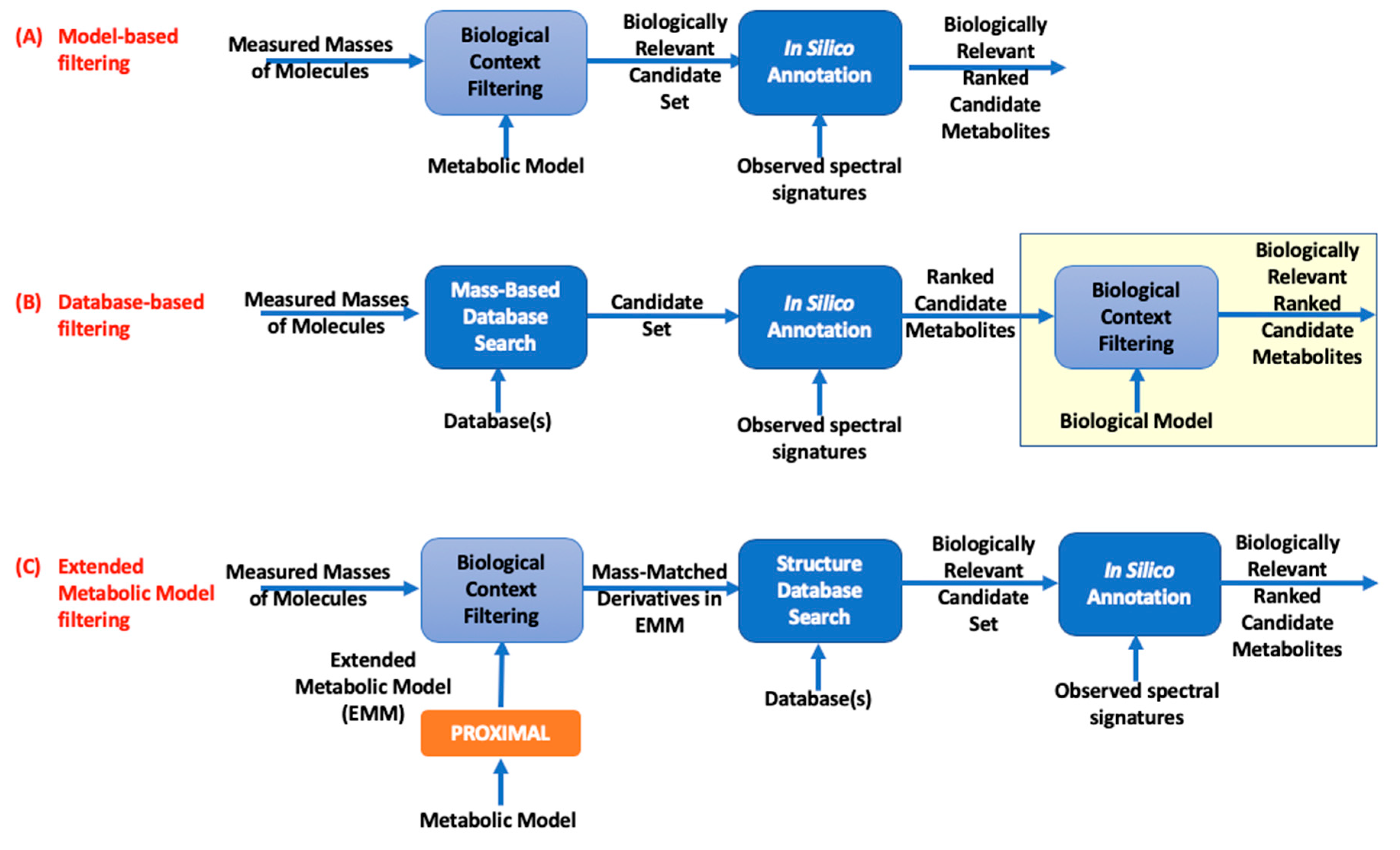
2. Methods
3. Results
3.1. Datasets, Reference Metabolic Models, and EMMs
3.2. Annotation Opportunities
3.3. Computational Time Required for Annotation
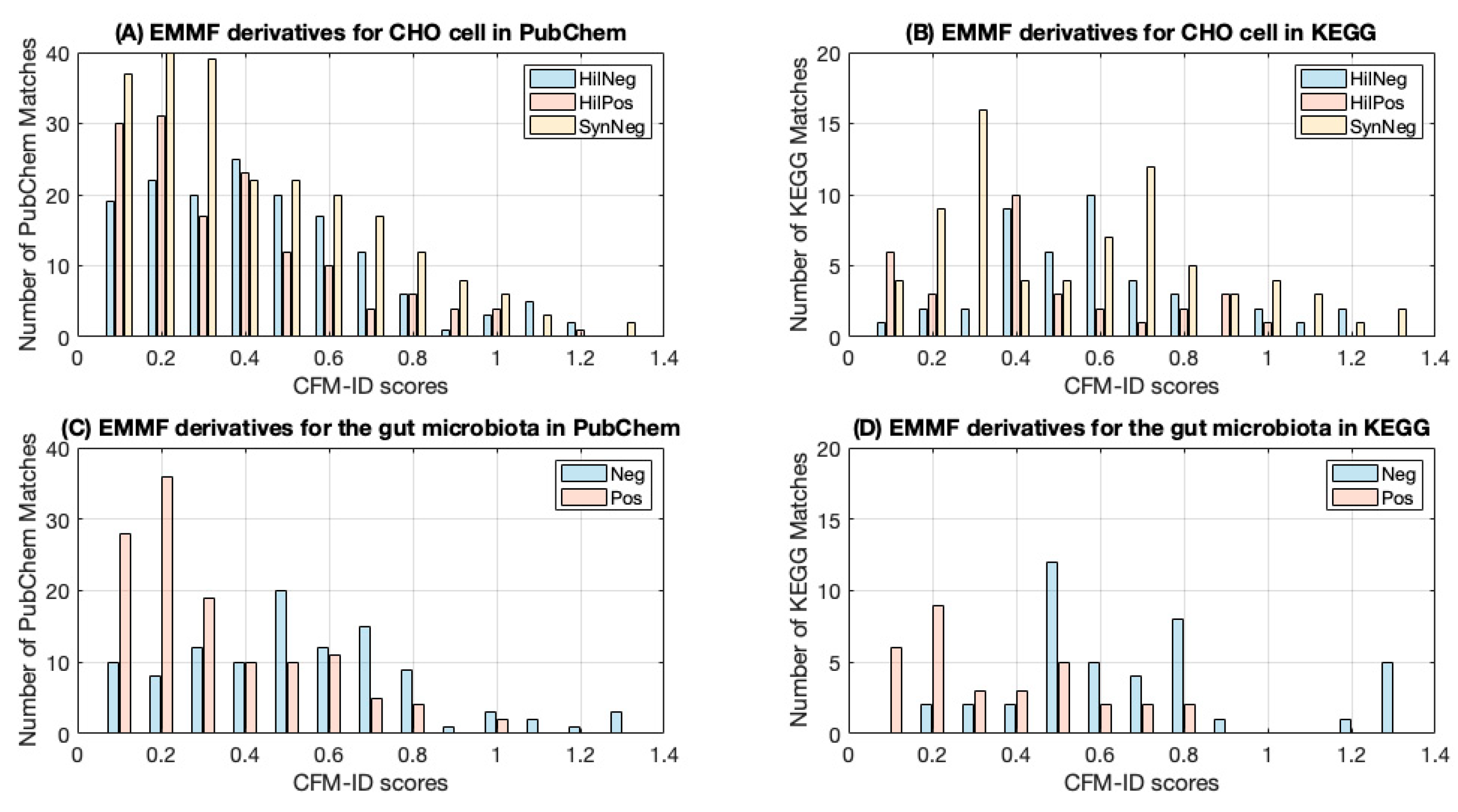
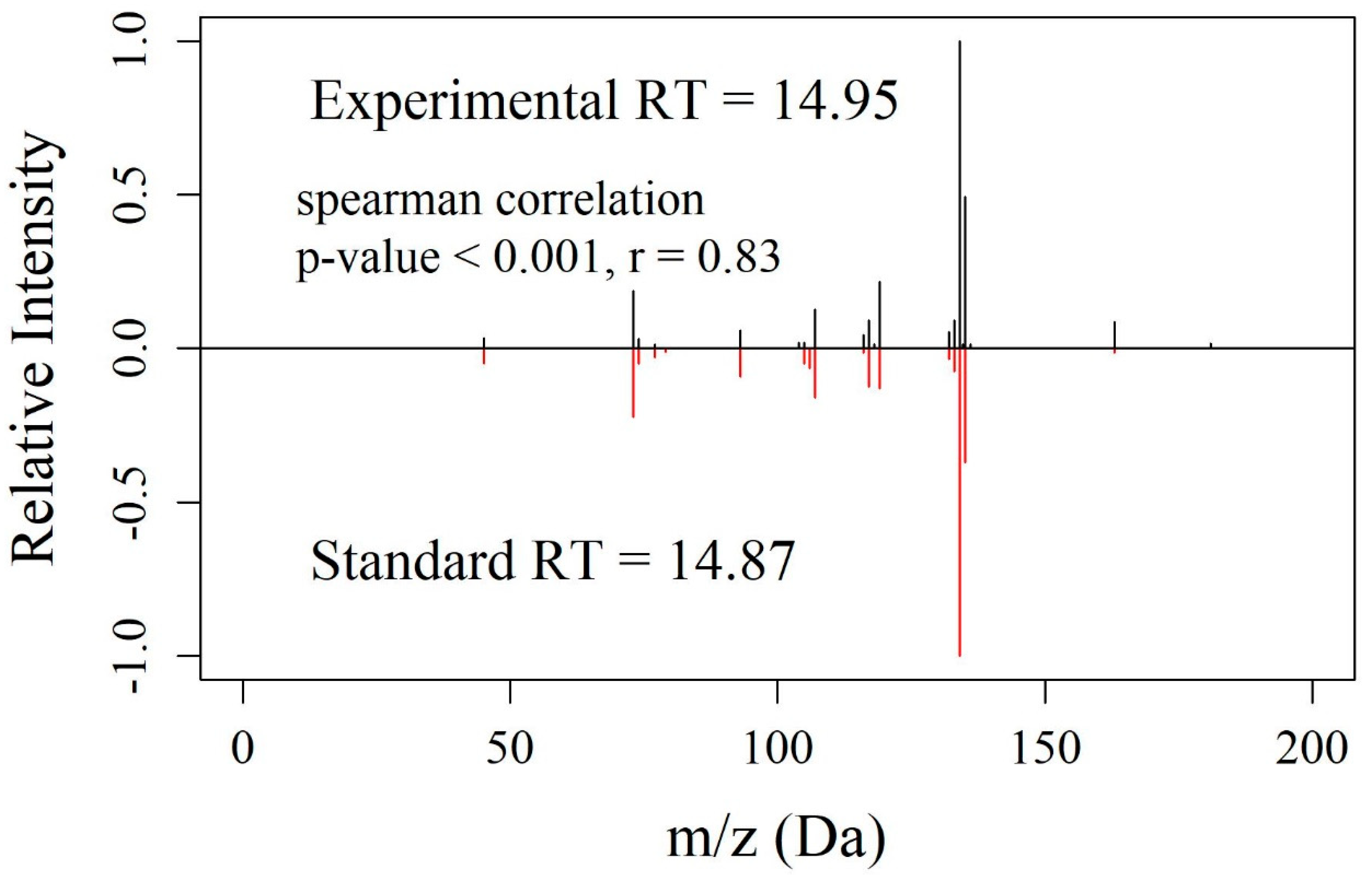
3.4. Experimental Validation of EMMF
4. Discussion and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Fiehn, O. Metabolomics—The link between genotypes and phenotypes. In Functional Genomics; Springer: Berlin/Heidelberg, Germany, 2002; pp. 155–171. [Google Scholar]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263. [Google Scholar] [CrossRef]
- Raamsdonk, L.M.; Teusink, B.; Broadhurst, D.; Zhang, N.; Hayes, A.; Walsh, M.C.; Berden, J.A.; Brindle, K.M.; Kell, D.B.; Rowland, J.J. A functional genomics strategy that uses metabolome data to reveal the phenotype of silent mutations. Nat. Biotechnol. 2001, 19, 45. [Google Scholar] [CrossRef] [PubMed]
- Alonso, A.; Marsal, S.; Julià, A. Analytical methods in untargeted metabolomics: State of the art in 2015. Front. Bioeng. Biotechnol. 2015, 3, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- May, J.C.; McLean, J.A. Advanced Multidimensional Separations in Mass Spectrometry: Navigating the Big Data Deluge. Annu. Rev. Anal. Chem. 2016, 9, 387–409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guijas, C.; Montenegro-Burke, J.R.; Domingo-Almenara, X.; Palermo, A.; Warth, B.; Hermann, G.; Koellensperger, G.; Huan, T.; Uritboonthai, W.; Aisporna, A.E.; et al. METLIN: A Technology Platform for Identifying Knowns and Unknowns. Anal. Chem. 2018, 90, 3156–3164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Neta, P.; Stein, S.E. Extending a Tandem Mass Spectral Library to Include MS(2) Spectra of Fragment Ions Produced In-Source and MS(n) Spectra. J. Am. Soc. Mass Spectrom. 2017, 28, 2280–2287. [Google Scholar] [CrossRef]
- Lynn, K.-S.; Cheng, M.-L.; Chen, Y.-R.; Hsu, C.; Chen, A.; Lih, T.M.; Chang, H.-Y.; Huang, C.-J.; Shiao, M.-S.; Pan, W.-H. Metabolite identification for mass spectrometry-based metabolomics using multiple types of correlated ion information. Anal. Chem. 2015, 87, 2143–2151. [Google Scholar] [CrossRef]
- Han, T.-L.; Yang, Y.; Zhang, H.; Law, K.P. Analytical challenges of untargeted GC-MS-based metabolomics and the critical issues in selecting the data processing strategy. F1000Research 2017, 6. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Muehlbauer, M.J.; O’Neal, S.K.; Newgard, C.B.; Hauser, E.R.; Bain, J.R.; Shah, S.H. Recommendations for Improving Identification and Quantification in Non-Targeted, GC-MS-Based Metabolomic Profiling of Human Plasma. Metabolites 2017, 7, 45. [Google Scholar] [CrossRef] [Green Version]
- Denihan, N.M.; Kirwan, J.A.; Walsh, B.H.; Dunn, W.B.; Broadhurst, D.I.; Boylan, G.B.; Murray, D.M. Untargeted metabolomic analysis and pathway discovery in perinatal asphyxia and hypoxic-ischaemic encephalopathy. J. Cereb. Blood Flow Metab. 2017. [Google Scholar] [CrossRef] [PubMed]
- Romo-Hualde, A.; Huerta, A.E.; González-Navarro, C.J.; Ramos-López, O.; Moreno-Aliaga, M.J.; Martínez, J.A. Untargeted metabolomic on urine samples after α-lipoic acid and/or eicosapentaenoic acid supplementation in healthy overweight/obese women. Lipids Health Dis. 2018, 17, 103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, K.E.; Harvey, J.; McCullagh, J.S. Targeted and Untargeted Metabolic Profiling of Wild Grassland Plants identifies Antibiotic and Anthelmintic Compounds Targeting Pathogen Physiology, Metabolism and Reproduction. Sci. Rep. 2018, 8, 1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kera, K.; Fine, D.D.; Wherritt, D.J.; Nagashima, Y.; Shimada, N.; Ara, T.; Ogata, Y.; Sumner, L.W.; Suzuki, H. Pathway-specific metabolome analysis with 18 O 2-labeled Medicago truncatula via a mass spectrometry-based approach. Metabolomics 2018, 14, 71. [Google Scholar] [CrossRef] [Green Version]
- Zhou, J.; Weber, R.J.; Allwood, J.W.; Mistrik, R.; Zhu, Z.; Ji, Z.; Chen, S.; Dunn, W.B.; He, S.; Viant, M.R. HAMMER: Automated operation of mass frontier to construct in silico mass spectral fragmentation libraries. Bioinformatics 2014, 30, 581–583. [Google Scholar] [CrossRef] [Green Version]
- Wolf, S.; Schmidt, S.; Müller-Hannemann, M.; Neumann, S. In silico fragmentation for computer assisted identification of metabolite mass spectra. BMC Bioinform. 2010, 11, 148. [Google Scholar] [CrossRef] [Green Version]
- Heinonen, M.; Rantanen, A.; Mielikäinen, T.; Pitkänen, E.; Kokkonen, J.; Rousu, J. FiD: New Software for De novo Identification of Metabolite Fragments from Tandem Mass Spectrometry Data. Rapid Commun. Mass Spectrom. 2008, 22, 3043–3052. [Google Scholar]
- Wegner, A.; Weindl, D.; Jager, C.; Sapcariu, S.C.; Dong, X.; Stephanopoulos, G.; Hiller, K. Fragment formula calculator (FFC): Determination of chemical formulas for fragment ions in mass spectrometric data. Anal. Chem.. 2014, 86, 2221–2228. [Google Scholar] [CrossRef] [Green Version]
- Allen, F.; Pon, A.; Wilson, M.; Greiner, R.; Wishart, D. CFM-ID: A web server for annotation, spectrum prediction and metabolite identification from tandem mass spectra. Nucleic Acids Res. 2014, 42. [Google Scholar] [CrossRef] [Green Version]
- Dührkop, K.; Shen, H.; Meusel, M.; Rousu, J.; Böcker, S. Searching molecular structure databases with tandem mass spectra using CSI:FingerID. Proc. Natl. Acad. Sci. USA 2015, 112, 12580–12585. [Google Scholar] [CrossRef] [Green Version]
- Rasche, F.; Svatos, A.; Maddula, R.K.; Bottcher, C.; Bocker, S. Computing fragmentation trees from tandem mass spectrometry data. Anal. Chem. 2011, 83, 1243–1251. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Dührkop, K.; Böcker, S.; Rousu, J. Metabolite identification through multiple kernel learning on fragmentation trees. Bioinformatics 2014, 30, i157–i164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dührkop, K.; Fleischauer, M.; Ludwig, M.; Aksenov, A.A.; Melnik, A.V.; Meusel, M.; Dorrestein, P.C.; Rousu, J.; Böcker, S. SIRIUS 4: A rapid tool for turning tandem mass spectra into metabolite structure information. Nat. Methods 2019, 16, 299–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinonen, M.; Shen, H.; Zamboni, N.; Rousu, J. Metabolite identification and molecular fingerprint prediction through machine learning. Bioinformatics 2012, 28, 2333–2341. [Google Scholar] [CrossRef] [PubMed]
- Ridder, L.; van der Hooft, J.J.; Verhoeven, S.; de Vos, R.C.; Bino, R.J.; Vervoort, J. Automatic chemical structure annotation of an LC-MS(n) based metabolic profile from green tea. Anal. Chem. 2013, 85, 6033–6040. [Google Scholar] [CrossRef]
- Wang, Y.; Kora, G.; Bowen, B.P.; Pan, C. MIDAS: A database-searching algorithm for metabolite identification in metabolomics. Anal. Chem. 2014, 86, 9496–9503. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S.; Kawashima, S.; Okuno, Y.; Hattori, M. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar] [CrossRef] [Green Version]
- Caspi, R.; Billington, R.; Keseler, I.M.; Kothari, A.; Krummenacker, M.; Midford, P.E.; Ong, W.K.; Paley, S.; Subhraveti, P.; Karp, P.D. The MetaCyc database of metabolic pathways and enzymes - a 2019 update. Nucleic Acids Res. 2020, 48, D445–D453. [Google Scholar] [CrossRef] [Green Version]
- King, Z.A.; Lu, J.; Drager, A.; Miller, P.; Federowicz, S.; Lerman, J.A.; Ebrahim, A.; Palsson, B.O.; Lewis, N.E. BiGG Models: A platform for integrating, standardizing and sharing genome-scale models. Nucleic Acids Res. 2016, 44, D515–D522. [Google Scholar] [CrossRef]
- Feist, A.M.; Herrgård, M.J.; Thiele, I.; Reed, J.L.; Palsson, B.Ø. Reconstruction of biochemical networks in microorganisms. Nat. Rev. Microbiol. 2009, 7, 129–143. [Google Scholar] [CrossRef] [Green Version]
- Schellenberger, J.; Park, J.O.; Conrad, T.M.; Palsson, B.Ø. BiGG: A Biochemical Genetic and Genomic knowledgebase of large scale metabolic reconstructions. BMC Bioinform. 2010, 11, 213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, T.Y.; Sohn, S.B.; Kim, Y.B.; Kim, W.J.; Lee, S.Y. Recent advances in reconstruction and applications of genome-scale metabolic models. Curr. Opin. Biotechnol. 2012, 23, 617–623. [Google Scholar] [CrossRef] [PubMed]
- Saha, R.; Chowdhury, A.; Maranas, C.D. Recent advances in the reconstruction of metabolic models and integration of omics data. Curr. Opin. Biotechnol. 2014, 29, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Metabolomics: From Small Molecules to Big Ideas. Nat. Methods 2011, 8, 117–121. [Google Scholar] [CrossRef]
- D’Ari, R.; Casadesus, J. Underground metabolism. Bioessays 1998, 20, 181–186. [Google Scholar] [CrossRef]
- Nobeli, I.; Favia, A.D.; Thornton, J.M. Protein promiscuity and its implications for biotechnology. Nat. Biotechnol. 2009, 27, 157–167. [Google Scholar] [CrossRef]
- Tawfik, O.K.; Dan, S. Enzyme Promiscuity: A Mechanistic and Evolutionary Perspective. Annu. Rev. Biochem. 2010, 79, 471–505. [Google Scholar] [CrossRef]
- Khersonsky, O.; Malitsky, S.; Rogachev, I.; Tawfik, D.S. Role of chemistry versus substrate binding in recruiting promiscuous enzyme functions. Biochemistry 2011, 50, 2683–2690. [Google Scholar] [CrossRef]
- Yousofshahi, M.; Manteiga, S.; Wu, C.; Lee, K.; Hassoun, S. PROXIMAL: A method for Prediction of Xenobiotic Metabolism. BMC Syst. Biol. 2015, 9, 94. [Google Scholar] [CrossRef] [Green Version]
- Amin, S.A.; Chavez, E.; Porokhin, V.; Nair, N.U.; Hassoun, S. Towards creating an extended metabolic model (EMM) for E. coli using enzyme promiscuity prediction and metabolomics data. Microb. Cell Factories 2019, 18, 109. [Google Scholar] [CrossRef] [Green Version]
- Oh, M.; Yamada, T.; Hattori, M.; Goto, S.; Kanehisa, M. Systematic analysis of enzyme-catalyzed reaction patterns and prediction of microbial biodegradation pathways. J. Chem. Inf. Model. 2007, 47, 1702–1712. [Google Scholar] [CrossRef] [PubMed]
- Weininger, D. SMILES, a chemical language and information system. 1. Introduction to methodology and encoding rules. J. Chem. Inf. Comput. Sci. 1988, 28, 31–36. [Google Scholar]
- Sridharan, G.V.; Choi, K.; Klemashevich, C.; Wu, C.; Prabakaran, D.; Pan, L.B.; Steinmeyer, S.; Mueller, C.; Yousofshahi, M.; Alaniz, R.C.; et al. Prediction and quantification of bioactive microbiota metabolites in the mouse gut. Nat. Commun. 2014, 5, 5492. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Carver, J.J.; Phelan, V.V.; Sanchez, L.M.; Garg, N.; Peng, Y.; Nguyen, D.D.; Watrous, J.; Kapono, C.A.; Luzzatto-Knaan, T. Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 2016, 34, 828–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wishart, D.S.; Feunang, Y.D.; Marcu, A.; Guo, A.C.; Liang, K.; Vázquez-Fresno, R.; Sajed, T.; Johnson, D.; Li, C.; Karu, N. HMDB 4.0: The human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [Google Scholar] [CrossRef] [PubMed]
- Ruttkies, C.; Schymanski, E.L.; Wolf, S.; Hollender, J.; Neumann, S. MetFrag relaunched: Incorporating strategies beyond in silico fragmentation. J. Cheminform. 2016, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Kind, T.; Scholz, M.; Fiehn, O. How large is the metabolome? A critical analysis of data exchange practices in chemistry. PloS ONE 2009, 4, e5440. [Google Scholar] [CrossRef] [Green Version]
- Kuhl, C.; Tautenhahn, R.; Bottcher, C.; Larson, T.R.; Neumann, S. CAMERA: An integrated strategy for compound spectra extraction and annotation of liquid chromatography/mass spectrometry data sets. Anal. Chem. 2012, 84, 283–289. [Google Scholar] [CrossRef] [Green Version]
- Aguilar-Mogas, A.; Sales-Pardo, M.; Navarro, M.; Tautenhahn, R.; Guimerà, R.; Yanes, O. iMet: A computational tool for structural annotation of unknown metabolites from tandem mass spectra. arXiv 2016, arXiv:1607.04122. [Google Scholar]
- Alden, N.; Krishnan, S.; Porokhin, V.; Raju, R.; McElearney, K.; Gilbert, A.; Lee, K. Biologically Consistent Annotation of Metabolomics Data. Anal. Chem. 2017, 89, 13097–13104. [Google Scholar] [CrossRef]
- Li, S.; Park, Y.; Duraisingham, S.; Strobel, F.H.; Khan, N.; Soltow, Q.A.; Jones, D.P.; Pulendran, B. Predicting network activity from high throughput metabolomics. PLoS Comput. Biol. 2013, 9, e1003123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morreel, K.; Saeys, Y.; Dima, O.; Lu, F.; Van de Peer, Y.; Vanholme, R.; Ralph, J.; Vanholme, B.; Boerjan, W. Systematic structural characterization of metabolites in Arabidopsis via candidate substrate-product pair networks. Plant Cell 2014, 26, 929–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulukutla, B.C.; Kale, J.; Kalomeris, T.; Jacobs, M.; Hiller, G.W. Identification and control of novel growth inhibitors in fed-batch cultures of Chinese hamster ovary cells. Biotechnol. Bioeng. 2017, 114, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Mulukutla, B.C.; Mitchell, J.; Geoffroy, P.; Harrington, C.; Krishnan, M.; Kalomeris, T.; Morris, C.; Zhang, L.; Pegman, P.; Hiller, G.W. Metabolic engineering of Chinese hamster ovary cells towards reduced biosynthesis and accumulation of novel growth inhibitors in fed-batch cultures. Metab. Eng. 2019, 54, 54–68. [Google Scholar] [CrossRef] [PubMed]
- Nam, H.; Lewis, N.E.; Lerman, J.A.; Lee, D.-H.; Chang, R.L.; Kim, D.; Palsson, B.O. Network context and selection in the evolution to enzyme specificity. Science 2012, 337, 1101–1104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Even, A.; Tawfik, D.S. Engineering specialized metabolic pathways—is there a room for enzyme improvements? Curr. Opin. Biotechnol. 2013, 24, 310–319. [Google Scholar] [CrossRef]
- Djoumbou-Feunang, Y.; Fiamoncini, J.; Gil-de-la-Fuente, A.; Greiner, R.; Manach, C.; Wishart, D.S. BioTransformer: A comprehensive computational tool for small molecule metabolism prediction and metabolite identification. J. Cheminform. 2019, 11, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Jeffryes, J.G.; Colastani, R.L.; Elbadawi-Sidhu, M.; Kind, T.; Niehaus, T.D.; Broadbelt, L.J.; Hanson, A.D.; Fiehn, O.; Tyo, K.E.; Henry, C.S. MINEs: Open access databases of computationally predicted enzyme promiscuity products for untargeted metabolomics. J. Cheminform. 2015, 7, 44. [Google Scholar] [CrossRef] [Green Version]
- Henry, C.S.; Jankowski, M.D.; Broadbelt, L.J.; Hatzimanikatis, V. Genome-scale thermodynamic analysis of Escherichia coli metabolism. Biophys. J. 2006, 90, 1453–1461. [Google Scholar] [CrossRef] [Green Version]
- Lai, Z.; Tsugawa, H.; Wohlgemuth, G.; Mehta, S.; Mueller, M.; Zheng, Y.; Ogiwara, A.; Meissen, J.; Showalter, M.; Takeuchi, K. Identifying metabolites by integrating metabolome databases with mass spectrometry cheminformatics. Nat. Methods 2018, 15, 53. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| (A) Experimental Data | (B) Metabolic Model | (C) Expanded Metabolic Model Using PROXIMAL | (D) Fold Change for EMM Relative to Metabolic Model | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Biological Sample | Dataset | MS Mode | Number of Measured Masses | Number of Reactions | Number of Metabolites | Number of Unique Masses | Number of Unique Operators | Number of Unique Derivatives | Number of Unique Derivative Masses | Number of Metabolites | Number of Unique Masses |
| CHO cell | HilNeg | negative | 2502 | 1619 | 1353 | 775 | 2392 | 76745 | 17930 | 56.72 | 23.14 |
| HilPos | positive | 3856 | |||||||||
| SynNeg | negative | 5336 | |||||||||
| gut microbiota | Neg | negative | 1651 | 1381 | 1307 | 779 | 2756 | 94186 | 23356 | 72.06 | 29.98 |
| Pos | positive | 1657 | |||||||||
| Biological Sample | (A) Metabolites in Metabolic Model | (B) All EMM Derivatives | (C) EMM Derivatives with Previously Known Chemical IDs | (D) Using PubChem-based Filtering | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of Measured Masses Matched to Those in Metabolic Model | Percentage of Measured Masses Matched to Those in Metabolic Model | Number of Chemical Ids Associated with Measured Masses | Number of Masses Matched to Those in EMM | Percentage of Masses Matched to Those in EMM | Number of Unique Mass-Matched Derivatives in EMM But Not in The Model | Number of Masses Matched to Those with Previously Known Chemical IDs | Percentage of Masses Matched to Those with Previously Known Chemical IDs | Number of Previously Known Chemical IDs for EMM Derivatives that Mass-Match to Measurements | Number of Unique Mass Matches in PubChem | Number of Corresponding Chemical IDs Associated with Measured Masses | ||
| CHO cell | HilNeg | 118 | 4.72% | 178 | 678 | 27.10% | 2,725 | 174 | 6.95% | 386 | 3,951,635 | 7,657,564 |
| HilPos | 75 | 1.95% | 93 | 715 | 18.54% | 2,729 | 132 | 3.42% | 226 | 3,362,305 | 6,406,877 | |
| SynNeg | 198 | 3.71% | 229 | 1,490 | 27.92% | 4,944 | 293 | 5.49% | 527 | 7,058,696 | 14,133,885 | |
| gut microbiota | Neg | 51 | 3.09% | 131 | 445 | 26.95% | 2,470 | 77 | 4.66% | 207 | 2,448,238 | 5,192,205 |
| Pos | 36 | 2.17% | 43 | 316 | 19.07% | 1,236 | 84 | 5.07% | 149 | 2,774,074 | 5,572,587 | |
| Averages | 96 | 3.13% | 135 | 729 | 23.92% | 2,821 | 152 | 5.12% | 299 | 3,918,990 | 7,792,624 | |
| Biological Sample | KEGG | PubChem | |||||
|---|---|---|---|---|---|---|---|
| Number of EMMF Derivatives | Percentage of EMMF Derivatives with Nonzero CFM-ID scores | Average CFM-ID Score | Number of EMMF Derivatives | Percentage of EMMF Derivatives with Nonzero CFM-ID scores | Average CFM-ID Score | ||
| CHO cell | HilNeg | 65 | 65% | 0.557 | 280 | 55% | 0.415 |
| HilPos | 48 | 63% | 0.395 | 286 | 49% | 0.316 | |
| SynNeg | 114 | 64% | 0.501 | 446 | 51% | 0.370 | |
| gut microbiota | Neg | 252 | 16% | 0.631 | 197 | 53% | 0.484 |
| Pos | 56 | 55% | 0.292 | 428 | 29% | 0.270 | |
| Average | 53% | 0.475 | 47% | 0.396 | |||
| Biological Sample | (A) Experimental Data | (B) In EMM And in KEGG | (C) In EMM And PubChem, And Not in KEGG | (D) Lower-Bound Fold Increase of Pubchem over KEGG | ||||
|---|---|---|---|---|---|---|---|---|
| Dataset | Number of Measured Masses | Number of Matched Masses | Number of Candidate Chemical IDs | Number of Matched Masses | Number of Candidate Chemical IDs | Number of Matched Masses | Number of Candidate Chemical IDs | |
| CHO cell | HilNeg | 2502 | 56 | 93 | 118 | 200 | 2.11 | 2.15 |
| HilPos | 3856 | 26 | 39 | 106 | 148 | 4.08 | 3.79 | |
| SynNeg | 5336 | 88 | 122 | 205 | 283 | 2.33 | 2.32 | |
| gut microbiota | Neg | 1651 | 25 | 47 | 52 | 113 | 2.08 | 2.40 |
| Pos | 1657 | 23 | 28 | 61 | 93 | 2.65 | 3.32 | |
| Average | 2.65 | 2.80 | ||||||
| (A) Candidate Metabolites | (B) KEGG | (C) PubChem | (D) PROXIMAL | (E) | ||||
|---|---|---|---|---|---|---|---|---|
| Mass Measurement (Daltons) | Candidate Metabolite Identified by EMMF | Rank | Matches | Rank | Matches | Number of Reactions Used to Derive Operator | Number of ECs Associated with Reactions | Experimentally Validated? |
| 122.04 | Salicylaldehyde | 1 | 1 | 1 | 1 | 1 | 1 | No |
| 182.06 | 4-Hydroxyphenyllactate | 1 | 2 | 1 | 4 | 12 | 15 | Yes |
| 101.05 | Acetoacetamide | 1 | 1 | 2 | 3 | 1 | 1 | No |
| 117.79 | 5-Aminopentanoate | 1 | 2 | 1 | 5 | 4 | 4 | No |
| 132.04 | Glutarate | 1 | 1 | 3 | 6 | 12 | 11 | No |
| 167.06 | 3-Methoxyanthranilate | 1 | 1 | 2 | 3 | 8 | 2 | No |
| 152.05 | 2-Hydroxyphenylacetic acid | NA | 1 | 1 | 4 | 1 | 1 | No |
| 183.05 | 4-Pyridoxate | NA | 0 | 1 | 1 | 1 | 1 | No |
| (A) EMMF | (B) CFMID | (C) GNPS | (D) HMDB | (E) PubChem | (F) MetFrag | ||||
|---|---|---|---|---|---|---|---|---|---|
| Mass Measurement (Daltons) | Candidate Metabolite | Score | Matched Compound ( Score) | Matched Compound (Score) | Number of Matches | Rank of Compound Identified by EMMF | # of Peaks Explained/ # of Peaks Used | Top Ranked Candidate | # of Peaks Explained/ # of Peaks Used |
| 122.04 | Salicylal | 0.596 | No Match | No Match | 241 | 27 | 4/8 | 2-cyclopenta-1,3-dien-1-yl-2-oxo-acetaldehyde | 4/8 |
| 182.06 | 4-Hydroxyphenyllactate | 0.717 | No Match | Homovanillic acid (0.43) | 1694 | 218 | 10/22 | methyl 2-hydroxy-2-phenyl-peroxyacetate | 11/22 |
| 101.05 | Acetoacetamide | 0.682 | Aminocyclopropane (0.92), L-threonine (0.90) | No Match | 445 | 331 | 1/2 | hydroxy N-isopropenylmethanimidate | 1/2 |
| 117.79 | 5-Aminopentanoate | 0.979 | No Match | L-Valine (0.44), Betaine (0.34), 5-Aminopentanoic acid (0.31) | 858 | 12 | 2/5 | 2-[ethyl(methyl)amino]acetic acid | 2/5 |
| 132.04 | Glutarate | 0.600 | No Match | Ethylmalonic acid (0.41) | N/A | ||||
| 167.06 | 3-Methoxyanthranilate | 0.949 | No Match | Mandelic acid (0.55), 3-Hydroxyphenylacetic acid (0.44), p-Hydroxyphenylacetic acid (0.40), Ortho-Hydroxyphenylacetic acid (0.19) | 1962 | 972 | 2/7 | (2-aminophenyl) peroxyacetate | 2/7 |
| 152.05 | 2-Hydroxyphenylacetic acid | 0.716 | 4-hydroxyphenylacetic acid (0.81) | No Match | 841 | 129 | 1/4 | methyl-phenyl-silyl-silane | 1/4 |
| 183.05 | 4-Pyridoxate | 0.870 | 4-Pyridoxate (0.76) | No Match | 1252 | 149 | 2/5 | 2-[1-(3-furyl)ethylideneamino]oxyacetic acid | 2/5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hassanpour, N.; Alden, N.; Menon, R.; Jayaraman, A.; Lee, K.; Hassoun, S. Biological Filtering and Substrate Promiscuity Prediction for Annotating Untargeted Metabolomics. Metabolites 2020, 10, 160. https://doi.org/10.3390/metabo10040160
Hassanpour N, Alden N, Menon R, Jayaraman A, Lee K, Hassoun S. Biological Filtering and Substrate Promiscuity Prediction for Annotating Untargeted Metabolomics. Metabolites. 2020; 10(4):160. https://doi.org/10.3390/metabo10040160
Chicago/Turabian StyleHassanpour, Neda, Nicholas Alden, Rani Menon, Arul Jayaraman, Kyongbum Lee, and Soha Hassoun. 2020. "Biological Filtering and Substrate Promiscuity Prediction for Annotating Untargeted Metabolomics" Metabolites 10, no. 4: 160. https://doi.org/10.3390/metabo10040160
APA StyleHassanpour, N., Alden, N., Menon, R., Jayaraman, A., Lee, K., & Hassoun, S. (2020). Biological Filtering and Substrate Promiscuity Prediction for Annotating Untargeted Metabolomics. Metabolites, 10(4), 160. https://doi.org/10.3390/metabo10040160