Optimized Protocol for the In Situ Derivatization of Glutathione with N-Ethylmaleimide in Cultured Cells and the Simultaneous Determination of Glutathione/Glutathione Disulfide Ratio by HPLC-UV-QTOF-MS

 ,
, 

Abstract
:
1. Introduction
2. Results and Discussion
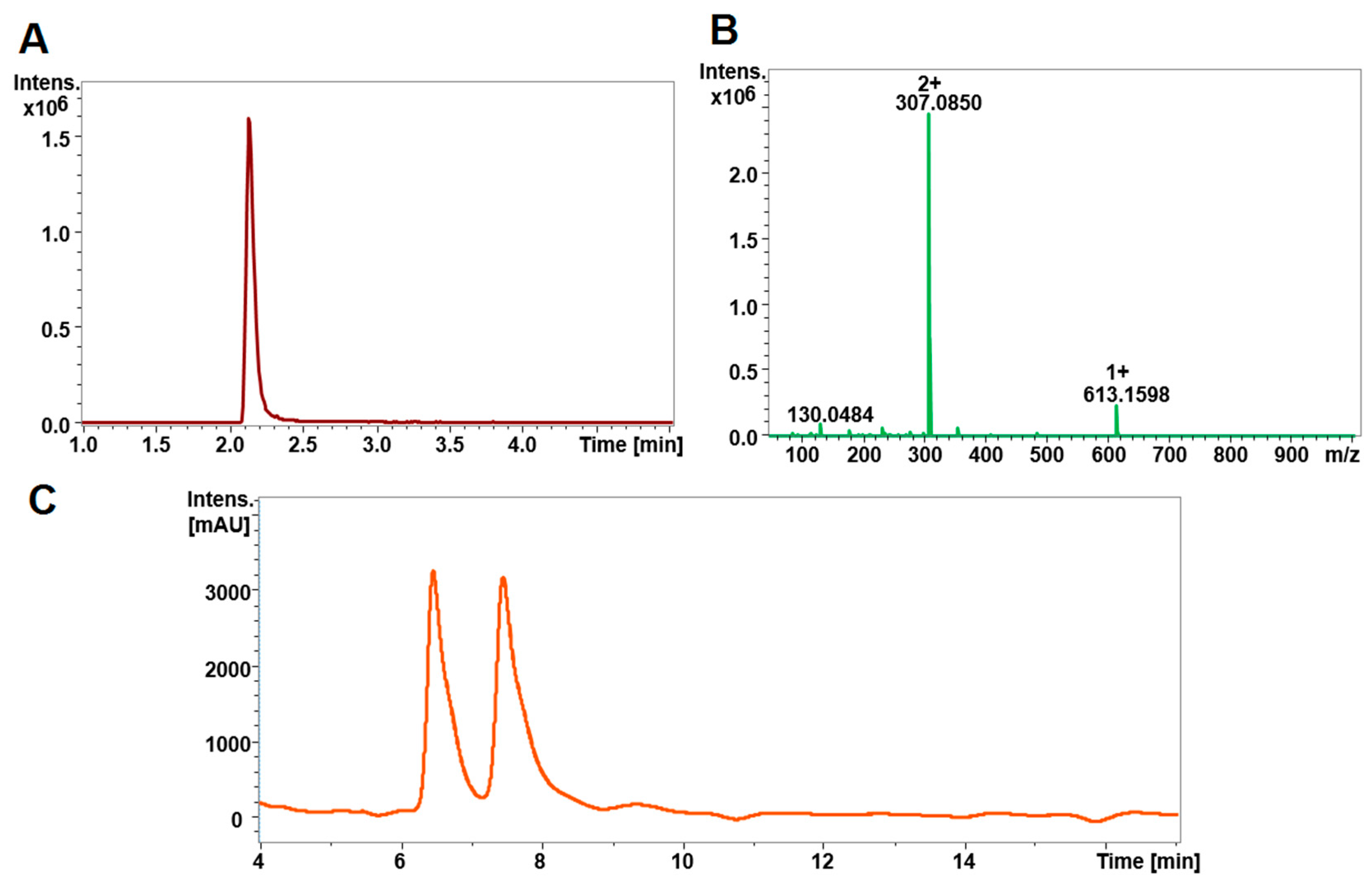
2.1. Chromatography and Mass Spectrometry
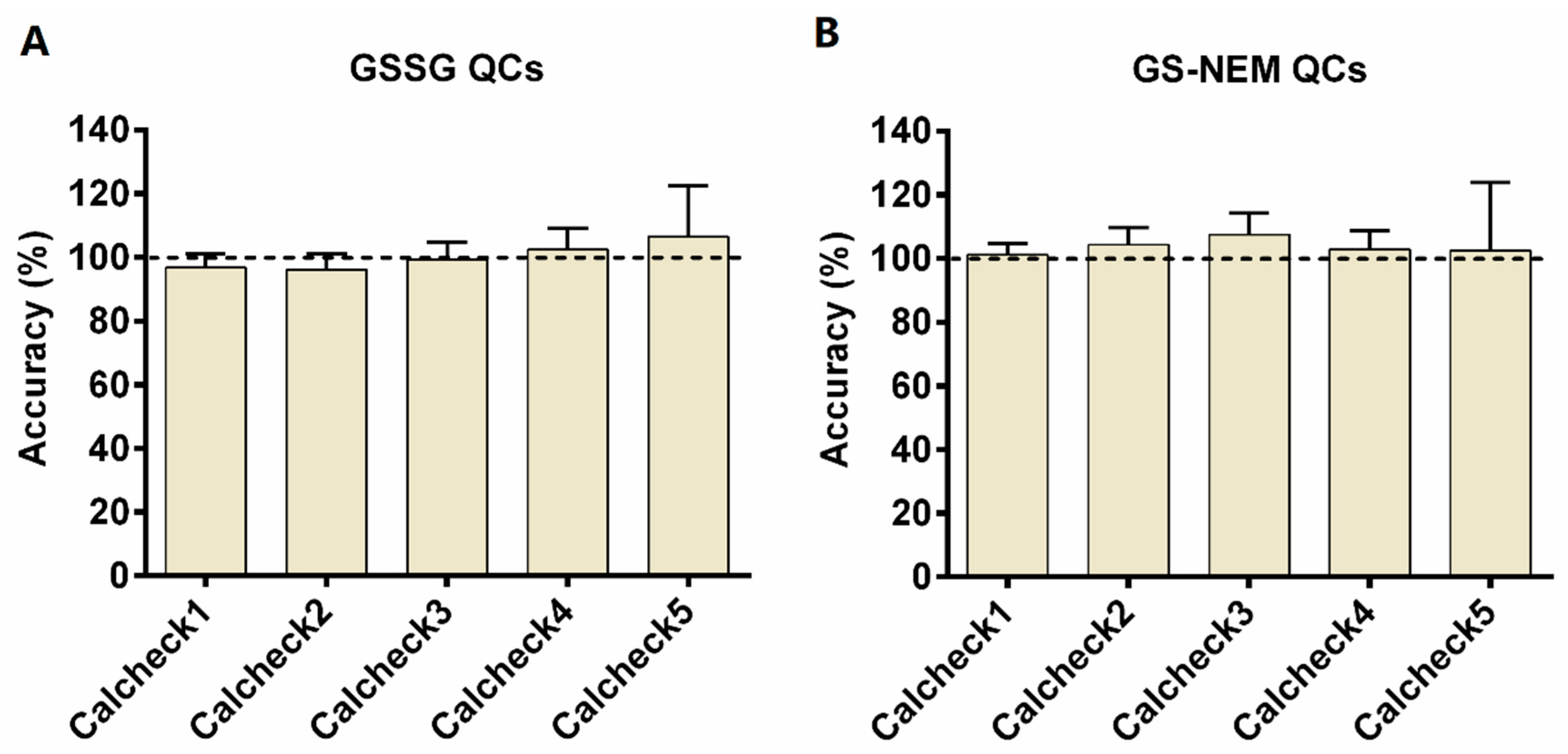
2.2. Method Validation
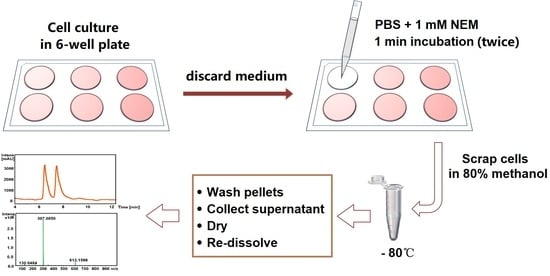
2.3. Cell Harvesting
- Procedure 1: As described above, the cell culture medium was removed, and the cells were washed twice for 1 min with PBS containing 1 mM NEM prior to cell harvesting in cold 80% methanol.
- Procedure 2: Cells were washed twice with 1 mL of PBS prior to addition of 400 µL of 1 mM NEM solution for 5 min, followed by addition of 1600 µL methanol (to a final concentration of 80%, v/v) to harvest cells in cold 80% methanol.
- Procedure 3: Cells were washed twice with 1 mL of PBS and then scrapped with 1 mL of cold 80% methanol containing 0.5 mM NEM.
- Procedure 4: NEM was added directly to the cell culture medium at a final concentration of ~1.5 mM for 2 min. Then, culture medium was discarded, cells were washed with PBS and scraped in 80% cold methanol.
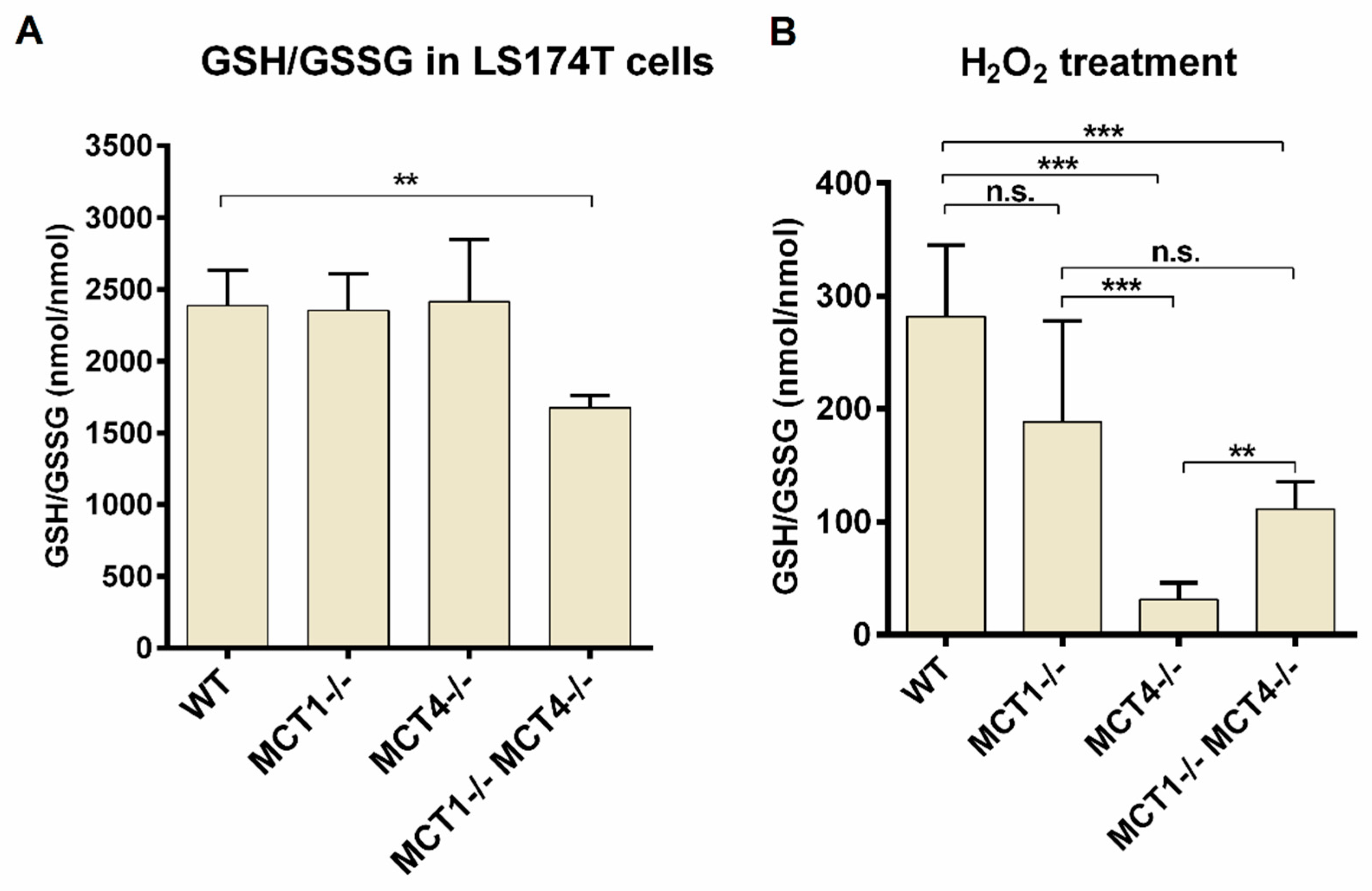
2.4. GSH and GSSG Determination in Monocarboxylate Transporter Deficient Cells
2.5. GSH and GSSG Determination in Isocitrate Dehydrogenase Wild Type and Mutant Cells
3. Experimental
3.1. Chemicals and Reagents
3.2. Stock Solutions
3.3. Cell Culture and Sample Preparation
3.4. HPLC-UV-QTOFMS Analysis of GSH and GSSG
3.5. Method Validation
3.5.1. Figures of Merit
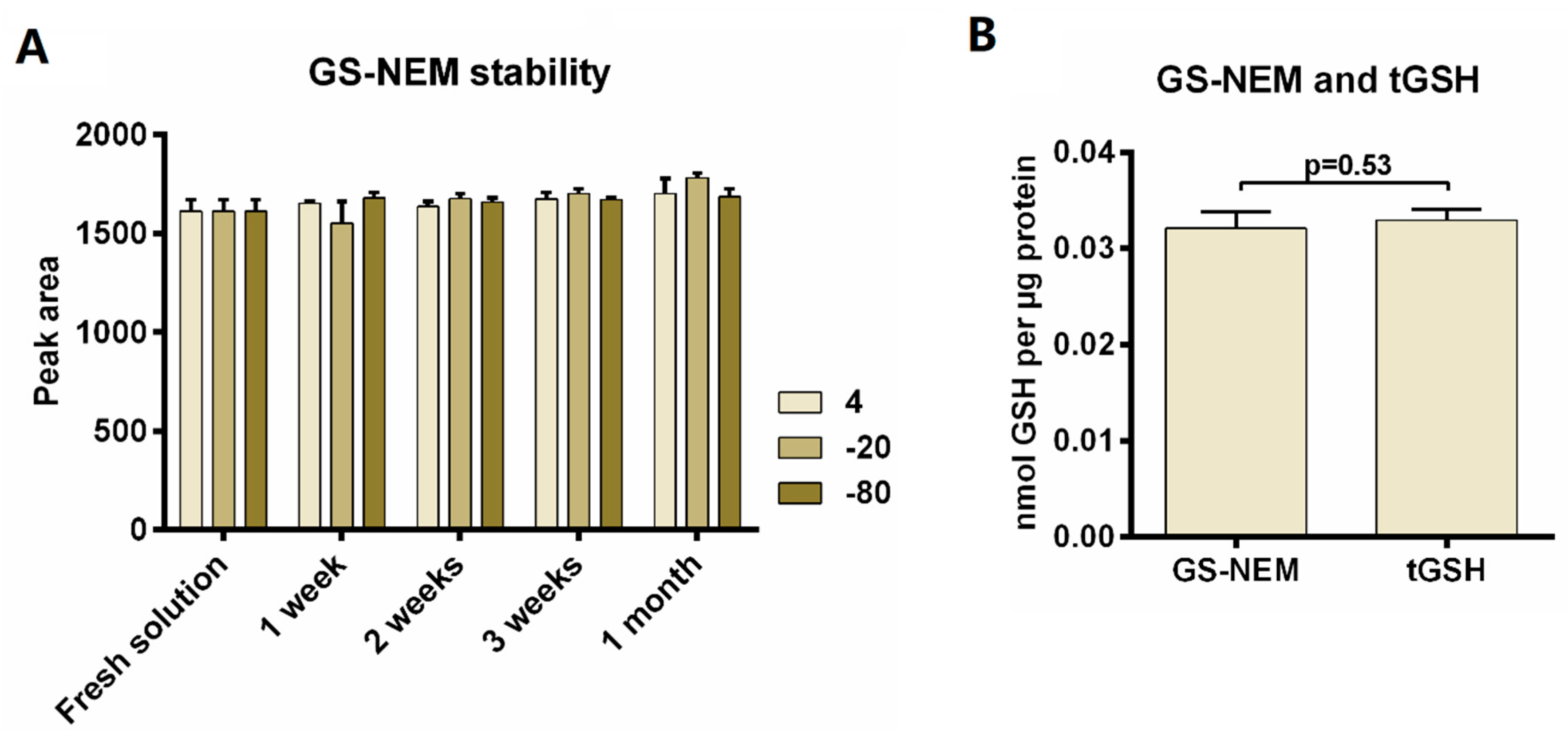
3.5.2. Stability of GS-NEM
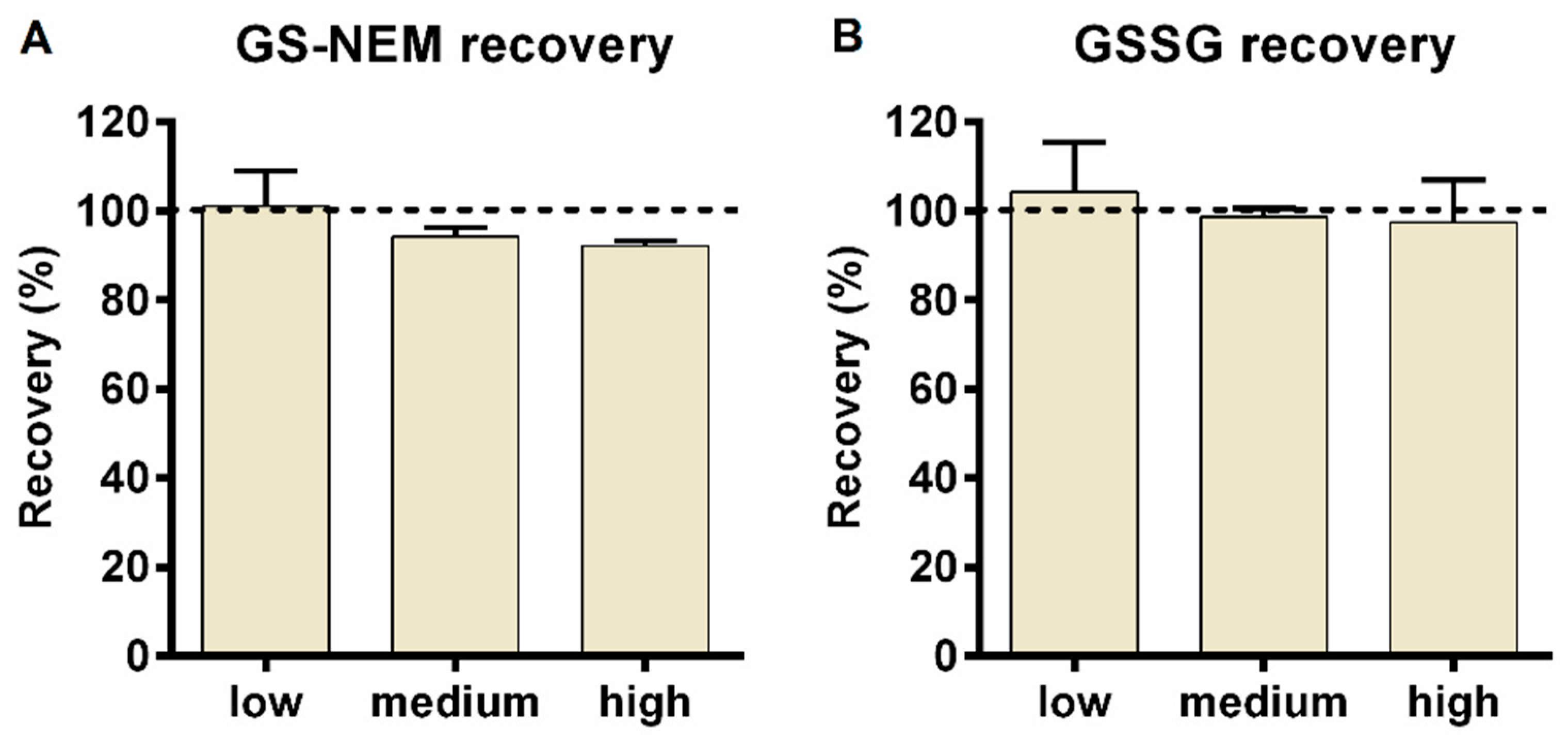
3.5.3. Recovery
3.6. Data Analysis and Statistics
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sies, H. Glutathione and its role in cellular functions. Free Radic. Biol. Med. 1999, 27, 916–921. [Google Scholar] [CrossRef]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef]
- Giustarini, D.; Dalle-Donne, I.; Milzani, A.; Fanti, P.; Rossi, R. Analysis of GSH and GSSG after derivatization with N-ethylmaleimide. Nat. Protoc. 2013, 8, 1660–1669. [Google Scholar] [CrossRef] [PubMed]
- Rahman, I.; Kode, A.; Biswas, S.K. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat. Protoc. 2006, 1, 3159–3165. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Dalle-Donne, I.; Colombo, R.; Milzani, A.; Rossi, R. An improved HPLC measurement for GSH and GSSG in human blood. Free Radic. Biol. Med. 2003, 35, 1365–1372. [Google Scholar] [CrossRef]
- Lipsa, D.; Cacho, C.; Leva, P.; Barrero-Moreno, J.; Aguar, P. Development of a HPLC-UV method for the simultaneous determination of intracellular glutathione species in human cells. J. Anal. Bioanal. Tech. 2015, 6, 1–8. [Google Scholar]
- Hakuna, L.; Doughan, B.; Escobedo, J.O.; Strongin, R.M. A simple assay for glutathione in whole blood. Analyst 2015, 140, 3339–3342. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Chi, D.; Song, D.; Su, G.; Li, L.; Shao, L. Quantification of Glutathione in Plasma Samples by HPLC Using 4-Fluoro-7-nitrobenzofurazan as a Fluorescent Labeling Reagent. J. Chromatogr. Sci. 2012, 50, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Nolin, T.D.; McMenamin, M.E.; Himmelfarb, J. Simultaneous determination of total homocysteine, cysteine, cysteinylglycine, and glutathione in human plasma by high-performance liquid chromatography: Application to studies of oxidative stress. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 852, 554–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo-Veyrat, S.; Hopfgartner, G. High-throughput liquid chromatography differential mobility spectrometry mass spectrometry for bioanalysis: Determination of reduced and oxidized form of glutathione in human blood. Anal. Bioanal. Chem. 2018, 410, 7153–7161. [Google Scholar] [CrossRef]
- Fahrenholz, T.; Wolle, M.M.; Kingston, H.M.; Faber, S.; Kern, J.C., 2nd; Pamuku, M.; Miller, L.; Chatragadda, H.; Kogelnik, A. Molecular speciated isotope dilution mass spectrometric methods for accurate, reproducible and direct quantification of reduced, oxidized and total glutathione in biological samples. Anal. Chem. 2015, 87, 1232–1240. [Google Scholar] [CrossRef] [PubMed]
- Blahova, L.; Kohoutek, J.; Lebedova, J.; Blaha, L.; Vecera, Z.; Buchtova, M.; Misek, I.; Hilscherova, K. Simultaneous determination of reduced and oxidized glutathione in tissues by a novel liquid chromatography-mass spectrometry method: Application in an inhalation study of Cd nanoparticles. Anal. Bioanal. Chem. 2014, 406, 5867–5876. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.; Le, A.; Niemi, A.-K.; Kwan, T.; Cusmano-Ozog, K.; Enns, G.M.; Cowan, T.M. A new LC-MS/MS method for the clinical determination of reduced and oxidized glutathione from whole blood. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 929, 51–55. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Colombo, G.; Garavaglia, M.L.; Astori, E.; Portinaro, N.M.; Reggiani, F.; Badalamenti, S.; Aloisi, A.M.; Santucci, A.; Rossi, R.; et al. Assessment of glutathione/glutathione disulphide ratio and S-glutathionylated proteins in human blood, solid tissues, and cultured cells. Free Radic. Biol. Med. 2017, 112, 360–375. [Google Scholar] [CrossRef]
- Giustarini, D.; Galvagni, F.; Tesei, A.; Farolfi, A.; Zanoni, M.; Pignatta, S.; Milzani, A.; Marone, I.M.; Dalle-Donne, I.; Nassini, R.; et al. Glutathione, glutathione disulfide, and S-glutathionylated proteins in cell cultures. Free Radic. Biol. Med. 2015, 89, 972–981. [Google Scholar] [CrossRef] [PubMed]
- Florholmen-Kjær, Å.; Lyså, R.A.; Fuskevåg, O.-M.; Goll, R.; Revhaug, A.; Mortensen, K.E. A sensitive method for the analysis of glutathione in porcine hepatocytes. Scand. J. Gastroenterol. 2014, 49, 1359–1366. [Google Scholar] [CrossRef] [PubMed]
- Sutton, T.R.; Minnion, M.; Barbarino, F.; Koster, G.; Fernandez, B.O.; Cumpstey, A.F.; Wischmann, P.; Madhani, M.; Frenneaux, M.P.; Postle, A.D.; et al. A robust and versatile mass spectrometry platform for comprehensive assessment of the thiol redox metabolome. Redox Biol. 2018, 16, 359–380. [Google Scholar] [CrossRef] [PubMed]
- Tomin, T.; Schittmayer, M.; Birner-Gruenberger, R. Addressing glutathione redox status in clinical samples by two-step alkylation with N-ethylmaleimide isotopologues. Metabolites 2020, 10, 71. [Google Scholar] [CrossRef] [Green Version]
- Shaik, I.H.; Mehvar, R. Rapid determination of reduced and oxidized glutathione levels using a new thiol-masking reagent and the enzymatic recycling method: Application to the rat liver and bile samples. Anal. Bioanal. Chem. 2006, 385, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Garcia, S.C.; Schott, K.; Charao, M.; Moro, A.; Bulcao, R.; Grotto, D.; Valentini, J.; Bohrer, D.; Cardoso, S.; Pomblum, V. Quantification of reduced glutathione by HPLC-UV in erythrocytes of hemodialysis patients. Biomed. Chromatogr. 2008, 22, 460–468. [Google Scholar] [CrossRef]
- Sun, X.; Heinrich, P.; Berger, R.S.; Oefner, P.J.; Dettmer, K. Quantification and (13)C-Tracer analysis of total reduced glutathione by HPLC-QTOFMS/MS. Anal. Chim. Acta 2019, 8, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Marchiq, I.; Le Floch, R.; Roux, D.; Simon, M.P.; Pouyssegur, J. Genetic disruption of lactate/H+ symporters (MCTs) and their subunit CD147/BASIGIN sensitizes glycolytic tumor cells to phenformin. Cancer Res. 2015, 75, 171–180. [Google Scholar] [CrossRef] [PubMed]
- Doherty, J.R.; Yang, C.; Scott, K.E.; Cameron, M.D.; Fallahi, M.; Li, W.; Hall, M.A.; Amelio, A.L.; Mishra, J.K.; Li, F.; et al. Blocking lactate export by inhibiting the Myc target MCT1 Disables glycolysis and glutathione synthesis. Cancer Res. 2014, 74, 908–920. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, C.S.; Graham, N.A.; Gu, W.; Espindola Camacho, C.; Mah, V.; Maresh, E.L.; Alavi, M.; Bagryanova, L.; Krotee, P.A.L.; Gardner, B.K.; et al. MCT1 Modulates Cancer Cell Pyruvate Export and Growth of Tumors that Co-express MCT1 and MCT4. Cell Rep. 2016, 14, 1590–1601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anastasiou, D.; Poulogiannis, G.; Asara, J.M.; Boxer, M.B.; Jiang, J.K.; Shen, M.; Bellinger, G.; Sasaki, A.T.; Locasale, J.W.; Auld, D.S.; et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011, 334, 1278–1283. [Google Scholar] [CrossRef] [Green Version]
- Gruning, N.M.; Du, D.; Keller, M.A.; Luisi, B.F.; Ralser, M. Inhibition of triosephosphate isomerase by phosphoenolpyruvate in the feedback-regulation of glycolysis. Open Biol. 2014, 4, 130232–130242. [Google Scholar] [CrossRef] [Green Version]
- Contreras-Baeza, Y.; Sandoval, P.Y.; Alarcon, R.; Galaz, A.; Cortes-Molina, F.; Alegria, K.; Baeza-Lehnert, F.; Arce-Molina, R.; Guequen, A.; Flores, C.A.; et al. Monocarboxylate transporter 4 (MCT4) is a high affinity transporter capable of exporting lactate in high-lactate microenvironments. J. Biol. Chem. 2019, 294, 20135–20147. [Google Scholar] [CrossRef]
- Stincone, A.; Prigione, A.; Cramer, T.; Wamelink, M.M.; Campbell, K.; Cheung, E.; Olin-Sandoval, V.; Gruning, N.M.; Kruger, A.; Tauqeer Alam, M.; et al. The return of metabolism: Biochemistry and physiology of the pentose phosphate pathway. Biol. Rev. Camb. Philos. Soc. 2015, 90, 927–963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, D.; Guan, K.L.; Xiong, Y. Metabolism, Activity, and Targeting of D- and L-2-Hydroxyglutarates. Trends Cancer 2018, 4, 151–165. [Google Scholar] [CrossRef] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef]
- Dang, L.; White, D.W.; Gross, S.; Bennett, B.D.; Bittinger, M.A.; Driggers, E.M.; Fantin, V.R.; Jang, H.G.; Jin, S.; Keenan, M.C.; et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 2009, 462, 739–744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, R.S. Development and Application of Analytical Tools to Study the Origin, Fate and Impact of the Oncometabolite 2-Hydroxyglutarate and its Lactone. Ph.D. Thesis, University of Regensburg, Regensburg, Germany, 2019. [Google Scholar]
- Guidance for Industry: Bioanalytical Method Validation; U.S. Department of Health and Human Services, Food and Drug Administration, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CVM): Rockville, MD, USA, 2018.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Method | Sample | GSH LOD * | GSH LOQ * | GSSG LOD * | GSSG LOQ * | Derivatization | Ref. |
|---|---|---|---|---|---|---|---|
| Enzymatic recycling | Rat liver/bile | -- | 6.25 pmol | -- | 2.17 pmol | M4VP | [19] |
| HPLC-UV | Erythrocytes | 820 pmol (0.041 mM) | 2700 pmol (0.135 mM) | -- | -- | DTNB | [20] |
| HPLC-fluorescence | Plasma | 0.6 pmol (0.03 µM) | 2 pmol (0.10 µM) | -- | -- | NBD-F | [8] |
| LC-MS/MS | Whole blood | 4 pmol (0.4 µM) | 15 pmol (1.5 µM) | 1.5 pmol (0.1 µM) | 1.5 pmol (0.1 µM) | NEM | [13] |
| HPLC-UV | Cultured cells | -- | -- | -- | -- | NEM/DTT/mBrB | [15] |
| HPLC-UV | Cultured cells | 78.1 pmol (7.81 µM) | 156.5 pmol (15.65 µM) | -- | -- | NEM | This study |
| HPLC-QTOF-MS | -- | -- | 0.01 pmol | 0.1 pmol | -- |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sun, X.; Berger, R.S.; Heinrich, P.; Marchiq, I.; Pouyssegur, J.; Renner, K.; Oefner, P.J.; Dettmer, K. Optimized Protocol for the In Situ Derivatization of Glutathione with N-Ethylmaleimide in Cultured Cells and the Simultaneous Determination of Glutathione/Glutathione Disulfide Ratio by HPLC-UV-QTOF-MS. Metabolites 2020, 10, 292. https://doi.org/10.3390/metabo10070292
Sun X, Berger RS, Heinrich P, Marchiq I, Pouyssegur J, Renner K, Oefner PJ, Dettmer K. Optimized Protocol for the In Situ Derivatization of Glutathione with N-Ethylmaleimide in Cultured Cells and the Simultaneous Determination of Glutathione/Glutathione Disulfide Ratio by HPLC-UV-QTOF-MS. Metabolites. 2020; 10(7):292. https://doi.org/10.3390/metabo10070292
Chicago/Turabian StyleSun, Xueni, Raffaela S. Berger, Paul Heinrich, Ibtissam Marchiq, Jacques Pouyssegur, Kathrin Renner, Peter J. Oefner, and Katja Dettmer. 2020. "Optimized Protocol for the In Situ Derivatization of Glutathione with N-Ethylmaleimide in Cultured Cells and the Simultaneous Determination of Glutathione/Glutathione Disulfide Ratio by HPLC-UV-QTOF-MS" Metabolites 10, no. 7: 292. https://doi.org/10.3390/metabo10070292
APA StyleSun, X., Berger, R. S., Heinrich, P., Marchiq, I., Pouyssegur, J., Renner, K., Oefner, P. J., & Dettmer, K. (2020). Optimized Protocol for the In Situ Derivatization of Glutathione with N-Ethylmaleimide in Cultured Cells and the Simultaneous Determination of Glutathione/Glutathione Disulfide Ratio by HPLC-UV-QTOF-MS. Metabolites, 10(7), 292. https://doi.org/10.3390/metabo10070292