Promoter Architecture and Promoter Engineering in Saccharomyces cerevisiae


 , and
, and
Abstract
:1. Introduction
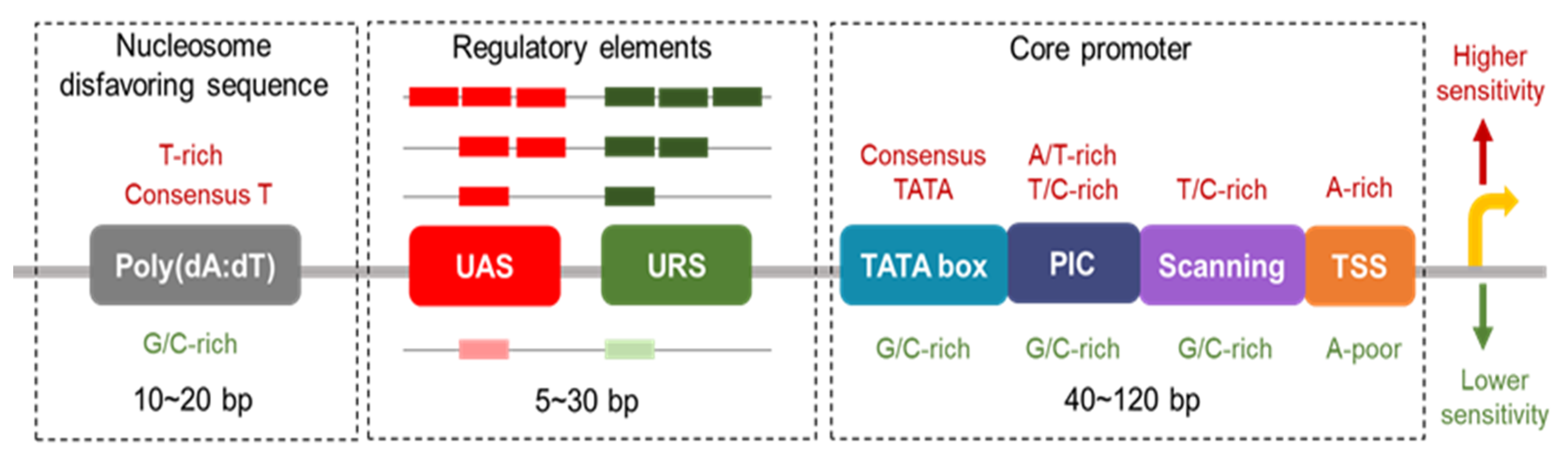
2. Promoter Architecture and Function
2.1. Core Promoters
2.2. UAS and URS
2.3. Nucleosomes Disfavoring Sequences at Gene Promoters
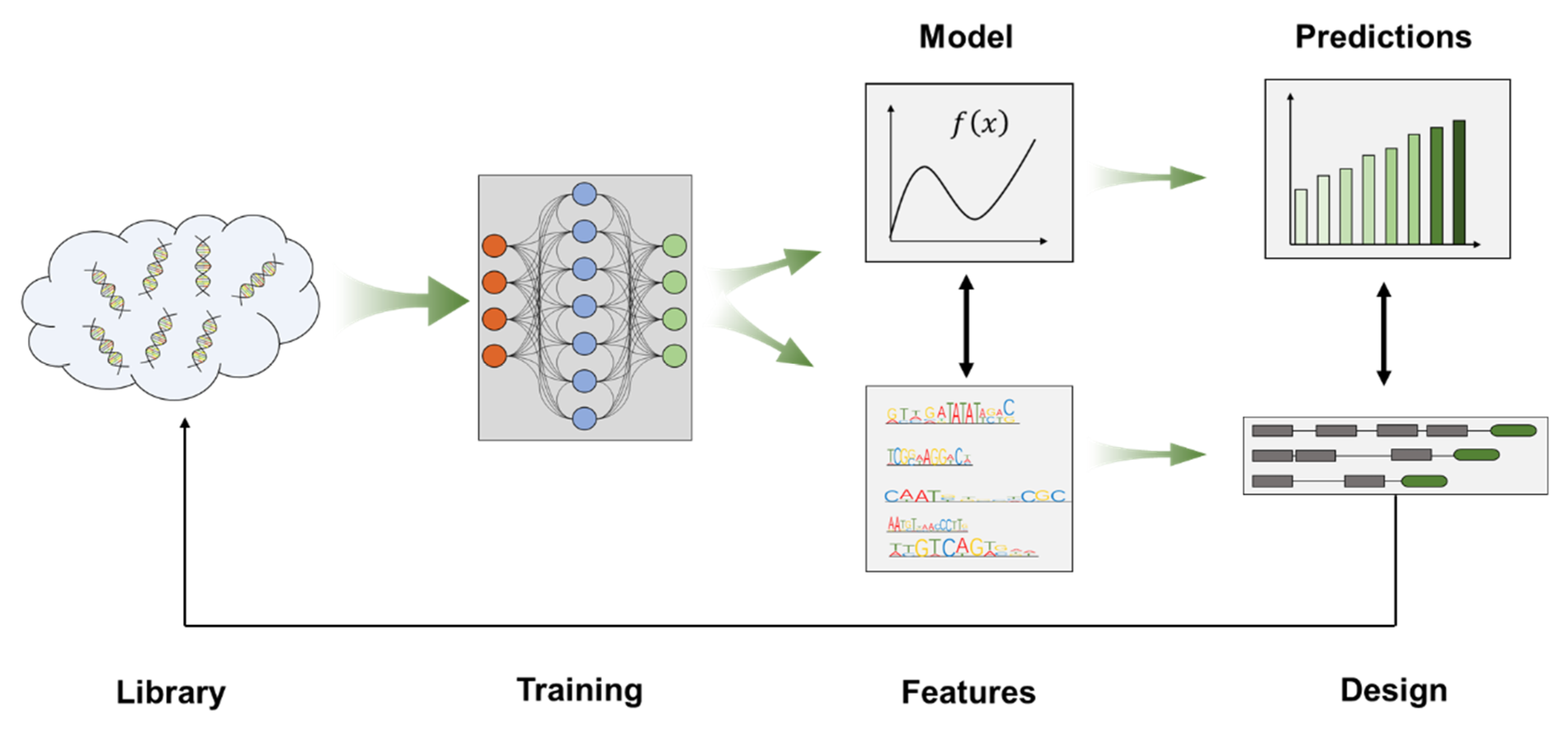
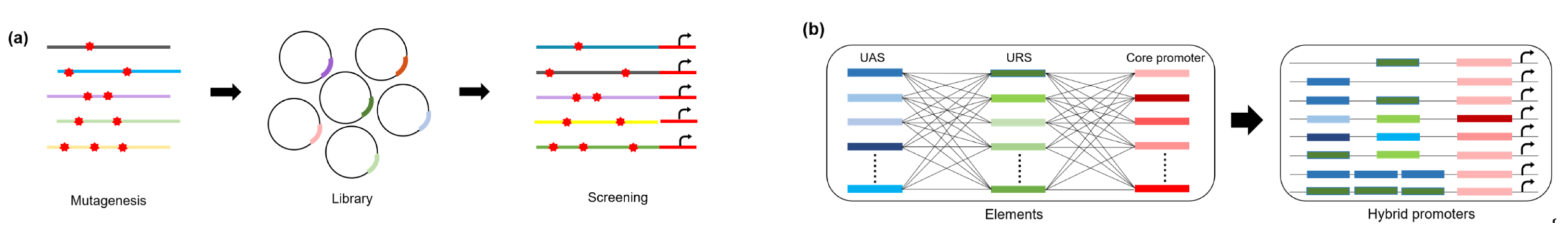
3. Promoter Engineering Approaches
4. Promoter Engineering for Diverse Synthetic Promoters and Their Applications
4.1. Synthetic Promoters for Expanding Dynamic Ranges
4.2. Synthetic Promoters for Reducing Homologous Recombination
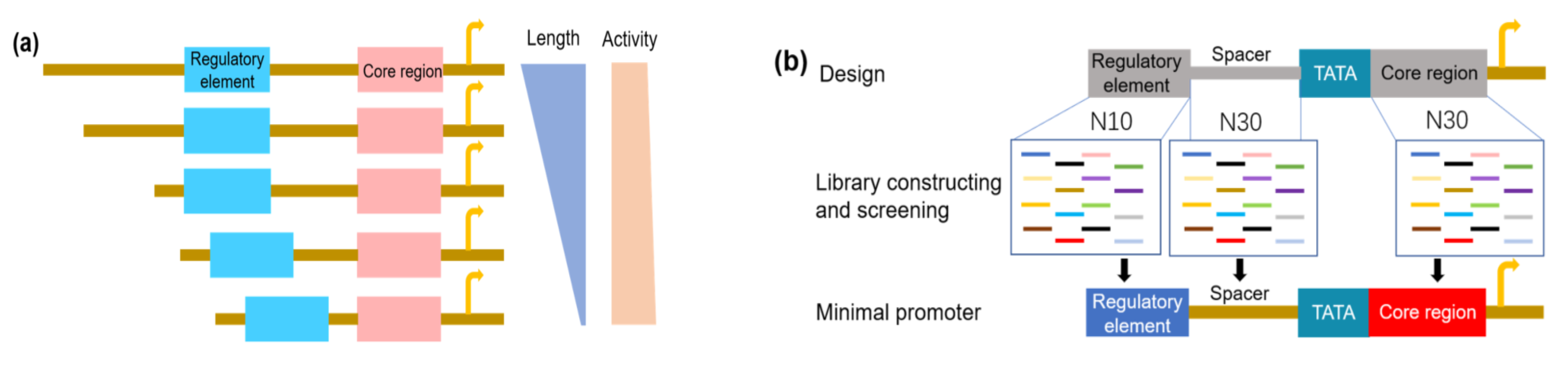
4.3. Synthetic Promoters with Minimal Size
4.4. Synthetic Promoters for Multi-Host Application
4.5. Synthetic Promoters for Constructing Biosensors
5. Perspective
Author Contributions
Funding
Conflicts of Interest
References
- Nevoigt, E. Progress in Metabolic Engineering of Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 2008, 72, 379–412. [Google Scholar] [CrossRef] [Green Version]
- Scalcinati, G.; Knuf, C.; Partow, S.; Chen, Y.; Maury, J.; Schalk, M.; Daviet, L.; Nielsen, J.; Siewers, V. Dynamic control of gene expression in Saccharomyces cerevisiae engineered for the production of plant sesquitepene α-santalene in a fed-batch mode. Metab. Eng. 2012, 14, 91–103. [Google Scholar] [CrossRef] [PubMed]
- Latimer, L.N.; Lee, M.E.; Medina-Cleghorn, D.; Kohnz, R.A.; Nomura, D.K.; Dueber, J.E. Employing a combinatorial expression approach to characterize xylose utilization in Saccharomyces cerevisiae. Metab. Eng. 2014, 25, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Keren, L.; Zackay, O.; Lotan-Pompan, M.; Barenholz, U.; Dekel, E.; Sasson, V.; Aidelberg, G.; Bren, A.; Zeevi, D.; Weinberger, A.; et al. Promoters maintain their relative activity levels under different growth conditions. Mol. Syst. Biol. 2013, 9, 701. [Google Scholar] [CrossRef] [PubMed]
- Lipson, D.; Raz, T.; Kieu, A.; Jones, D.R.; Giladi, E.; Thayer, E.; Thompson, J.F.; Letovsky, S.; Milos, P.; Causey, M. Quantification of the yeast transcriptome by single-molecule sequencing. Nat. Biotechnol. 2009, 27, 652–658. [Google Scholar] [CrossRef] [PubMed]
- Partow, S.; Siewers, V.; Bjørn, S.; Nielsen, J.; Maury, J. Characterization of different promoters for designing a new expression vector in Saccharomyces cerevisiae. Yeast 2010, 27, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Shao, Z.; Zhao, H.; Nair, N.; Wen, F.; Xu, J.-H.; Zhao, H. Cloning and characterization of a panel of constitutive promoters for applications in pathway engineering in Saccharomyces cerevisiae. Bitechnol. Bioeng. 2012, 109, 2082–2092. [Google Scholar] [CrossRef]
- Reider Apel, A.; D’Espaux, L.; Wehrs, M.; Sachs, D.; Li, R.A.; Tong, G.J.; Garber, M.; Nnadi, O.; Zhuang, W.; Hillson, N.J.; et al. A Cas9-based toolkit to program gene expression in Saccharomyces cerevisiae. Nucleic Acids Res. 2016, 45, 496–508. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.; Lee, K.; Bae, S.-J.; Hahn, J.-S. Promoters inducible by aromatic amino acids and γ-aminobutyrate (GABA) for metabolic engineering applications in Saccharomyces cerevisiae. Appl. Mircrobiol. Bitechnol. 2015, 99, 2705–2714. [Google Scholar] [CrossRef]
- Weinhandl, K.; Winkler, M.; Glieder, A.; Camattari, A. Carbon source dependent promoters in yeasts. Microb. Cell Factories 2014, 13, 5. [Google Scholar] [CrossRef] [Green Version]
- Ottoz, D.S.; Rudolf, F. Constitutive and Regulated Promoters in Yeast: How to Design and Make Use of Promoters in S. cerevisiae. Synth. Biol. Parts Devices Appl. 2018, 10, 14. [Google Scholar]
- Ro, D.-K.; Paradise, E.M.; Ouellet, M.; Fisher, K.J.; Newman, K.L.; Ndungu, J.M.; Ho, K.A.; Eachus, R.A.; Ham, T.S.; Kirby, J.; et al. Production of the antimalarial drug precursor artemisinic acid in engineered yeast. Nature 2006, 440, 940–943. [Google Scholar] [CrossRef]
- Luo, X.; Reiter, M.A.; d’Espaux, L.; Wong, J.; Denby, C.M.; Lechner, A.; Zhang, Y.; Grzybowski, A.T.; Harth, S.; Lin, W.; et al. Complete biosynthesis of cannabinoids and their unnatural analogues in yeast. Nature 2019, 567, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Lv, X.; Xie, W.; Zhou, P.; Zhu, Y.; Yao, Z.; Yang, C.; Yang, X.; Ye, L.; Yu, H. Combining Gal4p-mediated expression enhancement and directed evolution of isoprene synthase to improve isoprene production in Saccharomyces cerevisiae. Metab. Eng. 2017, 39, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Alper, H.; Fischer, C.; Nevoigt, E.; Stephanopoulos, G. Tuning genetic control through promoter engineering. Proc. Natl. Acad.Sci. USA 2005, 102, 12678–12683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazeck, J.; Garg, R.; Reed, B.; Alper, H.S. Controlling promoter strength and regulation in Saccharomyces cerevisiae using synthetic hybrid promoters. Bitechnol. Bioeng. 2012, 109, 2884–2895. [Google Scholar] [CrossRef]
- Curran, K.A.; Crook, N.C.; Karim, A.S.; Gupta, A.; Wagman, A.M.; Alper, H.S. Design of synthetic yeast promoters via tuning of nucleosome architecture. Nat. Commun. 2014, 5, 4002. [Google Scholar] [CrossRef]
- Zhu, J.; Zhang, M.Q. SCPD: A promoter database of the yeast Saccharomyces cerevisiae. Bioinformatics 1999, 15, 607–611. [Google Scholar] [CrossRef]
- Hertzberg, L.; Zuk, O.; Getz, G.; Domany, E. Finding Motifs in Promoter Regions. J. Comput. Biol. 2005, 12, 314–330. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, M.C.; Monteiro, P.; Jain, P.; Tenreiro, S.; Fernandes, A.R.; Mira, N.P.; Alenquer, M.; Freitas, A.T.; Oliveira, A.L.; Sá-Correia, I. The YEASTRACT database: A tool for the analysis of transcription regulatory associations in Saccharomyces cerevisiae. Nucleic Acids Res. 2006, 34, D446–D451. [Google Scholar] [CrossRef] [Green Version]
- Teixeira, M.C.; Monteiro, P.T.; Palma, M.; Costa, C.; Godinho, C.P.; Pais, P.; Cavalheiro, M.; Antunes, M.; Lemos, A.; Pedreira, T.; et al. YEASTRACT: An upgraded database for the analysis of transcription regulatory networks in Saccharomyces cerevisiae. Nucleic Acids Res. 2017, 46, D348–D353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basehoar, A.D.; Zanton, S.J.; Pugh, B.F. Identification and distinct regulation of yeast TATA box-containing genes. Cell 2004, 116, 699–709. [Google Scholar] [CrossRef] [Green Version]
- Pugh, B.F.; Tjian, R. Transcription from a TATA-less promoter requires a multisubunit TFIID complex. Genes Dev. 1991, 5, 1935–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mogno, I.; Vallania, F.; Mitra, R.D.; Cohen, B.A. TATA is a modular component of synthetic promoters. Genome Res. 2010, 20, 1391–1397. [Google Scholar] [CrossRef] [Green Version]
- Bae, S.-H.; Han, H.W.; Moon, J. Functional Analysis of the Molecular Interactions of TATA Box-Containing Genes and Essential Genes. PLoS ONE 2015, 10, e0120848. [Google Scholar] [CrossRef]
- Wobbe, C.R.; Struhl, K. Yeast and human TATA-binding proteins have nearly identical DNA sequence requirements for transcription in vitro. Mol. Cell. Biol. 1990, 10, 3859–3867. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.J.; Stargell, L.A. The stability of the TFIIA-TBP-DNA complex is dependent on the sequence of the TATAAA element. J. Biol. Chem. 2001, 276, 30078–30084. [Google Scholar] [CrossRef] [Green Version]
- Chen, W.; Struhl, K. Saturation mutagenesis of a yeast his3” TATA element”: genetic evidence for a specific TATA-binding protein. Proc. Natl. Acad. Sci. USA 1988, 85, 2691–2695. [Google Scholar] [CrossRef] [Green Version]
- Lubliner, S.; Regev, I.; Lotan-Pompan, M.; Edelheit, S.; Weinberger, A.; Segal, E. Core promoter sequence in yeast is a major determinant of expression level. Genome Res. 2015, 25, 1008–1017. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Dietrich, F.S. Mapping of transcription start sites in Saccharomyces cerevisiae using 5′ SAGE. Nucleic Acids Res. 2005, 33, 2838–2851. [Google Scholar] [CrossRef] [Green Version]
- Buratowski, S.; Hahn, S.; Guarente, L.; Sharp, P.A. Five intermediate complexes in transcription initiation by RNA polymerase II. Cell 1989, 56, 549–561. [Google Scholar] [CrossRef]
- Struhl, K. Promoters, activator proteins, and the mechanism of transcriptional initiation in yeast. Cell 1987, 49, 295–297. [Google Scholar] [CrossRef]
- Giardina, C.; Lis, J. DNA melting on yeast RNA polymerase II promoters. Science 1993, 261, 759–762. [Google Scholar] [CrossRef]
- Lubliner, S.; Keren, L.; Segal, E. Sequence features of yeast and human core promoters that are predictive of maximal promoter activity. Nucleic Acids Res. 2013, 11, 5569–5581. [Google Scholar] [CrossRef] [PubMed]
- Redden, H.R. Developing Synthetic, Minimal Promoters in Saccharomyces Cerevisiae; The University of Texas at Austin: Austin, TX, USA, 2017. [Google Scholar]
- Bitter, G.A.; Chang, K.K.; Egan, K.M. A multi-component upstream activation sequence of the Saccharomyces cerevisiae glyceraldehyde-3-phosphate dehydrogenase gene promoter. Mol. Gen. Genet. MGG 1991, 231, 22–32. [Google Scholar] [CrossRef]
- West, R.; Yocum, R.R.; Ptashne, M. Saccharomyces cerevisiae GAL1-GAL10 divergent promoter region: Location and function of the upstream activating sequence UASG. Mol. Cell. Biol. 1984, 4, 2467–2478. [Google Scholar] [CrossRef] [Green Version]
- Giniger, E.; Varnum, S.M.; Ptashne, M. Specific DNA binding of GAL4, a positive regulatory protein of yeast. Cell 1985, 40, 767–774. [Google Scholar] [CrossRef]
- Gancedo, J.M. Yeast carbon catabolite repression. Microbiol. Mol. Biol. Rev. 1998, 62, 334–361. [Google Scholar] [CrossRef] [Green Version]
- Bu, Y.; Schmidt, M.C. Identification of cis-acting elements in the SUC2 promoter of Saccharomyces cerevisiae required for activation of transcription. Nucleic Acids Res. 1998, 26, 1002–1009. [Google Scholar] [CrossRef] [Green Version]
- Ruiz, A.; Serrano, R.; Ariño, J. Direct regulation of genes involved in glucose utilization by the calcium/calcineurin pathway. J. Biol. Chem. 2008, 283, 13923–13933. [Google Scholar] [CrossRef] [Green Version]
- Nehlin, J.O.; Carlberg, M.; Ronne, H. Control of yeast GAL genes by MIG1 repressor: A transcriptional cascade in the glucose response. EMBO J. 1991, 10, 3373–3377. [Google Scholar] [CrossRef] [PubMed]
- Bojunga, N.; Entian, K.-D. Cat8p, the activator of gluconeogenic genes in Saccharomyces cerevisiae, regulates carbon source-dependent expression of NADP-dependent cytosolic isocitrate dehydrogenase (Idp2p) and lactate permease (Jen1p). Mol. Gen. Gent. MGG 1999, 262, 869–875. [Google Scholar] [CrossRef] [PubMed]
- Stewart, A.J.; Hannenhalli, S.; Plotkin, J.B. Why transcription factor binding sites are ten nucleotides long. Genetics 2012, 192, 973–985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawal, Y.; Chereji, R.V.; Valabhoju, V.; Qiu, H.; Ocampo, J.; Clark, D.J.; Hinnebusch, A.G. Gcn4 Binding in Coding Regions Can Activate Internal and Canonical 5’ Promoters in Yeast. Mol. Cell 2018, 70, 297–311.e294. [Google Scholar] [CrossRef] [Green Version]
- Fernandes, L.; Rodrigues-Pousada, C.; Struhl, K. Yap, a novel family of eight bZIP proteins in Saccharomyces cerevisiae with distinct biological functions. Mol. Cell. Biol. 1997, 17, 6982–6993. [Google Scholar] [CrossRef] [Green Version]
- Stathopoulos, A.M.; Cyert, M.S. Calcineurin acts through the CRZ1/TCN1-encoded transcription factor to regulate gene expression in yeast. Genes Dev. 1997, 11, 3432–3444. [Google Scholar] [CrossRef] [Green Version]
- Roque, A.; Petrezsélyová, S.; Serra-Cardona, A.; Ariño, J. Genome-wide recruitment profiling of transcription factor Crz1 in response to high pH stress. BMC Genom. 2016, 17, 662. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi-Iwai, Y.; Stearman, R.; Dancis, A.; Klausner, R.D. Iron-regulated DNA binding by the AFT1 protein controls the iron regulon in yeast. EMBO J. 1996, 15, 3377–3384. [Google Scholar] [CrossRef]
- Oehlen, L.; Cross, F. The mating factor response pathway regulates transcription of TEC1, a gene involved in pseudohyphal differentiation of Saccharomyces cerevisiae. FEBS Lett. 1998, 429, 83–88. [Google Scholar] [CrossRef] [Green Version]
- Dorrity, M.W.; Cuperus, J.T.; Carlisle, J.A.; Fields, S.; Queitsch, C. Preferences in a trait decision determined by transcription factor variants. Proc. Natl. Acad. Sci. USA 2018, 115, E7997–E8006. [Google Scholar] [CrossRef] [Green Version]
- Mori, K.; Kawahara, T.; Yoshida, H.; Yanagi, H.; Yura, T. Signalling from endoplasmic reticulum to nucleus: Transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells 1996, 1, 803–817. [Google Scholar] [CrossRef] [PubMed]
- Eastmond, D.L.; Nelson, H.C.M. Genome-wide analysis reveals new roles for the activation domains of the Saccharomyces cerevisiae heat shock transcription factor (Hsf1) during the transient heat shock response. J. Biol. Chem. 2006, 281, 32909–32921. [Google Scholar] [CrossRef] [Green Version]
- Nehlin, J.O.; Ronne, H. Yeast MIG1 repressor is related to the mammalian early growth response and Wilms’ tumour finger proteins. EMBO J. 1990, 9, 2891–2898. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gimeno, M.A.; Struhl, K. Aca1 and Aca2, ATF/CREB activators in Saccharomyces cerevisiae, are important for carbon source utilization but not the response to stress. Mol. Cell. Biol. 2000, 20, 4340–4349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Y.; Karpichev, I.V.; Kohanski, R.A.; Small, G.M. Purification, identification, and properties of a Saccharomyces cerevisiae oleate-activated upstream activating sequence-binding protein that is involved in the activation of POX1. J. Biol. Chem. 1996, 271, 12068–12075. [Google Scholar] [CrossRef] [Green Version]
- Balzi, E.; Chen, W.; Ulaszewski, S.; Capieaux, E.; Goffeau, A. The multidrug resistance gene PDR1 from Saccharomyces cerevisiae. J. Biol. Chem. 1987, 262, 16871–16879. [Google Scholar]
- Subik, J.; Ulaszewski, S.; Goffeau, A. Genetic mapping of nuclear mucidin resistance mutations in Saccharomyces cerevisiae. A new pdr locus on chromosome II. Curr. Genet. 1986, 10, 665–670. [Google Scholar] [CrossRef]
- Coorey, N.V.C.; Matthews, J.H.; Bellows, D.S.; Atkinson, P.H. Pleiotropic drug-resistance attenuated genomic library improves elucidation of drug mechanisms. Mol. Biosyst. 2015, 11, 3129–3136. [Google Scholar] [CrossRef]
- Shirozu, R.; Yashiroda, H.; Murata, S. Identification of minimum Rpn4-responsive elements in genes related to proteasome functions. FEBS Lett. 2015, 589, 933–940. [Google Scholar] [CrossRef] [Green Version]
- Guerra-Moreno, A.; Hanna, J. Induction of proteotoxic stress by the mycotoxin patulin. Toxicol. Lett. 2017, 276, 85–91. [Google Scholar] [CrossRef]
- Fingerman, I.M.; Sutphen, K.; Montano, S.P.; Georgiadis, M.M.; Vershon, A.K. Characterization of critical interactions between Ndt80 and MSE DNA defining a novel family of Ig-fold transcription factors. Nucleic Acids Res. 2004, 32, 2947–2956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stebbins, J.L.; Triezenberg, S.J. Identification, mutational analysis, and coactivator requirements of two distinct transcriptional activation domains of the Saccharomyces cerevisiae Hap4 protein. Eukaryot. Cell 2004, 3, 339–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Proft, M.; Gibbons, F.D.; Copeland, M.; Roth, F.P.; Struhl, K. Genomewide identification of Sko1 target promoters reveals a regulatory network that operates in response to osmotic stress in Saccharomyces cerevisiae. Eukaryotic. Cell 2005, 4, 1343–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mai, B.; Breeden, L. Xbp1, a stress-induced transcriptional repressor of the Saccharomyces cerevisiae Swi4/Mbp1 family. Mol. Cell. Biol. 1997, 17, 6491–6501. [Google Scholar] [CrossRef] [Green Version]
- Barbaric, S.; Münsterkötter, M.; Goding, C.; Hörz, W. Cooperative Pho2-Pho4 Interactions at thePHO5 Promoter Are Critical for Binding of Pho4 to UASp1 and for Efficient Transactivation by Pho4 at UASp2. Mol. Cell. Biol. 1998, 18, 2629–2639. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Butler, E.; Rodgers, J.; Spizzo, T.; Duesterhoeft, S.; Eide, D. Regulation of zinc homeostasis in yeast by binding of the ZAP1 transcriptional activator to zinc-responsive promoter elements. J. Biol. Chem. 1998, 273, 28713–28720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Nadal, E.; Casadome, L.; Posas, F. Targeting the MEF2-like transcription factor Smp1 by the stress-activated Hog1 mitogen-activated protein kinase. Mol. Cell. Biol. 2003, 23, 229–237. [Google Scholar] [CrossRef] [Green Version]
- Dodou, E.; Treisman, R. The Saccharomyces cerevisiae MADS-box transcription factor Rlm1 is a target for the Mpk1 mitogen-activated protein kinase pathway. Mol. Cell. Biol. 1997, 17, 1848–1859. [Google Scholar] [CrossRef] [Green Version]
- Tachibana, C.; Yoo, J.Y.; Tagne, J.-B.; Kacherovsky, N.; Lee, T.I.; Young, E.T. Combined Global Localization Analysis and Transcriptome Data Identify Genes That Are Directly Coregulated by Adr1 and Cat8. Mol. Cell. Biol. 2005, 25, 2138–2146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weider, M.; Machnik, A.; Klebl, F.; Sauer, N. Vhr1p, a new transcription factor from budding yeast, regulates biotin-dependent expression of VHT1 and BIO5. J. Biol. Chem. 2006, 281, 13513–13524. [Google Scholar] [CrossRef] [Green Version]
- Blaiseau, P.L.; Thomas, D. Multiple transcriptional activation complexes tether the yeast activator Met4 to DNA. EMBO J. 1998, 17, 6327–6336. [Google Scholar] [CrossRef] [Green Version]
- Estruch, F.; Carlson, M. Two homologous zinc finger genes identified by multicopy suppression in a SNF1 protein kinase mutant of Saccharomyces cerevisiae. Mol. Cell. Biol. 1993, 13, 3872–3881. [Google Scholar] [CrossRef] [Green Version]
- Vincent, O.; Carlson, M. Sip4, a Snf1 kinase-dependent transcriptional activator, binds to the carbon source-responsive element of gluconeogenic genes. EMBO J. 1998, 17, 7002–7008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozcan, S.; Leong, T.; Johnston, M. Rgt1p of Saccharomyces cerevisiae, a key regulator of glucose-induced genes, is both an activator and a repressor of transcription. Mol. Cell. Biol. 1996, 16, 6419–6426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.-H.; Polish, J.; Johnston, M. Specificity and regulation of DNA binding by the yeast glucose transporter gene repressor Rgt1. Mol. Cell. Biol. 2003, 23, 5208–5216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tornow, J.; Zeng, X.; Gao, W.; Santangelo, G.M. GCR1, a transcriptional activator in Saccharomyces cerevisiae, complexes with RAP1 and can function without its DNA binding domain. EMBO J. 1993, 12, 2431–2437. [Google Scholar] [CrossRef] [PubMed]
- Lowry, C.V.; Cerdán, M.E.; Zitomer, R.S. A hypoxic consensus operator and a constitutive activation region regulate the ANB1 gene of Saccharomyces cerevisiae. Mol. Cell. Biol. 1990, 10, 5921–5926. [Google Scholar] [CrossRef] [Green Version]
- Fields, D.S.; He, Y.Y.; Al-Uzri, A.Y.; Stormo, G.D. Quantitative specificity of the Mnt repressor. J. Mol. Biol. 1997, 271, 178–194. [Google Scholar] [CrossRef] [Green Version]
- Vashee, S.; Xu, H.; Johnston, S.; Kodadek, T. How do “Zn2 cys6” proteins distinguish between similar upstream activation sites? Comparison of the DNA-binding specificity of the GAL4 protein in vitro and in vivo. J. Biol. Chem. 1993, 268, 24699–24706. [Google Scholar]
- Skowron, P.M.; Harasimowicz, R.; Rutkowska, S.M. GCN4 eukaryotic transcription factor/FokI endonuclease-mediated ‘Achilles’ heel cleavage’: Quantitative study of protein-DNA interaction. Gene 1996, 170, 1–8. [Google Scholar] [CrossRef]
- Hill, D.E.; Hope, I.A.; Macke, J.P.; Struhl, K. Saturation mutagenesis of the yeast his3 regulatory site: Requirements for transcriptional induction and for binding by GCN4 activator protein. Science 1986, 234, 451–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharon, E.; Kalma, Y.; Sharp, A.; Raveh-Sadka, T.; Levo, M.; Zeevi, D.; Keren, L.; Yakhini, Z.; Weinberger, A.; Segal, E. Inferring gene regulatory logic from high-throughput measurements of thousands of systematically designed promoters. Nat. Bitechnol. 2012, 30, 521. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giniger, E.; Ptashne, M. Cooperative DNA binding of the yeast transcriptional activator GAL4. Proc. Natl. Acad. Sci. USA 1988, 85, 382–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hahn, S.; Young, E.T. Transcriptional regulation in Saccharomyces cerevisiae: Transcription factor regulation and function, mechanisms of initiation, and roles of activators and coactivators. Genetics 2011, 189, 705–736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobi, K.C.; Winston, F. Analysis of transcriptional activation at a distance in Saccharomyces cerevisiae. Mol. Cell. Biol. 2007, 27, 5575–5586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escalante-Chong, R.; Savir, Y.; Carroll, S.M.; Ingraham, J.B.; Wang, J.; Marx, C.J.; Springer, M. Galactose metabolic genes in yeast respond to a ratio of galactose and glucose. Proc. Natl. Acad. Sci. USA 2015, 112, 1636–1641. [Google Scholar] [CrossRef] [Green Version]
- Mustonen, V.; Kinney, J.; Callan, C.G.; Lässig, M. Energy-dependent fitness: A quantitative model for the evolution of yeast transcription factor binding sites. Proc. Natl. Acad. Sci. USA 2008, 105, 12376–12381. [Google Scholar] [CrossRef] [Green Version]
- Moses, A.M.; Chiang, D.Y.; Kellis, M.; Lander, E.S.; Eisen, M.B. Position specific variation in the rate of evolution in transcription factor binding sites. BMC Evol. Biol. 2003, 3, 19. [Google Scholar] [CrossRef] [Green Version]
- Nagalakshmi, U.; Wang, Z.; Waern, K.; Shou, C.; Raha, D.; Gerstein, M.; Snyder, M. The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 2008, 320, 1344–1349. [Google Scholar] [CrossRef] [Green Version]
- Hu, M.; Yu, J.; Taylor, J.M.; Chinnaiyan, A.M.; Qin, Z.S. On the detection and refinement of transcription factor binding sites using ChIP-Seq data. Nucleic Acids Res. 2010, 38, 2154–2167. [Google Scholar] [CrossRef] [Green Version]
- Shultzaberger, R.K.; Maerkl, S.J.; Kirsch, J.F.; Eisen, M.B. Probing the Informational and regulatory plasticity of a transcription factor DNA–binding domain. PLoS Gent. 2012, 8, e1002614. [Google Scholar] [CrossRef]
- Lee, W.; Tillo, D.; Bray, N.; Morse, R.H.; Davis, R.W.; Hughes, T.R.; Nislow, C. A high-resolution atlas of nucleosome occupancy in yeast. Nat. Gent. 2007, 39, 1235–1244. [Google Scholar] [CrossRef]
- Juan, L.-J.; Walter, P.; Taylor, I.; Kingston, R.; Workman, J. Nucleosome cores and histone H1 in the binding of GAL4 derivatives and the reactivation of transcription from nucleosome templates in vitro. In Cold Spring Harbor Symposia on Quantitative Biology; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1993; Volume 58, pp. 213–223. [Google Scholar]
- Ercan, S.; Simpson, R.T. Global chromatin structure of 45,000 base pairs of chromosome III in a-and α-cell yeast and during mating-type switching. Mol. Cell. Biol. 2004, 24, 10026–10035. [Google Scholar] [CrossRef] [Green Version]
- Ercan, S.; Carrozza, M.J.; Workman, J.L. Global nucleosome distribution and the regulation of transcription in yeast. Genome Biol. 2004, 5, 243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernstein, B.E.; Liu, C.L.; Humphrey, E.L.; Perlstein, E.O.; Schreiber, S.L. Global nucleosome occupancy in yeast. Genome Biol. 2004, 5, R62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segal, E.; Fondufe-Mittendorf, Y.; Chen, L.; Thåström, A.; Field, Y.; Moore, I.K.; Wang, J.-P.Z.; Widom, J. A genomic code for nucleosome positioning. Nature 2006, 442, 772–778. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Carey, M.; Workman, J.L. The role of chromatin during transcription. Cell 2007, 128, 707–719. [Google Scholar] [CrossRef] [Green Version]
- Liang, S.D.; Marmorstein, R.; Harrison, S.C.; Ptashne, M. DNA sequence preferences of GAL4 and PPR1: How a subset of Zn2 Cys6 binuclear cluster proteins recognizes DNA. Mol. Cell. Biol. 1996, 16, 3773–3780. [Google Scholar] [CrossRef] [Green Version]
- Cote, J.; Quinn, J.; Workman, J.L.; Peterson, C.L. Stimulation of GAL4 derivative binding to nucleosomal DNA by the yeast SWI/SNF complex. Science 1994, 265, 53–60. [Google Scholar] [CrossRef]
- Yu, L.; Morse, R.H. Chromatin Opening and Transactivator Potentiation by RAP1 in Saccharomyces cerevisiae. Mol. Cell. Biol. 1999, 19, 5279–5288. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Bryant, G.O.; Floer, M.; Spagna, D.; Ptashne, M. An effect of DNA sequence on nucleosome occupancy and removal. Nat. Struct. Mol. Biol. 2011, 18, 507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Workman, J.L. Nucleosome displacement in transcription. Genes Dev. 2006, 20, 2009–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svaren, J.; Schmitz, J.; Hörz, W. The transactivation domain of Pho4 is required for nucleosome disruption at the PHO5 promoter. EMBO J. 1994, 13, 4856–4862. [Google Scholar] [CrossRef] [PubMed]
- Venter, U.; Svaren, J.; Schmitz, J.; Schmid, A.; Hörz, W. A nucleosome precludes binding of the transcription factor Pho4 in vivo to a critical target site in the PHO5 promoter. EMBO J. 1994, 13, 4848–4855. [Google Scholar] [CrossRef] [PubMed]
- Segal, E.; Widom, J. Poly (dA: dT) tracts: Major determinants of nucleosome organization. Curr. Opin. Struct. Biol. 2009, 19, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Verdone, L.; Camilloni, G.; Di Mauro, E.; Caserta, M. Chromatin remodeling during Saccharomyces cerevisiae ADH2 gene activation. Mol. Cell. Biol. 1996, 16, 1978–1988. [Google Scholar] [CrossRef] [Green Version]
- Filetici, P.; Aranda, C.; Gonzalez, A.; Ballario, P. GCN5, a Yeast Transcriptional Coactivator, Induces Chromatin Reconfiguration ofHIS3Promoter in Vivo. Biochem. Biophys. Res. Commun. 1998, 242, 84–87. [Google Scholar] [CrossRef]
- Lascaris, R.F.; Groot, E.d.; Hoen, P.-B.t.; Mager, W.H.; Planta, R.J. Different roles for abf1p and a T-rich promoter element in nucleosome organization of the yeast RPS28A gene. Nucleic Acids Res. 2000, 28, 1390–1396. [Google Scholar] [CrossRef] [Green Version]
- Tanaka, S.; Livingstone-Zatchej, M.; Thoma, F. Chromatin structure of the yeast URA3 gene at high resolution provides insight into structure and positioning of nucleosomes in the chromosomal context. J. Mol. Biol. 1996, 257, 919–934. [Google Scholar] [CrossRef]
- Iyer, V.; Struhl, K. Poly (dA: dT), a ubiquitous promoter element that stimulates transcription via its intrinsic DNA structure. EMBO J. 1995, 14, 2570–2579. [Google Scholar] [CrossRef]
- Raveh-Sadka, T.; Levo, M.; Shabi, U.; Shany, B.; Keren, L.; Lotan-Pompan, M.; Zeevi, D.; Sharon, E.; Weinberger, A.; Segal, E. Manipulating nucleosome disfavoring sequences allows fine-tune regulation of gene expression in yeast. Nat. Gent. 2012, 44, 743. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.-Q.; Jin, W.-R.; Ma, Z.-C.; Shen, Q.; Cai, X.; Liu, Z.-Q.; Zheng, Y.-G. Promoter engineering strategies for the overproduction of valuable metabolites in microbes. Appl. Mircrobiol. Bitechnol. 2019, 103, 8725–8736. [Google Scholar] [CrossRef]
- Xu, N.; Wei, L.; Liu, J. Recent advances in the applications of promoter engineering for the optimization of metabolite biosynthesis. World J. Mircrobiol. Bitechnol. 2019, 35, 33. [Google Scholar] [CrossRef]
- Blazeck, J.; Alper, H.S. Promoter engineering: Recent advances in controlling transcription at the most fundamental level. Bitechnol. J. 2013, 8, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Bhardwaj, N.; Langlois, R.E.; Zhao, G.; Lu, H. Kernel-based machine learning protocol for predicting DNA-binding proteins. Nucleic Acids Res. 2005, 33, 6486–6493. [Google Scholar] [CrossRef] [PubMed]
- Abeel, T.; Saeys, Y.; Bonnet, E.; Rouzé, P.; Van de Peer, Y. Generic eukaryotic core promoter prediction using structural features of DNA. Genome Res. 2008, 18, 310–323. [Google Scholar] [CrossRef] [Green Version]
- Zakov, S.; Goldberg, Y.; Elhadad, M.; Ziv-Ukelson, M. Rich parameterization improves RNA structure prediction. J. Comput. Biol. 2011, 18, 1525–1542. [Google Scholar] [CrossRef]
- Cuperus, J.T.; Groves, B.; Kuchina, A.; Rosenberg, A.B.; Jojic, N.; Fields, S.; Seelig, G. Deep learning of the regulatory grammar of yeast 5′ untranslated regions from 500,000 random sequences. Genome Res. 2017, 27, 2015–2024. [Google Scholar] [CrossRef] [Green Version]
- De Mey, M.; Maertens, J.; Lequeux, G.J.; Soetaert, W.K.; Vandamme, E.J. Construction and model-based analysis of a promoter library for E. coli: An indispensable tool for metabolic engineering. BMC Bitechnol. 2007, 7, 34. [Google Scholar] [CrossRef] [Green Version]
- Meng, H.; Wang, J.; Xiong, Z.; Xu, F.; Zhao, G.; Wang, Y. Quantitative design of regulatory elements based on high-precision strength prediction using artificial neural network. PLoS ONE 2013, 8, e60288. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, H.; Wei, L.; Li, S.; Liu, L.; Wang, X. Synthetic promoter design in Escherichia coli based on a deep generative network. Nucleic Acids Res 2020, 48, 6403–6412. [Google Scholar] [CrossRef] [PubMed]
- Kotopka, B.J.; Smolke, C.D. Model-driven generation of artificial yeast promoters. Nat. Commun. 2020, 11, 2113. [Google Scholar] [CrossRef]
- Chen, X.; Gao, C.; Guo, L.; Hu, G.; Luo, Q.; Liu, J.; Nielsen, J.; Chen, J.; Liu, L. DCEO Biotechnology: Tools To Design, Construct, Evaluate, and Optimize the Metabolic Pathway for Biosynthesis of Chemicals. Chem. Rev. 2018, 118, 4–72. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Li, G.; Dong, J.; Xing, X.-h.; Dai, J.; Zhang, C. MiYA, an efficient machine-learning workflow in conjunction with the YeastFab assembly strategy for combinatorial optimization of heterologous metabolic pathways in Saccharomyces cerevisiae. Metab. Eng. 2018, 47, 294–302. [Google Scholar] [CrossRef] [PubMed]
- Nevoigt, E.; Kohnke, J.; Fischer, C.R.; Alper, H.; Stahl, U.; Stephanopoulos, G. Engineering of promoter replacement cassettes for fine-tuning of gene expression in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 2006, 72, 5266–5273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, Y.; Zhao, H. Directed evolution of a highly efficient cellobiose utilizing pathway in an industrial Saccharomyces cerevisiae strain. Bitechnol. Bioeng. 2013, 110, 2874–2881. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhai, H.; Rexida, R.; Shen, Y.; Hou, J.; Bao, X. Developing synthetic hybrid promoters to increase constitutive or diauxic shift-induced expression in Saccharomyces cerevisiae. FEMS Yeast Res. 2018, 18, foy098. [Google Scholar] [CrossRef]
- Peng, B.; Wood, R.J.; Nielsen, L.K.; Vickers, C.E. An Expanded Heterologous GAL Promoter Collection for Diauxie-Inducible Expression in Saccharomyces cerevisiae. ACS Synth. Biol. 2018, 7, 748–751. [Google Scholar] [CrossRef]
- Shabbir Hussain, M.; Gambill, L.; Smith, S.; Blenner, M.A. Engineering Promoter Architecture in Oleaginous Yeast Yarrowia lipolytica. ACS Synth. Biol. 2016, 5, 213–223. [Google Scholar] [CrossRef]
- Decoene, T.; De Maeseneire, S.L.; De Mey, M. Modulating transcription through development of semi-synthetic yeast core promoters. PLoS ONE 2019, 14, e0224476. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Liu, L.; Li, X.; Liu, D.; Yuan, Y. Engineering yeast artificial core promoter with designated base motifs. Microb. Cell Factories 2020, 19, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, L.; Nawab, S.; Xia, M.; Ma, X.; Huo, Y.-X. Context-dependency of synthetic minimal promoters in driving gene expression: A case study. Microb. Bitechnol. 2019, 12, 1476–1486. [Google Scholar] [CrossRef]
- Redden, H.; Alper, H.S. The development and characterization of synthetic minimal yeast promoters. Nat. Commun. 2015, 6, 7810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katzke, N.; Knapp, A.; Loeschcke, A.; Drepper, T.; Jaeger, K.-E. Novel Tools for the Functional Expression of Metagenomic DNA. In Metagenomics: Methods and Protocols, Streit, W.R., Daniel, R., Eds.; Springer New York: Midtown Manhattan, NY, USA, 2017; pp. 159–196. [Google Scholar] [CrossRef]
- Yang, S.; Liu, Q.; Zhang, Y.; Du, G.; Chen, J.; Kang, Z. Construction and Characterization of Broad-Spectrum Promoters for Synthetic Biology. ACS Synth. Biol. 2018, 7, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Portela, R.M.C.; Vogl, T.; Kniely, C.; Fischer, J.E.; Oliveira, R.; Glieder, A. Synthetic Core Promoters as Universal Parts for Fine-Tuning Expression in Different Yeast Species. ACS Synth. Biol. 2017, 6, 471–484. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Sonnenschein, N.; Pihl, T.P.B.; Pedersen, K.R.; Jensen, M.K.; Keasling, J.D. Engineering an NADPH/NADP+ Redox Biosensor in Yeast. ACS Synth. Biol. 2016, 5, 1546–1556. [Google Scholar] [CrossRef] [PubMed]
- Rajkumar, A.S.; Liu, G.; Bergenholm, D.; Arsovska, D.; Kristensen, M.; Nielsen, J.; Jensen, M.K.; Keasling, J.D. Engineering of synthetic, stress-responsive yeast promoters. Nucleic Acids Res. 2016, 44, e136. [Google Scholar] [CrossRef]
- Gopinarayanan, V.E.; Nair, N.U. A semi-synthetic regulon enables rapid growth of yeast on xylose. Nat. Commun. 2018, 9, 1233. [Google Scholar] [CrossRef] [Green Version]
- Garí, E.; Piedrafita, L.; Aldea, M.; Herrero, E. A Set of Vectors with a Tetracycline-Regulatable Promoter System for Modulated Gene Expression in Saccharomyces cerevisiae. Yeast 1997, 13, 837–848. [Google Scholar] [CrossRef]
- Murphy, K.F.; Balázsi, G.; Collins, J.J. Combinatorial promoter design for engineering noisy gene expression. Proc. Natl. Acad. Sci. USA 2007, 104, 12726–12731. [Google Scholar] [CrossRef] [Green Version]
- Ikushima, S.; Zhao, Y.; Boeke, J.D. Development of a tightly controlled off switch for Saccharomyces cerevisiae regulated by camphor, a low-cost natural product. G3 Genes Genomes Gent. 2015, 5, 1983–1990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teo, W.S.; Hee, K.S.; Chang, M.W. Bacterial F ad R and synthetic promoters function as modular fatty acid sensor-regulators in S accharomyces cerevisiae. Eng. Life Sci. 2013, 13, 456–463. [Google Scholar] [CrossRef]
- Li, S.; Si, T.; Wang, M.; Zhao, H. Development of a synthetic malonyl-CoA sensor in Saccharomyces cerevisiae for intracellular metabolite monitoring and genetic screening. ACS Synth. Biol. 2015, 4, 1308–1315. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, M.; McMillen, D.R. Design and characterization of a dual-mode promoter with activation and repression capability for tuning gene expression in yeast. Nucleic Acids Res. 2014, 42, 9514–9522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Williams, T.C.; Nielsen, L.K.; Vickers, C.E. Engineered Quorum Sensing Using Pheromone-Mediated Cell-to-Cell Communication in Saccharomyces cerevisiae. ACS Synth. Biol. 2013, 2, 136–149. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UAS Sequence (5′-3′) | Transcription Factor | Promoters | Inducer | Function | Reference |
|---|---|---|---|---|---|
| CGGRNNRCYNYNCNCCG | Gal4p | GAL1/2/7/10, MEL1 | Galactose | Regulation of galactose metabolism | [38] |
| ATGACTCTT | Gcn4p | ARG1, ARG4, HIS4, CPA2 | Amino acid starvation | Regulation of amino acid biosynthetic genes | [45] |
| TTACTAA | Yap1p/2p | GSH1, TRX2, YCF1, GLR1 | Oxidative stress such as H2O2 | Regulation of genes expressed in response to environmental changes | [46] |
| TG(A/C)GCCNC | Crz1p | PMC1, PMR1, FKS2 | Calcium | Calcineurin-responsive transcription factor | [47,48] |
| CGGNBNVMHGGA | Cat8p | FBP1, PCK1, ACR1, IDP2, JEN1 | Non-fermentative growth conditions | Derepression of gene expression under non-fermentative growth conditions | [43] |
| PyPuCACCCPu | Aft1p | FRE1, FTR1, FTH1 | Iron deprivation | Iron utilization and homeostasis | [49] |
| TGAAAC | Ste12p | TEC1, FLO11 | Pheromone | Involved in mating and invasive growth | [50] |
| GAATGT | Tec1p | TEC1, FLO11 | n/A | Ste12p cofactor | [50,51] |
| CAGCGTG | Hac1p | KAR2, PDI1, EUG1, FKB2 | Unfolded/misfolded proteins | Regulates the unfolded protein response | [52] |
| NGAAN | Hsf1p | HSP82, HSP26, HSP104, HSP26, | Heat shock | Trimeric heat shock transcription factor | [53] |
| SYGGGG | Mig1p | GAL1, HXT2, SUC2, JEN1 | Glucose | Involved in glucose repression | [54] |
| TGACGTCA | Aca1p | GRE2, COS8 | n/A | Basic leucine zipper (bZIP) transcription factor involved in carbon source utilization | [55] |
| CGGN3TNAN9-12CCG | Oaf1p | CTA1, FOX1/2/3, FAA2, PAS8, PAS10 | Oleate | Involved in fatty acid beta-oxidation | [56] |
| TCCGCGGA | Pdr1/3p | SNQ2, PDR5 | Pleiotropic drug | Pleiotropic drug response | [57,58,59] |
| GGTGGCAAA | Rpn4p | RPT2/3/6 | Patulin | Regulation of proteasome genes | [60,61] |
| DNCRCAAAW | Ndt80p | SMK1, SPR3 | Sporulation | Required for full meiotic recombination and middle sporulation | [62] |
| CCAAT | Hap4p | CYC1 | Heme | Global regulator of respiratory gene expression | [63] |
| TGACGTCA | Sko1p | SUC2, MSN2, ROX1, PTP3 | Osmotic stress | Involved in osmotic and oxidative stress responses | [40,64] |
| GcCTCGA(G/A)G(C/A)g(a/g) | Xbp1p | CLN1, CYS3, SMF2 | Stress or starvation | Transcriptional repressor | [65] |
| CAC(A/G)T(T/G) | Pho4p | HIS4, PHO5 | Phosphate limitation | Regulation of the purine and histidine biosynthesis pathways | [66] |
| ACCYYNAAGGT | Zap1p | ZRT1, ZRT2 | Zinc | Zinc-regulated transcription factor | [67] |
| ACTACTA(T/A)4TAG | Smp1p | STL1, CWP1 | Osmotic stress | Osmotic stress response | [68] |
| CTA(T/A)4TAG | Rlm1p | HKR1, KTR2, HSP150, FLO1 | n/A | Maintenance of cell integrity | [69] |
| TTGGRG | Adr1p | ADH2, ALD4, ALD6, POX1 | n/A | Carbon source responsive transcription factor | [70] |
| AATCA-N8-TGAYT | Vhr1p | VHT1, BIO5 | Biotin | Response to low biotin concentrations | [71] |
| AAACTGTGG | Met31p | MET25, MET14, MET3 | n/A | Sulfur amino acid metabolism | [72] |
| CCCCT | Msn2/4p | CTT1, DDR2, HSP12 | Various stress | Response to multiple stress conditions | [73] |
| CCRTYCRTCCG | Sip4p | FBP1, PKC1, ICL1 | n/A | Positive regulation of gluconeogenesis | [74] |
| CGGANNA | Rgt1p | HXT2, HXT4 | Glucose | Glucose-responsive transcription factor | [75,76] |
| CTTCC | Gcr1p | ENO1, TPI1, TDH3 | n/A | Transcriptional activator involved in the regulation of glycolysis | [77] |
| RRRTAACAAGAG | Rox1p | HEM13, COX5B, ANB1, CYC7 | Heme | Heme-dependent repressor of hypoxic genes | [78] |
| Application | Note (Elements or Parts) | Approach | Expression Range (fold) | Product/Inducers | Reference |
|---|---|---|---|---|---|
| Expanding dynamic ranges | PTEF1 | Random mutation | 0–2.0 | n/A | [15] |
| PTEF1 | Random mutation | 0.08–1.2 | Increase glycerol 3-phosphate dehydrogenase activity | [127] | |
| PENO2; PPDC1 | Random mutation | 24.4; 3.0 | Obtain a higher cellobiose consumption rate (6.41-fold) and ethanol productivity (6.36-fold) | [128] | |
| UASCLB(3X)-PTDH3; UASGAL1-PLEUM/PCYC/PGAL1 | Hybrid | 2.5; 50-fold dynamic range | n/A | [16] | |
| UASENO2(3X)-PTEF1; UASHXK2-PTEF1/UASHSP30-PTEF1 | Hybrid | 2.0; 8-fold induction range | UASHXK2-PTEF1 and UASHSP30-PTEF1 are post-diauxic phase-induced promoters | [129] | |
| PHIS3 | Manipulating poly(dA:dT) tracts | 3-fold dynamic range | n/A | [113] | |
| PCYC1 | Tuning of nucleosome architecture | 6.0 | n/A | [17] | |
| PTDH3 | Machine learning | 1.37 | n/A | [124] | |
| Reducing homologous recombination | Galactose-inducible promoters | Heterologous expression | 2.5-fold to 99-fold induction ratio | Producing 11.5 mg/L lycopene | [130] |
| Psynth promoters | De novo | 20-fold dynamic range | n/A | [17] | |
| Minimal promoters | UASA/UASC/UASFEC, PTEF1 | Truncation and hybrid | 20-fold dynamic range | n/A | [132] |
| UASEXP1/UASGPD, PN30 | Saturation mutagenesis and hybrid | n/A | 5.5-fold enhancement of lycopene–carotene transformation; producing β-carotene 7.4 mg/g DCW | [133] | |
| UASN10; PN30 | De novo by saturation mutagenesis | 0.7 | achieve 70% of the strength of the strongest TDH3 promoter | [135] | |
| Multi-host suitable | Pmin | Random mutation and hybrid | n/A | Pbs was much stronger than E. coli PJ23119; 75% of that of Pcdd in B. subtilis; lower than that of the strong promoter PTDH3 | [137] |
| CRM; PAOX1 | Computational design and hybrid | 200-fold dynamic range | 0.3% to 70.6% of the wild type PAOX1 level | [138] | |
| Biosensor | PTRX2 | Hybrid | 100-fold dynamic range | NADPH/NADP+ ratio | [139] |
| PYGP1; PCCW14 | Hybrid | 6.0; 16.0 | Enabling a 10-fold increased production of lactic acid; low pH | [140] | |
| PCYC1 | Hybrid | 1000-fold induction ratio | Tetracycline | [142] | |
| PCYC1 | Hybrid | n/A | Camphor | [141] | |
| PGAL1 | Hybrid | n/A | Fatty acid/fatty acyl-CoA | [145] | |
| PGPM1 | Hybrid | 1-fold to 4.17-fold induction ratio | Enhancing 3-hydroxypropionic acid titer by 120%; Malonyl-CoA biosensor | [146] | |
| PCYC1 | Hybrid | 8-fold induction ratio | IPTG and testosterone dual induction | [147] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tang, H.; Wu, Y.; Deng, J.; Chen, N.; Zheng, Z.; Wei, Y.; Luo, X.; Keasling, J.D. Promoter Architecture and Promoter Engineering in Saccharomyces cerevisiae. Metabolites 2020, 10, 320. https://doi.org/10.3390/metabo10080320
Tang H, Wu Y, Deng J, Chen N, Zheng Z, Wei Y, Luo X, Keasling JD. Promoter Architecture and Promoter Engineering in Saccharomyces cerevisiae. Metabolites. 2020; 10(8):320. https://doi.org/10.3390/metabo10080320
Chicago/Turabian StyleTang, Hongting, Yanling Wu, Jiliang Deng, Nanzhu Chen, Zhaohui Zheng, Yongjun Wei, Xiaozhou Luo, and Jay D. Keasling. 2020. "Promoter Architecture and Promoter Engineering in Saccharomyces cerevisiae" Metabolites 10, no. 8: 320. https://doi.org/10.3390/metabo10080320
APA StyleTang, H., Wu, Y., Deng, J., Chen, N., Zheng, Z., Wei, Y., Luo, X., & Keasling, J. D. (2020). Promoter Architecture and Promoter Engineering in Saccharomyces cerevisiae. Metabolites, 10(8), 320. https://doi.org/10.3390/metabo10080320