Development of Tandem Mass Tag Labeling Method for Lipid Molecules Containing Carboxy and Phosphate Groups, and Their Stability in Human Serum

 ,
, 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Lipid Extraction
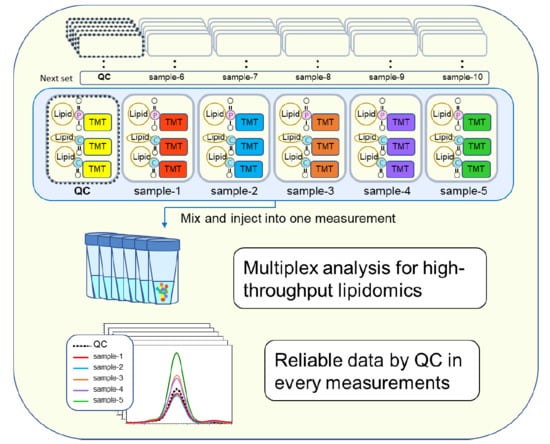
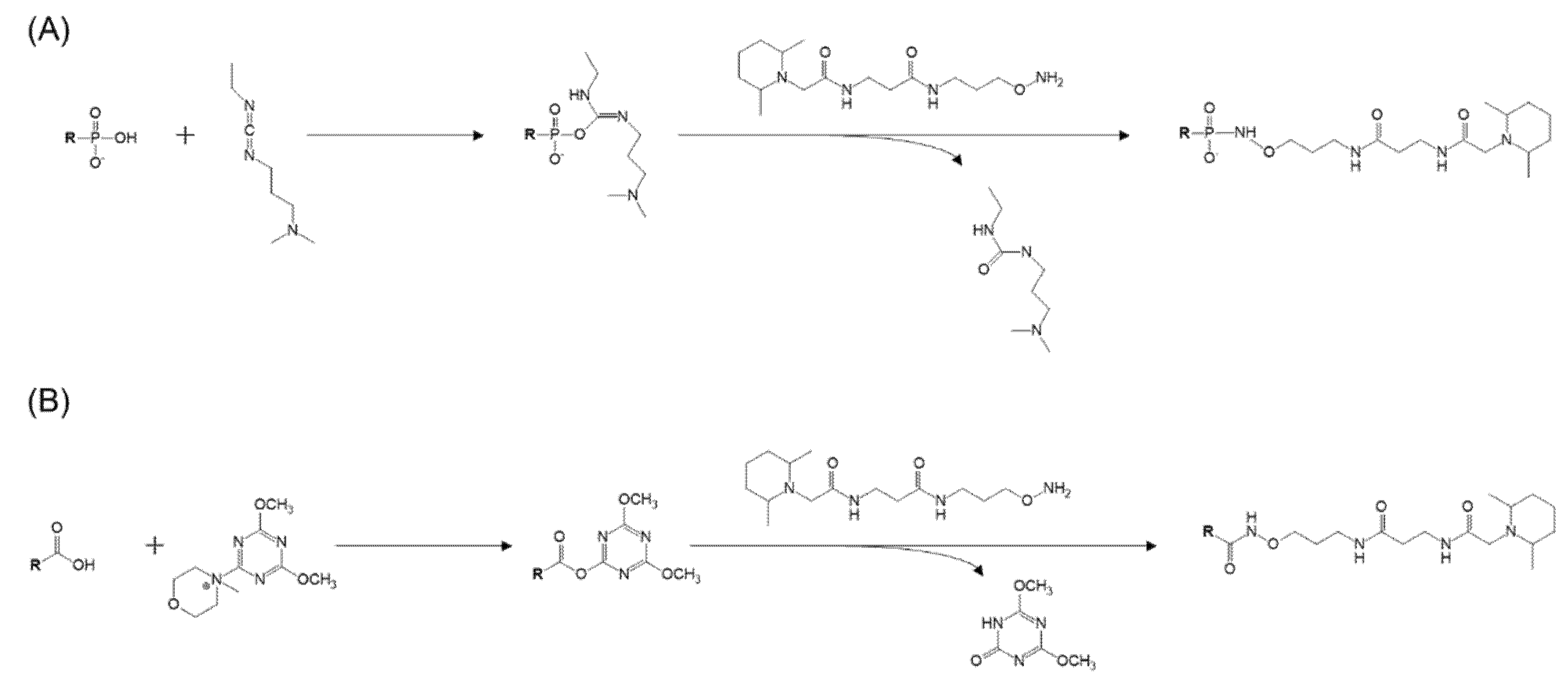
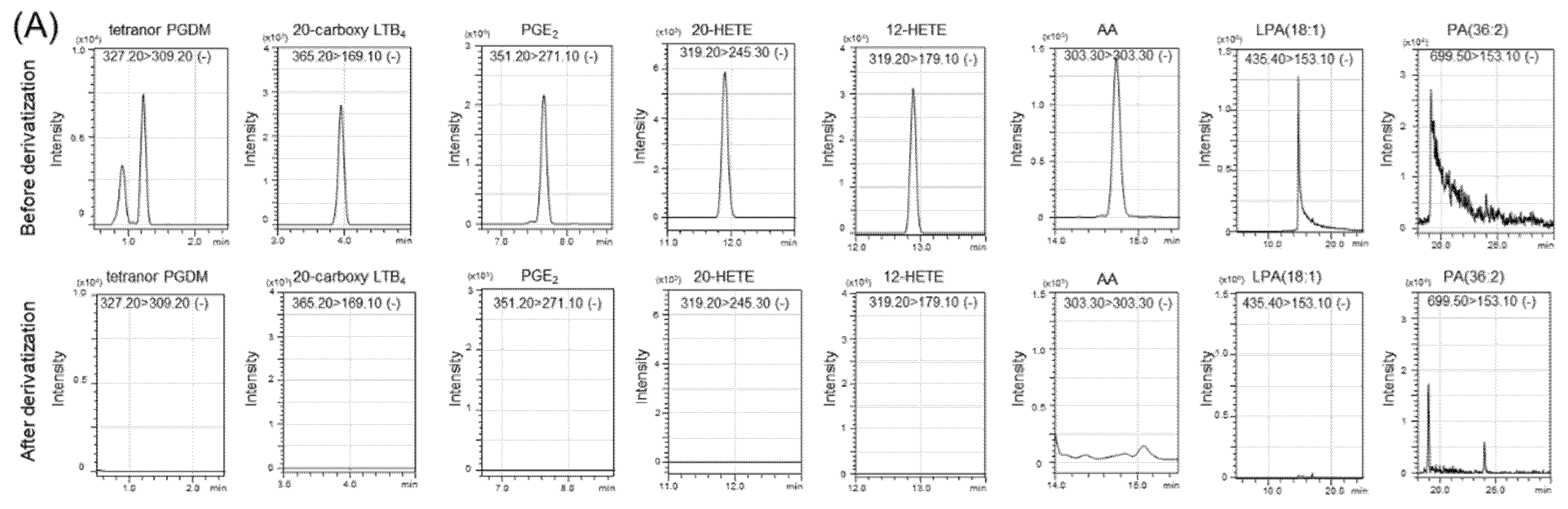
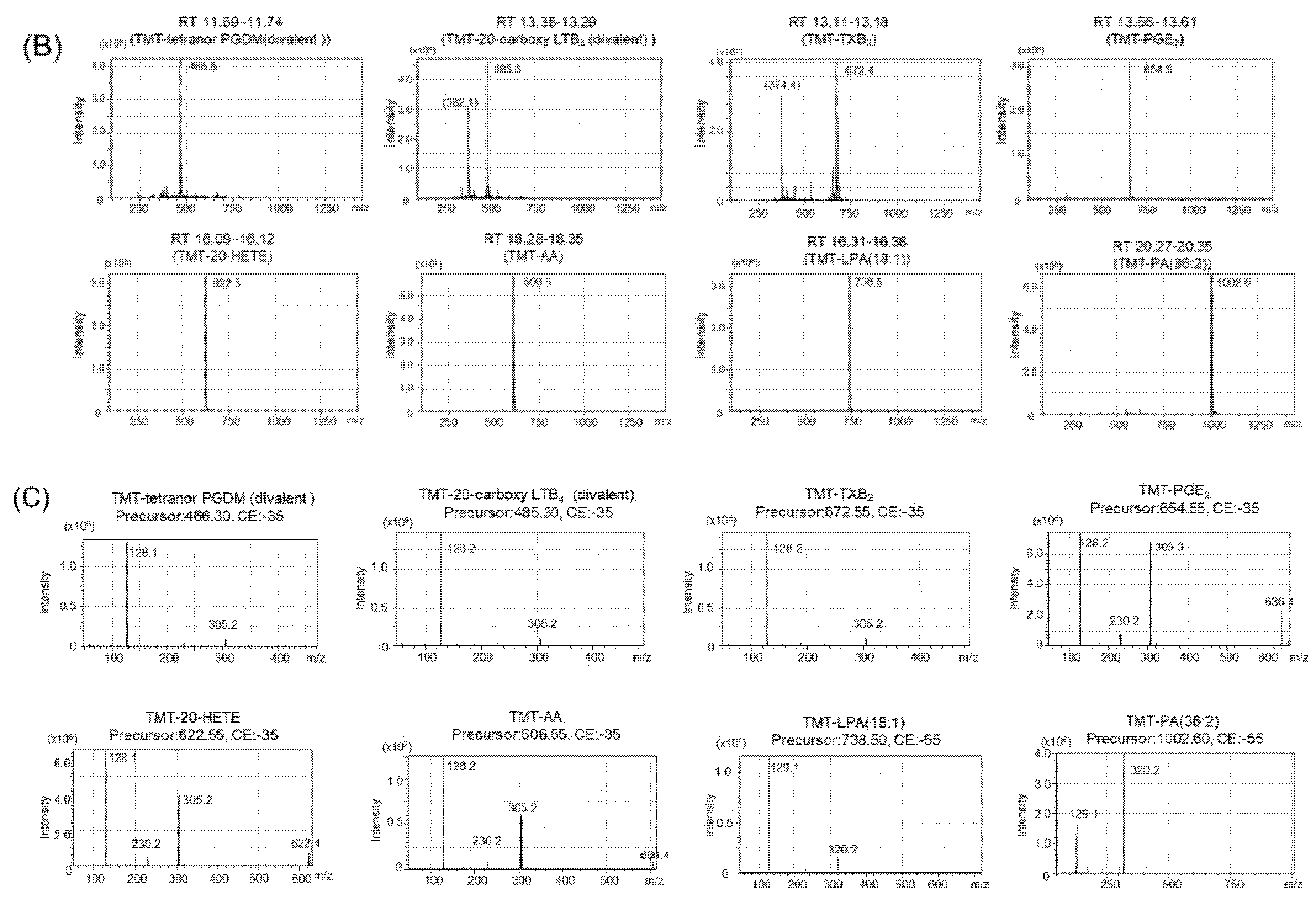
2.2. Tandem Mass Tag (TMT) Derivatization
2.3. Reproducibility
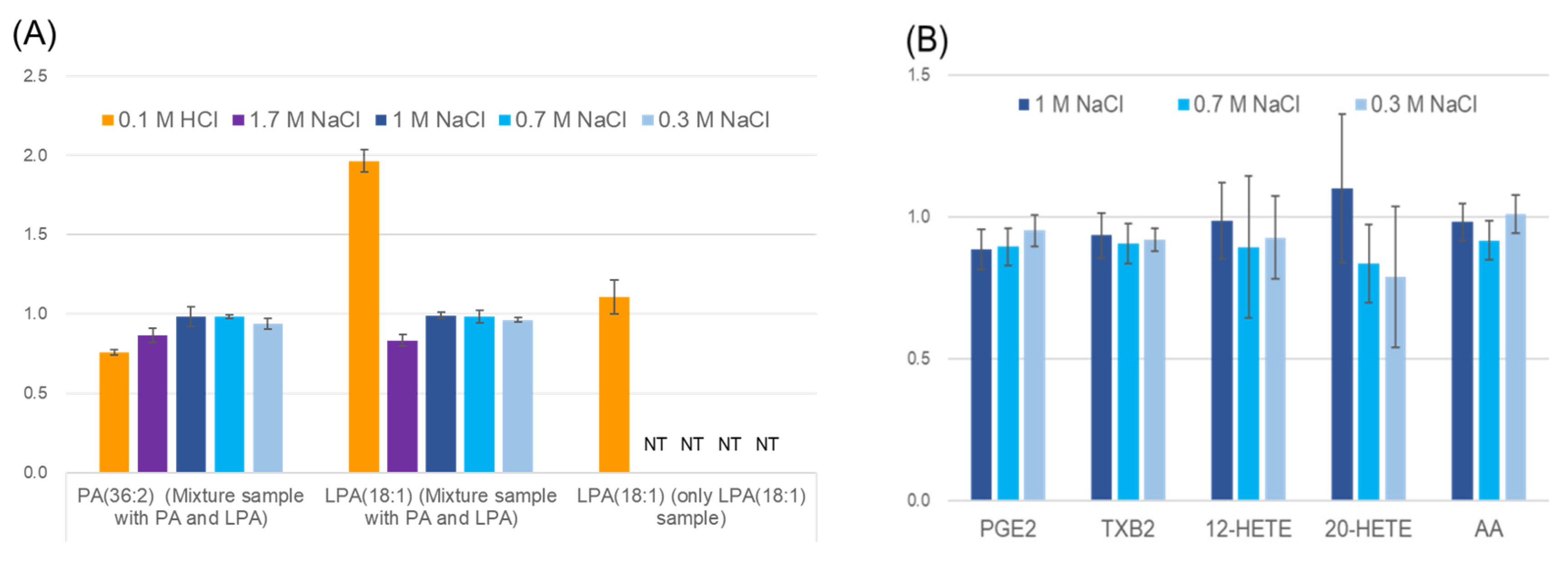
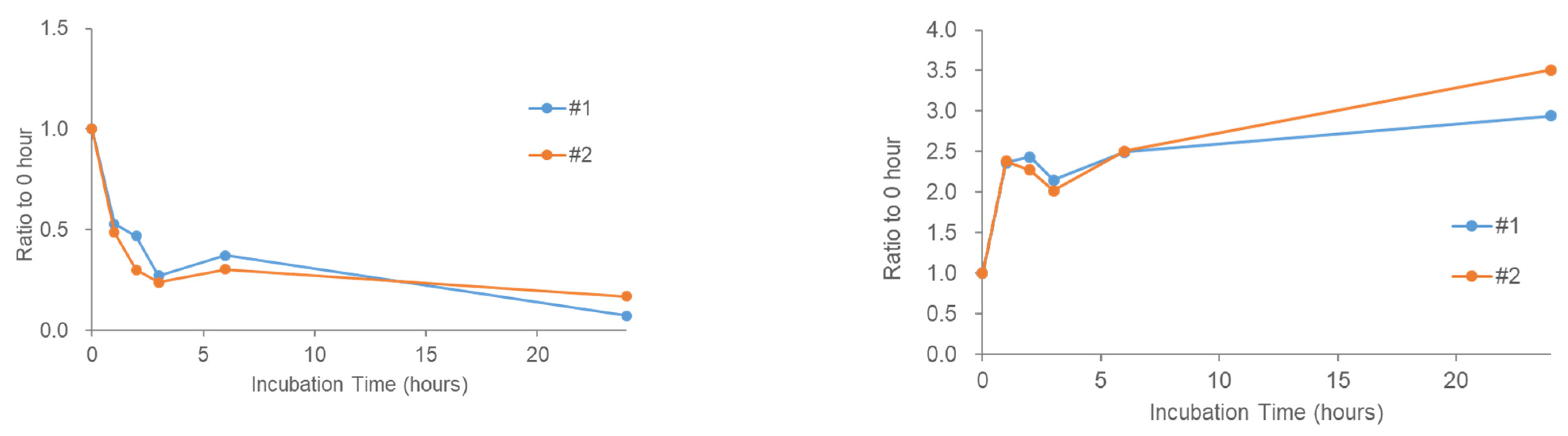
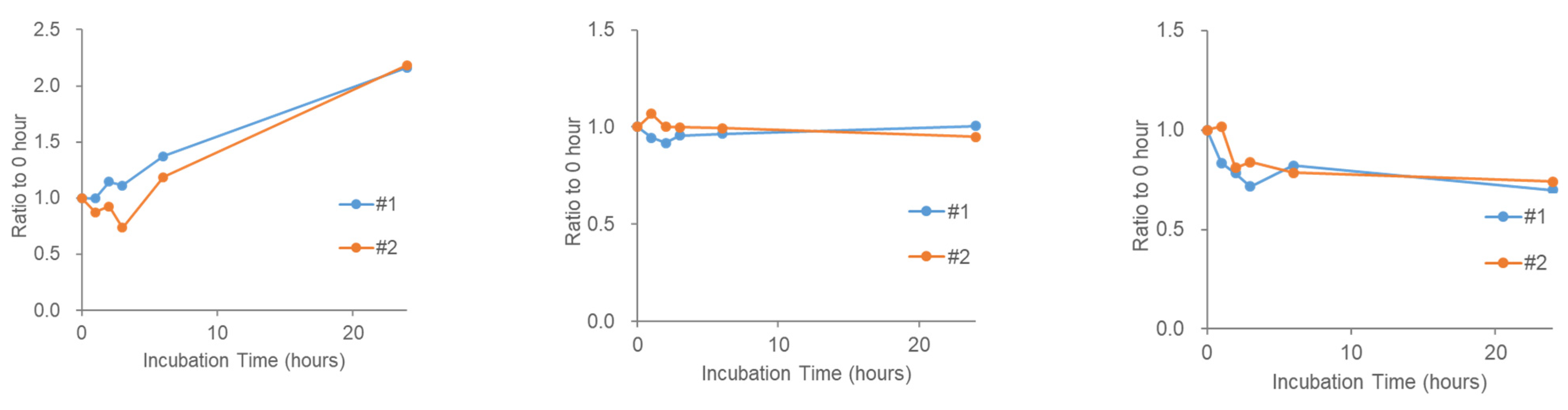
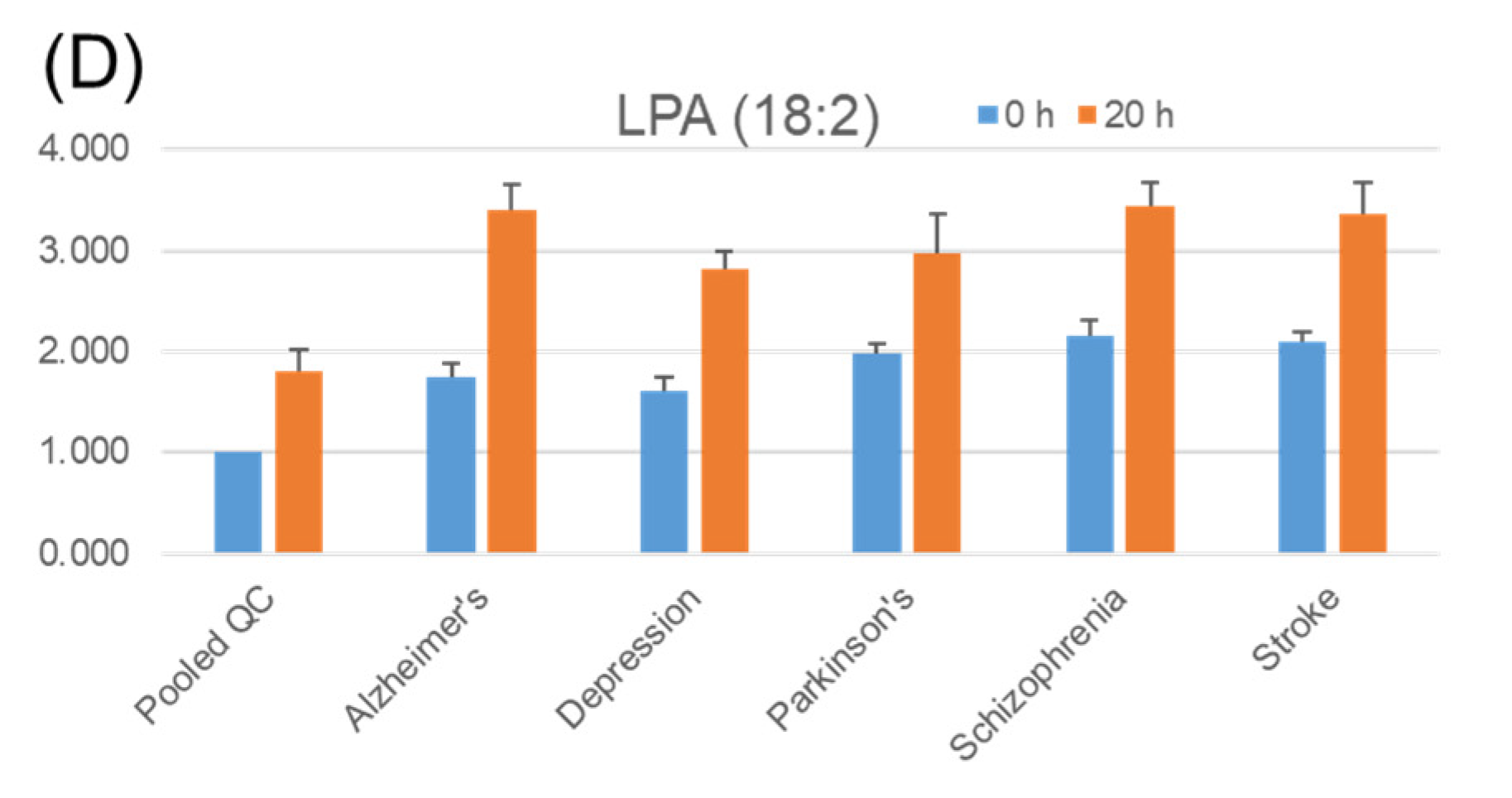
2.4. Stability of Phosphatidic Acid (PA) in Serum
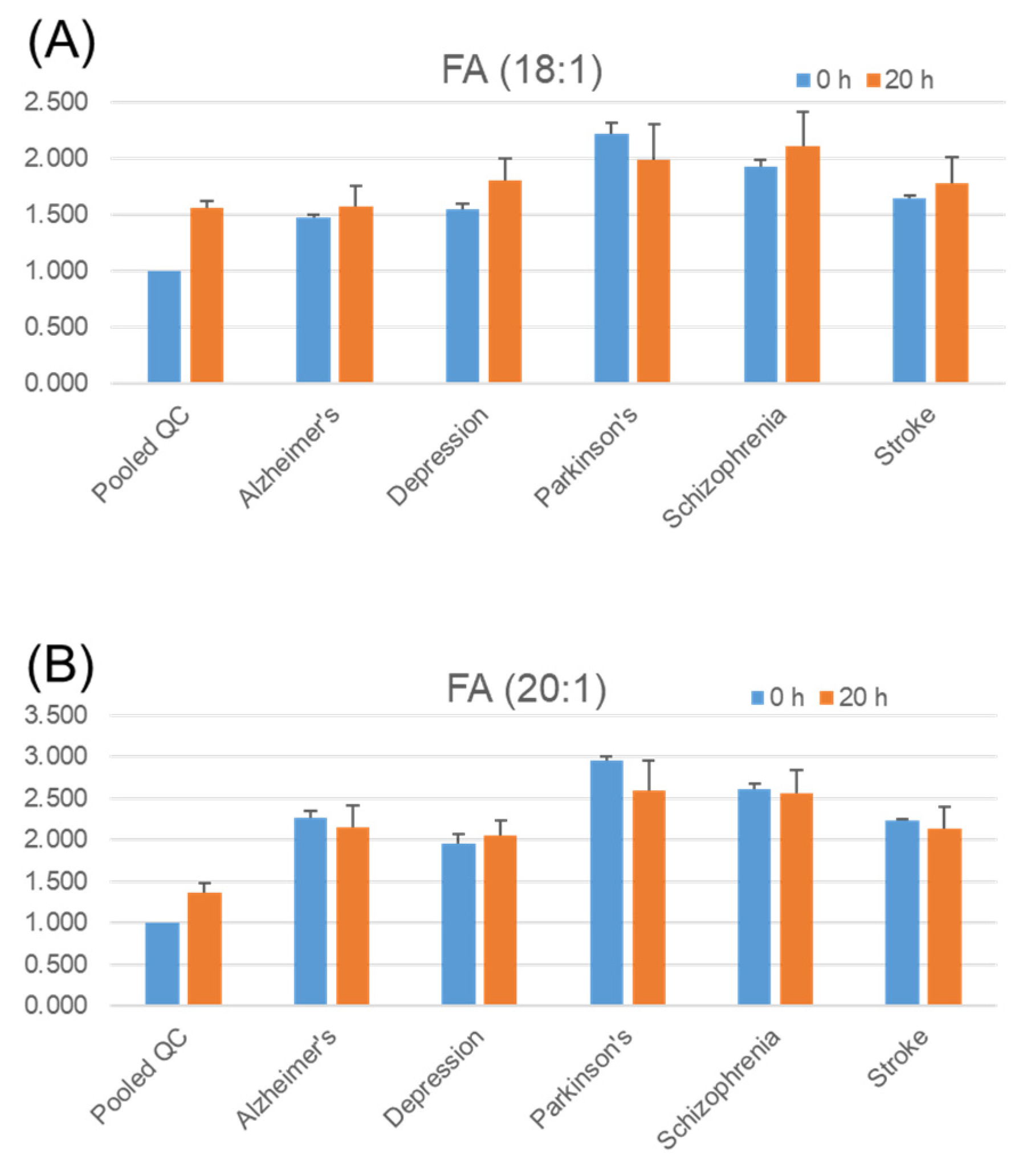
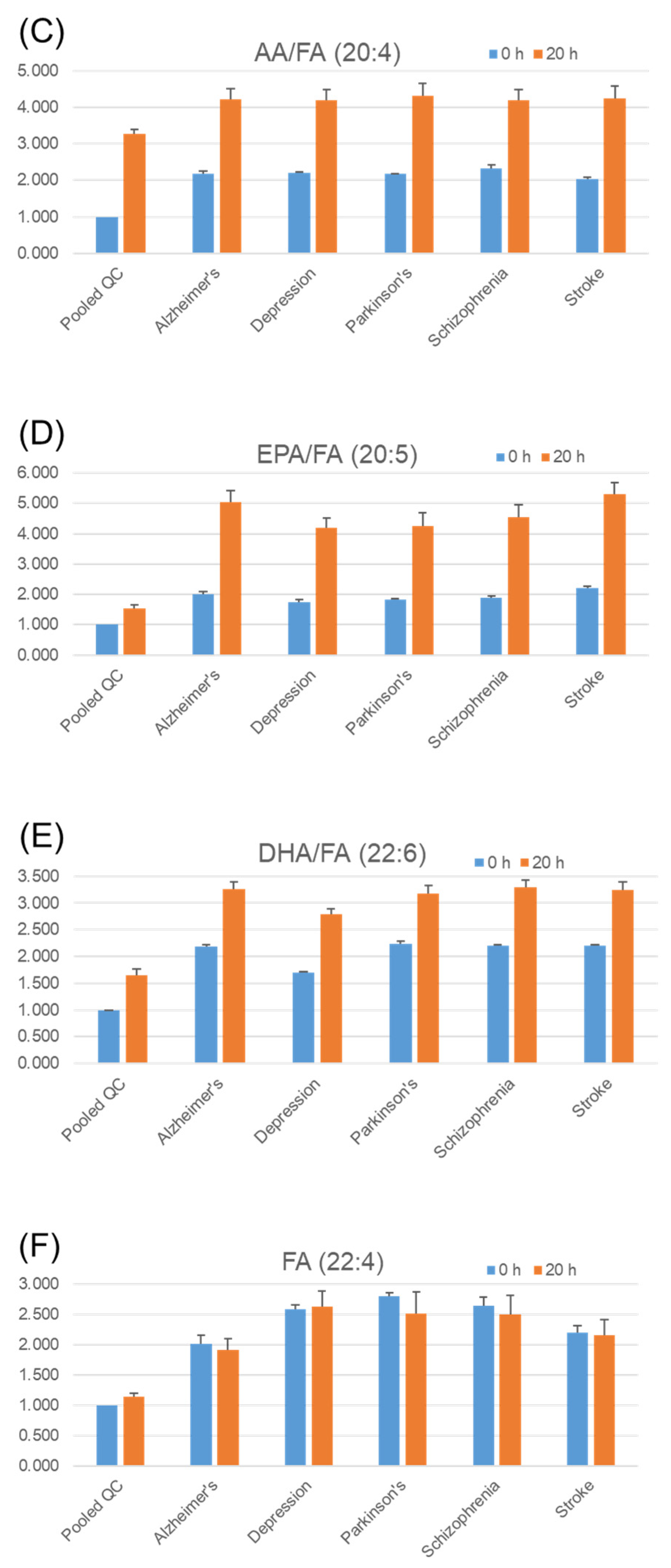
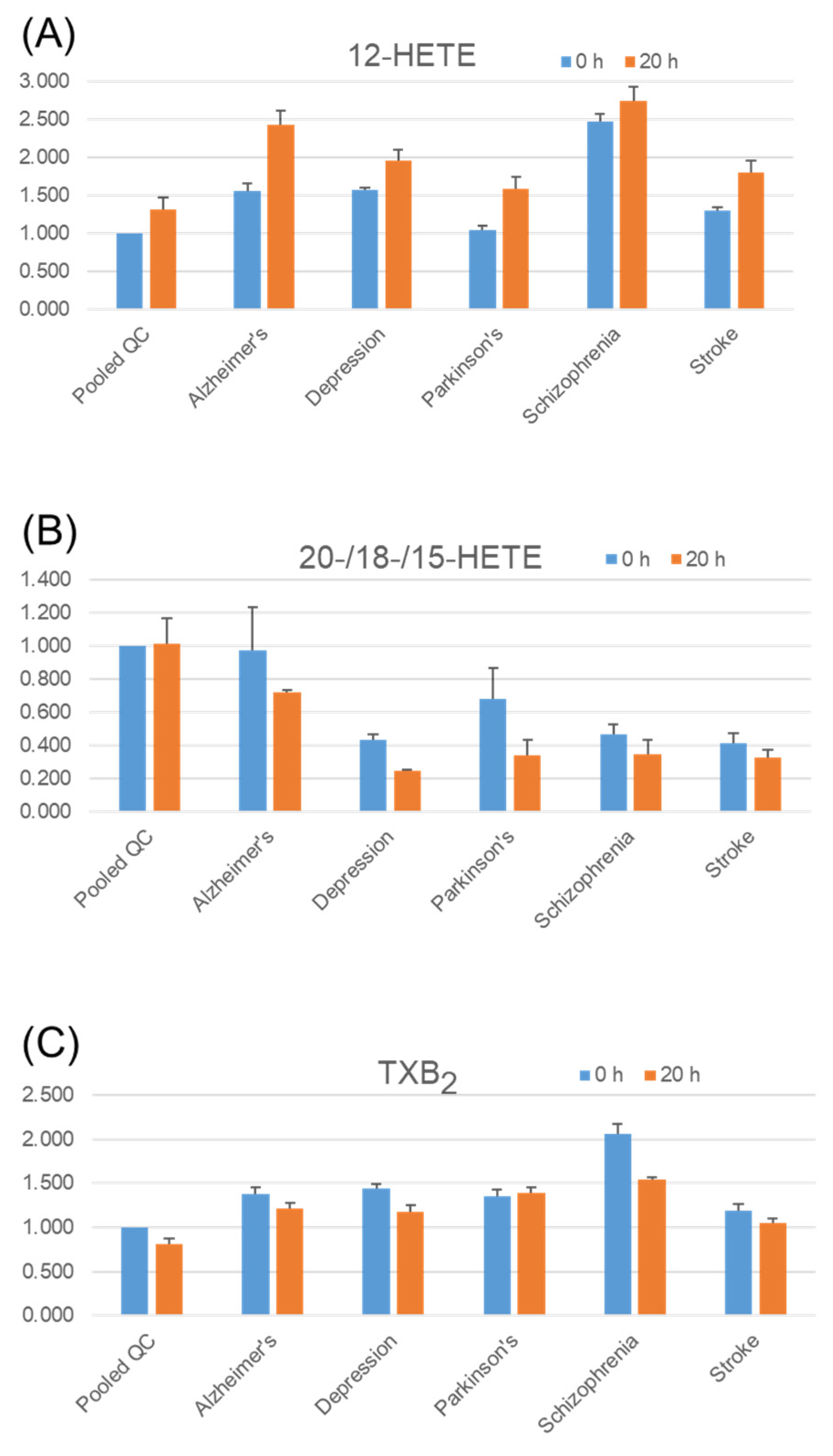
2.5. Stability of Fatty Acids and Their Metabolites in Serum
3. Materials and Methods
3.1. Reagents
3.2. Samples
3.3. Lipid Extraction
3.4. TMT Reaction
3.5. Liquid Chromatography/Mass Spectrometry (LC/MS) Measurements
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, D.; Dubois, R.N. Eicosanoids and cancer. Nat. Rev. Cancer 2010, 10, 181–193. [Google Scholar] [CrossRef] [PubMed]
- Korotkova, M.; Jakobsson, P.J. Persisting eicosanoid pathways in rheumatic diseases. Nat. Rev. Rheumatol. 2014, 10, 229–241. [Google Scholar] [CrossRef] [PubMed]
- Dennis, E.A.; Norris, P.C. Eicosanoid storm in infection and inflammation. Nat. Rev. Immunol. 2015, 15, 511–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liao, Y.; Xie, B.; Zhang, H.; He, Q.; Guo, L.; Subramaniapillai, M.; Fan, B.; Lu, C.; Mclntyer, R.S. Efficacy of omega-3 PUFAs in depression: A meta-analysis. Transl. Psychiatry 2019, 9, 190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tocher, D.R.; Betancor, M.B.; Sprague, M.; Olsen, R.E.; Napier, J.A. Omega-3 long-chain polyunsaturated fatty acids, EPA and DHA: Bridging the gap between supply and demand. Nutrients 2019, 11, 89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castejón, N.; Señoráns, F.J. Enzymatic modification to produce health-promoting lipids from fish oil, algae and other new omega-3 sources: A review. New Biotechnol. 2020, 57, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Holland, P.; Knævelsrud, H.; Søreng, K.; Mathai, B.J.; Lystad, A.H.; Pankiv, S.; Bjørndal, G.T.; Schultz, S.W.; Lobert, V.H.; Chan, R.B.; et al. HS1BP3 negatively regulates autophagy by modulation of phosphatidic acid levels. Nat. Commun. 2016, 7, 13889. [Google Scholar] [CrossRef]
- Park, S.Y.; Yang, J.S.; Li, Z.; Deng, P.; Zhu, X.; Young, D.; Ericsson, M.; Andringa, R.L.H.; Minnaard, A.J.; Zhu, C.; et al. The late stage of COPI vesicle fission requires shorter forms of phosphatidic acid and diacylglycerol. Nat. Commun. 2019, 10, 1–14. [Google Scholar] [CrossRef]
- Foster, D.A. Phosphatidic acid and lipid-sensing by mTOR. Trends Endocrinol. Metab. 2013, 24, 272–278. [Google Scholar] [CrossRef] [Green Version]
- Khayyo, V.I.; Hoffmann, R.M.; Wang, H.; Bell, J.A.; Burke, J.E.; Reue, K.; Airola, M.V.V. Crystal structure of a lipin/Pah phosphatidic acid phosphatase. Nat. Commun. 2020, 11, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Remane, D.; Wissenbach, D.K.; Meyer, M.R.; Maurer, H.H. Systematic investigation of ion suppression and enhancement effects of fourteen stable-isotope-labeled internal standards by their native analogues using atmospheric-pressure chemical ionization and electrospray ionization and the relevance for multi-anal. Rapid Commun. Mass Spectrom. 2010, 24, 859–867. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Huang, K.; Cross, F.R.; Cowburn, D.; Chait, B.T. Accurate quantitation of protein expression and site-specific phosphorylation. Proc. Natl. Acad. Sci. USA 1999, 96, 6591–6596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gygi, S.P.; Rist, B.; Gerber, S.A.; Turecek, F.; Gelb, M.H.; Aebersold, R.A. Access quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat. Biotechnol. 1999, 17, 994–999. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, A.; Kellermann, J.; Lottspeich, F. A novel strategy for quantitative proteomics using isotope-coded protein labels. Proteomics 2005, 5, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Reddy, N.R. Stable labeled isotopes as internal standards: A critical review. Mod. Appl. Pharm. Pharmacol. 2017, 1, 1–4. [Google Scholar] [CrossRef]
- Tokuoka, S.M.; Yasumoto, A.; Kita, Y.; Shimizu, T.; Yatomi, Y.; Oda, Y. Limitations of deuterium-labeled internal standards for quantitative electrospray ionization mass spectrometry analysis of fatty acid metabolites. Rapid Commun. Mass Spectrom. 2020, 34. [Google Scholar] [CrossRef] [PubMed]
- Ross, P.L.; Huang, Y.N.; Marchese, J.N.; Williamson, B.; Parker, K.; Hattan, S.; Khainovski, N.; Pillai, S.; Dey, S.; Daniels, S.; et al. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteom. 2004, 3, 1154–1169. [Google Scholar] [CrossRef] [Green Version]
- Thompson, A.; Schäfer, J.; Kuhn, K.; Kienle, S.; Schwarz, J.; Schmidt, G.; Neumann, T.; Hamon, C. Tandem mass tags: A novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003, 75, 1895–1904. [Google Scholar] [CrossRef]
- Zemski Berry, K.A.; Murphy, R.C. Analysis of cell membrane aminophospholipids as isotope-tagged derivatives. J. Lipid Res. 2005, 46, 1038–1046. [Google Scholar] [CrossRef] [Green Version]
- Murphy, J.P.; Everley, R.A.; Coloff, J.L.; Gygi, S.P. Combining amine metabolomics and quantitative proteomics of cancer cells using derivatization with isobaric tags. Anal. Chem. 2014, 86, 3585–3593. [Google Scholar] [CrossRef]
- Sun, F.; Choi, A.A.; Wu, R. Systematic analysis of fatty acids in human cells with a multiplexed isobaric tag (TMT)-based method. J. Proteome Res. 2018, 17, 1606–1614. [Google Scholar] [CrossRef] [PubMed]
- Tokuoka, S.M.; Kita, Y.; Shimizu, T.; Oda, Y. Isobaric mass tagging and triple quadrupole mass spectrometry to determine lipid biomarker candidates for Alzheimer’s disease. PLoS ONE 2019, 14, e0226073. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.L.; Desiderio, D.M.; Miller, D.D.; Tolley, B.; Tigyi, G.J. Direct quantitative analysis of lysophosphatidic acid molecular species by stable isotope dilution electrospray lonization liquid chromatography-mass spectrometry. Anal. Biochem. 2001, 292, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Triebl, A.; Trötzmüller, M.; Eberl, A.; Hanel, P.; Hartler, J.; Köfeler, H.C. Quantitation of phosphatidic acid and lysophosphatidic acid molecular species using hydrophilic interaction liquid chromatography coupled to electrospray ionization high resolution mass spectrometry. J. Chromatogr. A 2014, 1347, 104–110. [Google Scholar] [CrossRef]
- Scherer, M.; Schmitz, G.; Liebisch, G. High-throughput analysis of sphingosine 1-phostphate, sphinganine 1-phosphate, and lysophosphatidic acid in plasma samples by liquid chromatography—Tandem mass spectrometry. Clin. Chem. 2009, 55, 1218–1222. [Google Scholar] [CrossRef]
- Zhao, Z.; Xu, Y. An extremely simple method for extraction of lysophospholipids and phospholipids from blood samples. J. Lipid Res. 2010, 51, 652–659. [Google Scholar] [CrossRef] [Green Version]
- Wickramathilaka, M.P.; Tao, B.Y. Characterization of covalent crosslinking strategies for synthesizing DNA-based bioconjugates. J. Biol. Eng. 2019, 13, 8–17. [Google Scholar] [CrossRef] [Green Version]
- Carter, J.D.; LaBean, T.H. Coupling strategies for the synthesis of peptide-oligonucleotide conjugates for patterned synthetic biomineralization. J. Nucleic Acids 2011, 2011, 926595. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, K.; Ohkawa, R.; Okubo, S.; Tozuka, M.; Okada, M.; Aoki, S.; Aoki, J.; Arai, H.; Ikeda, H.; Yatomi, Y. Measurement of lysophospholipase D/autotaxin activity in human serum samples. Clin. Biochem. 2007, 40, 274–277. [Google Scholar] [CrossRef]
- Yasumoto, A.; Tokuoka, S.M.; Kita, Y.; Shimizu, T.; Yatomi, Y. Multiplex quantitative analysis of eicosanoid mediators in human plasma and serum: Possible introduction into clinical testing. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1068, 98–104. [Google Scholar] [CrossRef]
- Nakao, T.; Yasumoto, A.; Tokuoka, S.; Kita, Y.; Kawahara, T.; Daimon, M.; Yatomi, Y. The impact of night-shift work on platelet function in healthy medical staff. J. Occup. Health 2018, 60, 324–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murakami, M.; Sato, H.; Miki, Y.; Yamamoto, K.; Taketomi, Y. A new era of secreted phospholipase A2. J. Lipid Res. 2015, 56, 1248–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.H.; Hsiao, G.; Al-Shabrawey, M. Eicosanoids and oxidative stress in diabetic retinopathy. Antioxidants 2020, 9, 520. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, L.; Ceglarek, U.; Kortz, L.; Hoop, M.; Kirkby, K.; Thiery, J.; Himmerich, H. Mechanisms of involvement of eicosanoids and their precursors in the pathophysiology and treatment of schizophrenia. Med. Chem. 2013, 9, 763–773. [Google Scholar] [CrossRef]
- Onorato, J.M.; Shipkova, P.; Minnich, A.; Aubry, A.F.; Easter, J.; Tymiak, A. Challenges in accurate quantitation of lysophosphatidic acids in human biofluids. J. Lipid Res. 2014, 55, 1784–1796. [Google Scholar] [CrossRef] [Green Version]
- Yamada, M.; Kita, Y.; Kohira, T.; Yoshida, K.; Hamano, F.; Tokuoka, S.M.; Shimizu, T. A comprehensive quantification method for eicosanoids and related compounds by using liquid chromatography/mass spectrometry with high speed continuous ionization polarity switching. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2015, 995, 74–84. [Google Scholar] [CrossRef]
- HJM Jansen, E. Long term stability of parameters of lipid metabolism in frozen human serum: Triglycerides, free fatty acids, total-, HDL- and LDL-cholesterol, apolipoprotein-A1 and B. J. Mol. Biomark. Diagn. 2014, 5, 1000182. [Google Scholar] [CrossRef] [Green Version]
- Metherel, A.H.; Stark, K.D. The stability of blood fatty acids during storage and potential mechanisms of degradation: A review. Prostaglandins Leukot. Essent. Fat. Acids 2016, 104, 33–43. [Google Scholar] [CrossRef]
- Matthan, N.R.; Ip, B.; Resteghini, N.; Ausman, L.M.; Lichtenstein, A.H. Long-term fatty acid stability in human serum cholesteryl ester, triglyceride, and phospholipid fractions. J. Lipid Res. 2010, 51, 2826–2832. [Google Scholar] [CrossRef] [Green Version]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
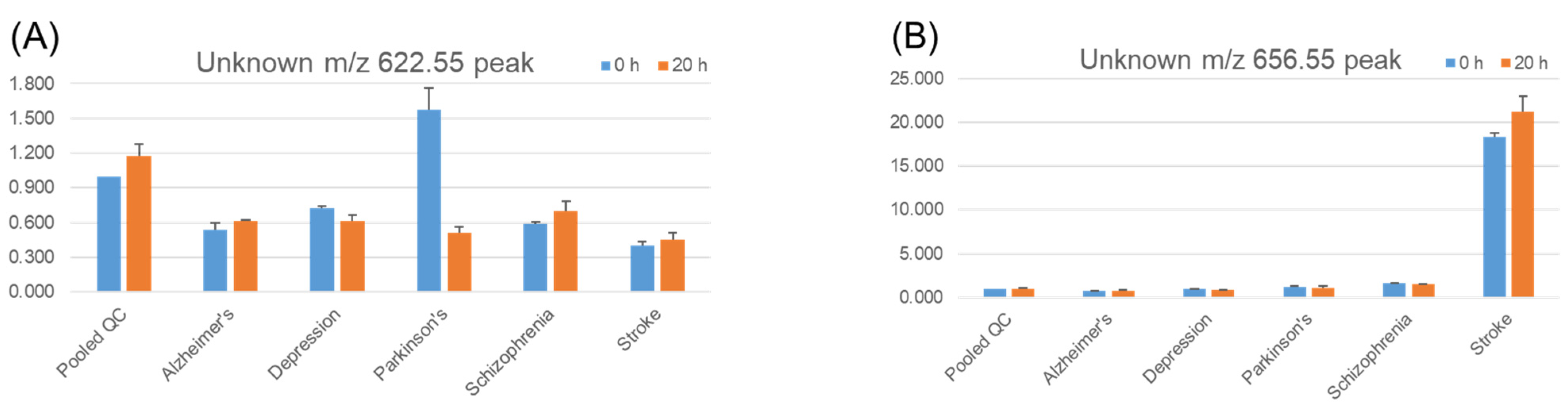{kind=link}
| 12-HETE × 16 | 12-HETE × 8 | 12-HETE × 4 | 12-HETE × 2 | 12-HETE × 1 | Correlation | |
| CV% in Buffer | 8.04 | 13.03 | 13.36 | 4.42 | 5.26 | 0.966 |
| CV% in Serum | 4.18 | 8.23 | 4.60 | 2.93 | 1.78 | 0.951 |
| 20-HETE × 16 | 20-HETE × 8 | 20-HETE × 4 | 20-HETE × 2 | 20-HETE × 1 | Correlation | |
| CV% in Buffer | 6.86 | 8.51 | 11.01 | 5.79 | 4.03 | 0.963 |
| CV% in Serum | 4.01 | 8.54 | 3.01 | 2.71 | 2.99 | 0.949 |
| PGE2 × 16 | PGE2 × 8 | PGE2 × 4 | PGE2 × 2 | PGE2 × 1 | Correlation | |
| CV% in Buffer | 5.14 | 8.97 | 8.20 | 4.34 | 5.91 | 0.962 |
| CV% in Serum | 4.01 | 8.54 | 3.01 | 2.71 | 2.99 | 0.949 |
| TXB2 × 16 | TXB2 × 8 | TXB2 × 4 | TXB2 × 2 | TXB2 × 1 | Correlation | |
| CV% in Buffer | 7.93 | 10.18 | 12.41 | 4.91 | 6.14 | 0.999 |
| CV% in Serum | 4.81 | 6.65 | 5.86 | 7.51 | 7.75 | 0.998 |
| 20-carboxy-LTB4 × 16 | 20-carboxy-LTB4 × 8 | 20-carboxy-LTB4 × 4 | 20-carboxy-LTB4 × 2 | 20-carboxy-LTB4 × 1 | Correlation | |
| CV% in Buffer | 11.30 | 10.59 | 27.28 | 14.91 | 12.27 | 0.994 |
| CV% in Serum | 15.26 | 6.23 | 5.42 | 5.70 | 11.09 | 0.996 |
| tetranor-PGDM × 16 | tetranor-PGDM × 8 | tetranor-PGDM × 4 | tetranor-PGDM × 2 | tetranor-PGDM × 1 | Correlation | |
| CV% in Buffer | 18.83 | 14.52 | 28.35 | 14.87 | 12.62 | 0.996 |
| CV% in Serum | 6.69 | 9.37 | 7.06 | 9.13 | 10.40 | 0.978 |
| LPA (16:0) × 16 | LPA (16:0) × 8 | LPA (16:0) × 4 | LPA (16:0) × 2 | LPA (16:0) × 1 | Correlation | |
| CV% in Buffer | 22.15 | 17.68 | 16.81 | 16.69 | 8.02 | 0.998 |
| CV% in Serum | 8.40 | 9.57 | 9.13 | 10.64 | 2.79 | 0.998 |
| LPA (18:1) × 16 | LPA (18:1) × 8 | LPA (18:1) × 4 | LPA (18:1) × 2 | LPA (18:1) × 1 | Correlation | |
| CV% in Buffer | 16.32 | 15.21 | 19.61 | 9.43 | 7.52 | 0.998 |
| CV% in Serum | 6.18 | 9.55 | 12.70 | 7.47 | 4.79 | 0.989 |
| PA (36:2) × 16 | PA (36:2) × 8 | PA (36:2) × 4 | PA (36:2) × 2 | PA (36:2) × 1 | Correlation | |
| CV% in Buffer | - | 12.29 | 12.55 | 5.27 | 4.03 | 0.999 |
| CV% in Serum | 2.55 | 7.21 | 5.23 | 5.22 | 2.02 | 0.995 |
| Alzheimer’s Disease | Depression | Parkinson’s Disease | Schizophrenia | Stroke | |
|---|---|---|---|---|---|
| N | 67 | 26 | 21 | 21 | 37 |
| Female % | 62.7% | 53.8% | 52.4% | 42.9% | 37.8% |
| Age (years) mean ± SD | 80.3 ± 7.18 | 75.3 ± 6.70 | 74.4 ± 5.50 | 72.7 ± 5.82 | 77.5 ± 6.07 |
| Age (years) Range | 62–95 | 65–88 | 66–90 | 65–82 | 65–90 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tokuoka, S.M.; Kita, Y.; Sato, M.; Shimizu, T.; Yatomi, Y.; Oda, Y. Development of Tandem Mass Tag Labeling Method for Lipid Molecules Containing Carboxy and Phosphate Groups, and Their Stability in Human Serum. Metabolites 2021, 11, 19. https://doi.org/10.3390/metabo11010019
Tokuoka SM, Kita Y, Sato M, Shimizu T, Yatomi Y, Oda Y. Development of Tandem Mass Tag Labeling Method for Lipid Molecules Containing Carboxy and Phosphate Groups, and Their Stability in Human Serum. Metabolites. 2021; 11(1):19. https://doi.org/10.3390/metabo11010019
Chicago/Turabian StyleTokuoka, Suzumi M., Yoshihiro Kita, Masaya Sato, Takao Shimizu, Yutaka Yatomi, and Yoshiya Oda. 2021. "Development of Tandem Mass Tag Labeling Method for Lipid Molecules Containing Carboxy and Phosphate Groups, and Their Stability in Human Serum" Metabolites 11, no. 1: 19. https://doi.org/10.3390/metabo11010019
APA StyleTokuoka, S. M., Kita, Y., Sato, M., Shimizu, T., Yatomi, Y., & Oda, Y. (2021). Development of Tandem Mass Tag Labeling Method for Lipid Molecules Containing Carboxy and Phosphate Groups, and Their Stability in Human Serum. Metabolites, 11(1), 19. https://doi.org/10.3390/metabo11010019