Involvements of Hyperhomocysteinemia in Neurological Disorders

 ,
,  ,
,  ,
,  , and
, and 
Abstract
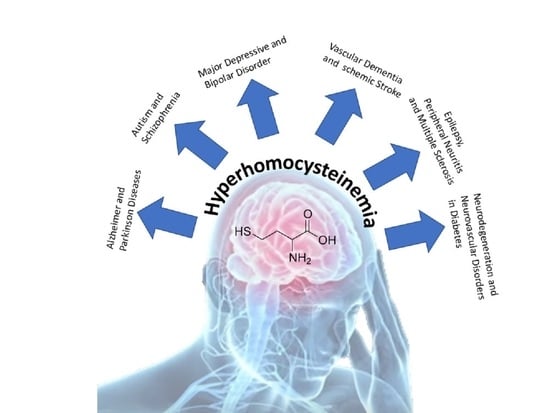
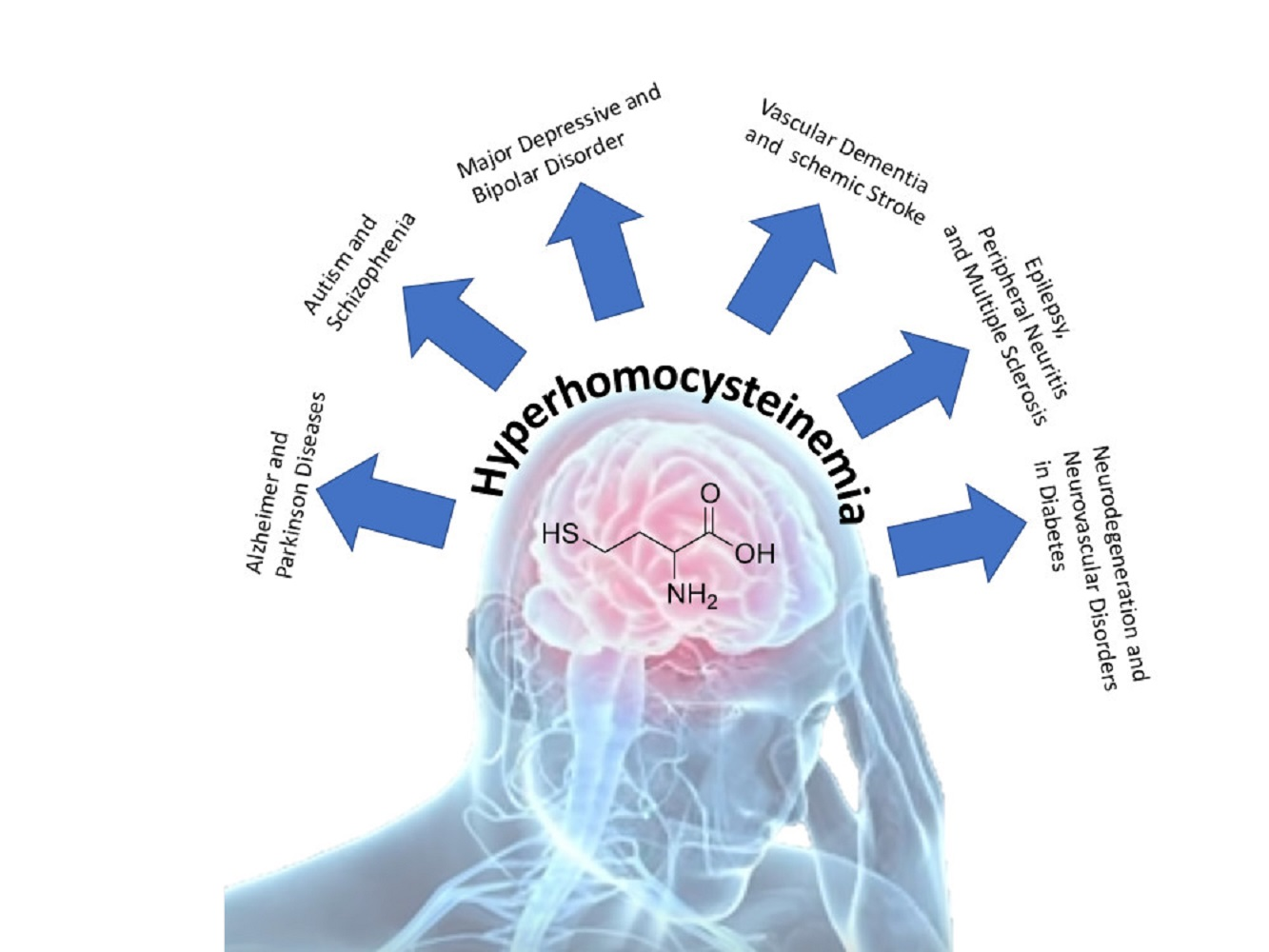
:
1. Introduction
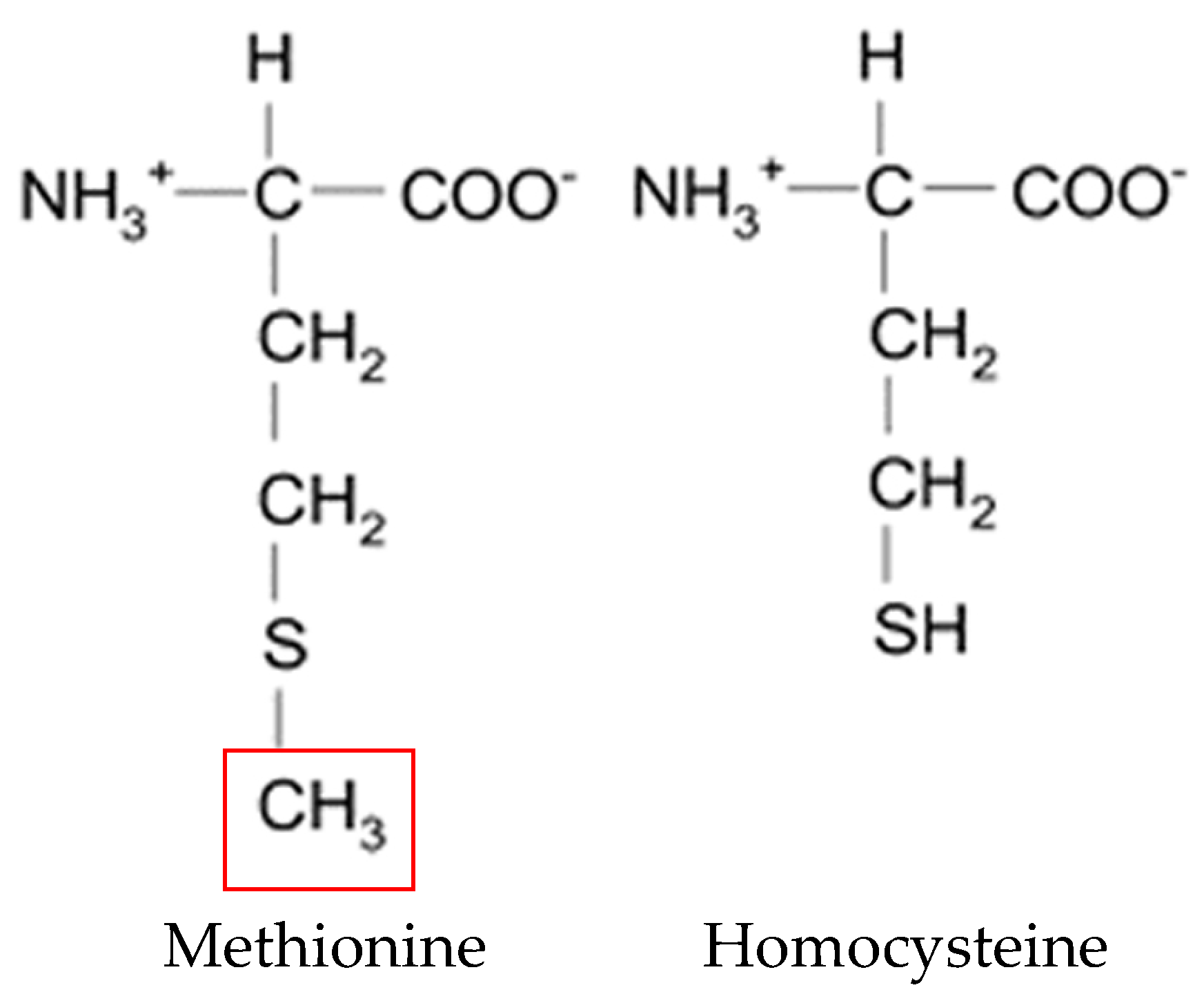
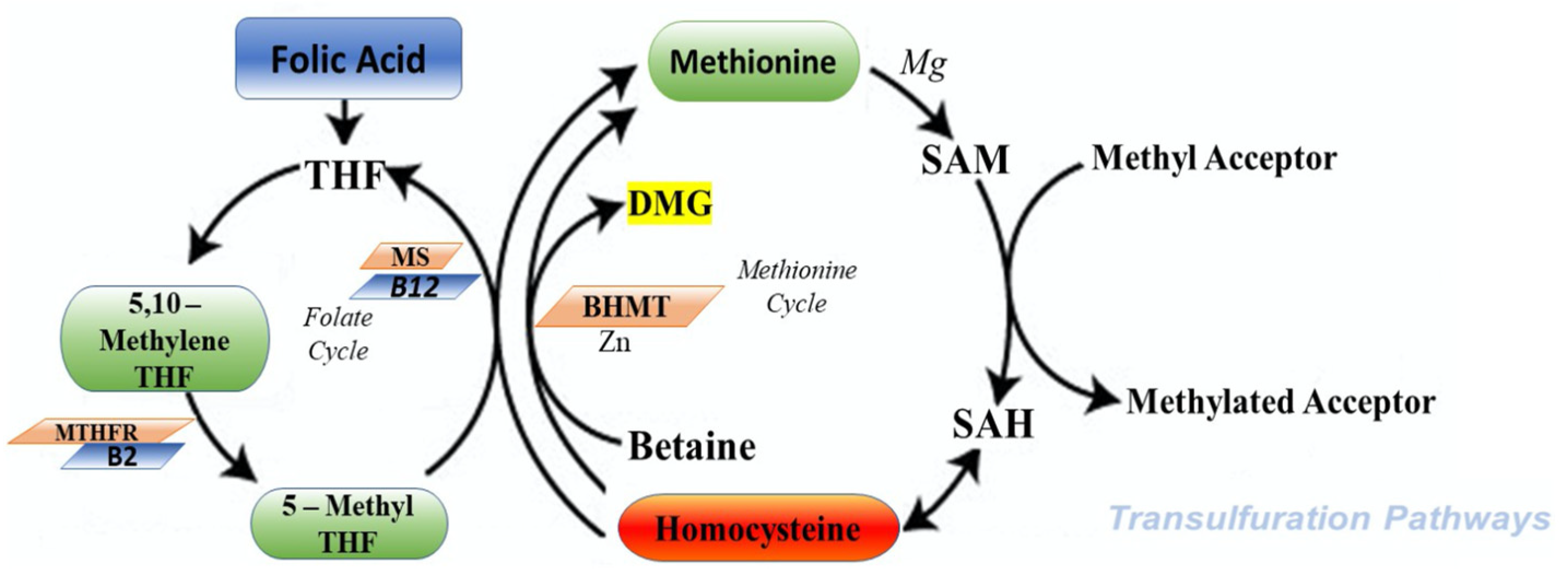
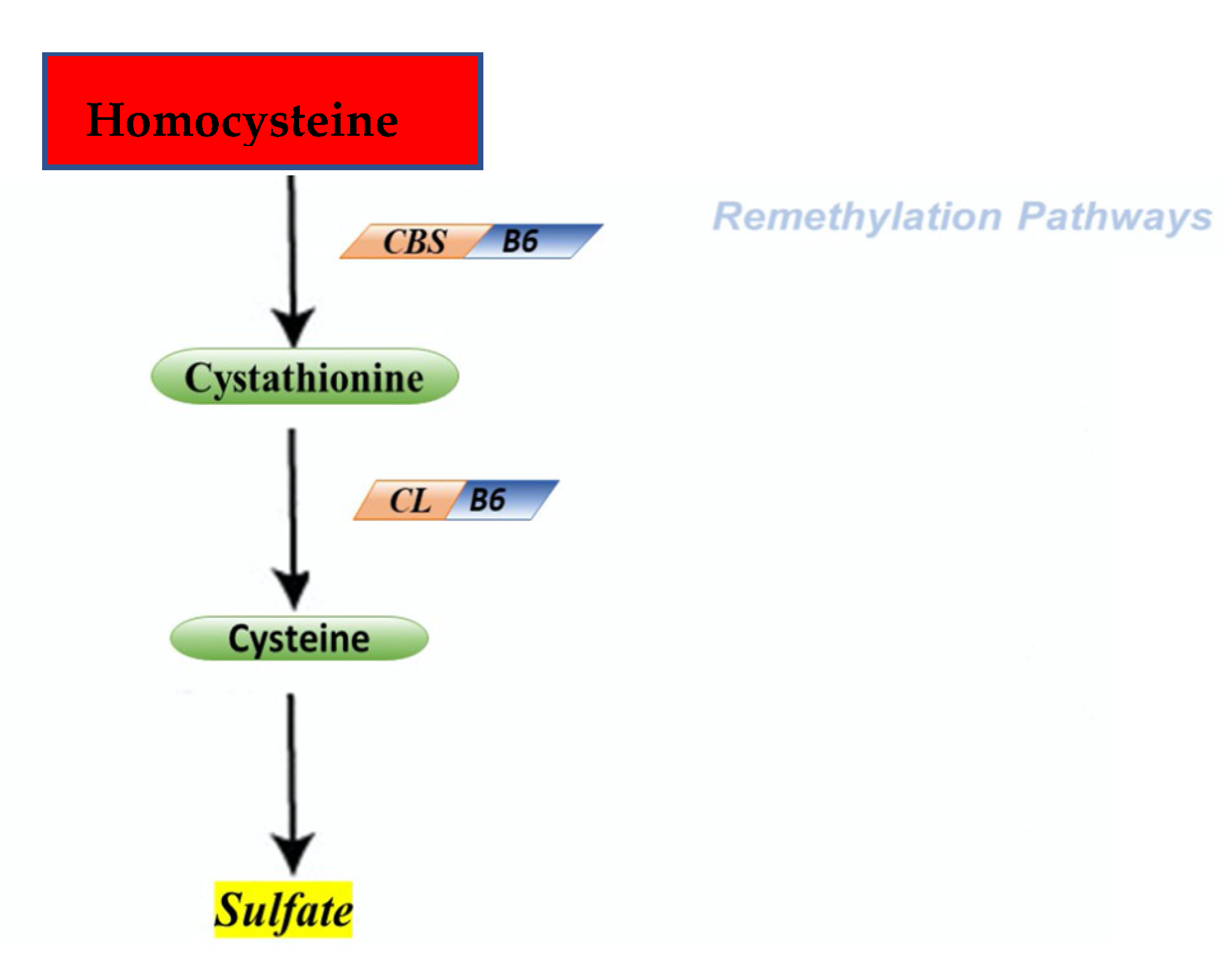
2. Physiological Role of Homocysteine
2.1. Cycle
2.2. Circulation and Concentration of HCY in Human Body
3. Pathological Role of Homocysteine
3.1. Alzheimer’s Disease
3.2. Parkinson’s Disease
3.3. Autism
3.4. Schizophrenia
3.5. Major Depressive and Bipolar Disorder
3.6. Vascular Dementia
3.7. Ischemic Stroke
3.8. Epilepsy
3.9. Peripheral Neuritis
3.10. Headache
3.11. Multiple Sclerosis
3.12. Neurodegeneration and Neurovascular Disorders in Diabetes
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kok, F.J. Folic acid, vitamins B6 and B12: Relation to homocysteine and cardiovascular disease. Bibl. Nutr. Dieta 2001, 189–191. [Google Scholar] [CrossRef]
- Miller, A.L. The methionine-homocysteine cycle and its effects on cognitive diseases. Altern. Med. Rev. 2003, 8, 7–19. [Google Scholar] [PubMed]
- Hopkins, P.N.; Wu, L.L.; Wu, J.; Hunt, S.C.; James, B.C.; Vincent, G.M.; Williams, R.R. Higher plasma homocyst(e)ine and increased susceptibility to adverse effects of low folate in early familial coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1314–1320. [Google Scholar] [CrossRef] [PubMed]
- Loehrer, F.M.; Angst, C.P.; Haefeli, W.E.; Jordan, P.P.; Ritz, R.; Fowler, B. Low whole-blood S-adenosylmethionine and correlation between 5-methyltetrahydrofolate and homocysteine in coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 1996, 16, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Boushey, C.J.; Beresford, S.A.; Omenn, G.S.; Motulsky, A.G. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA 1995, 274, 1049–1057. [Google Scholar] [CrossRef]
- Robinson, K.; Mayer, E.L.; Miller, D.P.; Green, R.; van Lente, F.; Gupta, A.; Kottke-Marchant, K.; Savon, S.R.; Selhub, J.; Nissen, S.E.; et al. Hyperhomocysteinemia and low pyridoxal phosphate. Common and independent reversible risk factors for coronary artery disease. Circulation 1995, 92, 2825–2830. [Google Scholar] [CrossRef] [PubMed]
- Petronijevic, N.D.; Radonjic, N.V.; Ivkovic, M.D.; Marinkovic, D.; Piperski, V.D.; Duricic, B.M.; Paunovic, V.R. Plasma homocysteine levels in young male patients in the exacerbation and remission phase of schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2008, 32, 1921–1926. [Google Scholar] [CrossRef]
- Ghosh, K.; Khare, A.; Shetty, S. Fasting plasma homocysteine levels are increased in young patients with acute myocardial infarction from Western India. Indian Heart J. 2007, 59, 242–245. [Google Scholar]
- Nikfardjam, M.; Graf, S.; Hornykewycz, S.; Zorn, G.; Huber-Beckmann, R.; Wojta, J.; Huber, K. Homocysteine plasma levels in young patients with coronary artery disease. Relation to history of acute myocardial infarction and anatomical extent of disease. Thromb. Res. 2001, 103 (Suppl. 1), S35–S39. [Google Scholar] [CrossRef]
- Targher, G.; Zenari, L.; Bertolini, L.; Falezza, G.; Muggeo, M.; Zoppini, G. Plasma total homocysteine levels are associated with von Willebrand factor, soluble intercellular adhesion molecule-1, and soluble tumor necrosis factor-alpha receptors in young type 1 diabetic patients without clinical evidence of macrovascular complications. Diabetes Care 2001, 24, 1496–1497. [Google Scholar] [CrossRef] [Green Version]
- Landgren, F.; Israelsson, B.; Lindgren, A.; Hultberg, B.; Andersson, A.; Brattstrom, L. Plasma homocysteine in acute myocardial infarction: Homocysteine-lowering effect of folic acid. J. Intern. Med. 1995, 237, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Ishihara, J.; Iso, H.; Inoue, M.; Iwasaki, M.; Okada, K.; Kita, Y.; Kokubo, Y.; Okayama, A.; Tsugane, S.; Group, J.S. Intake of folate, vitamin B6 and vitamin B12 and the risk of CHD: The Japan Public Health Center-Based Prospective Study Cohort I. J. Am. Coll. Nutr. 2008, 27, 127–136. [Google Scholar] [CrossRef]
- Chasan-Taber, L.; Selhub, J.; Rosenberg, I.H.; Malinow, M.R.; Terry, P.; Tishler, P.V.; Willett, W.; Hennekens, C.H.; Stampfer, M.J. A prospective study of folate and vitamin B6 and risk of myocardial infarction in US physicians. J. Am. Coll. Nutr. 1996, 15, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Knekt, P.; Alfthan, G.; Aromaa, A.; Heliovaara, M.; Marniemi, J.; Rissanen, H.; Reunanen, A. Homocysteine and major coronary events: A prospective population study amongst women. J. Intern. Med. 2001, 249, 461–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whincup, P.H.; Refsum, H.; Perry, I.J.; Morris, R.; Walker, M.; Lennon, L.; Thomson, A.; Ueland, P.M.; Ebrahim, S.B. Serum total homocysteine and coronary heart disease: Prospective study in middle aged men. Heart 1999, 82, 448–454. [Google Scholar] [CrossRef]
- Bots, M.L.; Launer, L.J.; Lindemans, J.; Hoes, A.W.; Hofman, A.; Witteman, J.C.; Koudstaal, P.J.; Grobbee, D.E. Homocysteine and short-term risk of myocardial infarction and stroke in the elderly: The Rotterdam Study. Arch. Intern. Med. 1999, 159, 38–44. [Google Scholar] [CrossRef] [Green Version]
- Brattstrom, L.; Lindgren, A.; Israelsson, B.; Malinow, M.R.; Norrving, B.; Upson, B.; Hamfelt, A. Hyperhomocysteinaemia in stroke: Prevalence, cause, and relationships to type of stroke and stroke risk factors. Eur. J. Clin. Investig. 1992, 22, 214–221. [Google Scholar] [CrossRef]
- Perry, I.J.; Refsum, H.; Morris, R.W.; Ebrahim, S.B.; Ueland, P.M.; Shaper, A.G. Prospective study of serum total homocysteine concentration and risk of stroke in middle-aged British men. Lancet 1995, 346, 1395–1398. [Google Scholar] [CrossRef]
- Faurschou, M.; Nielsen, O.J.; Jensen, M.K.; Hasselbalch, H.C. High prevalence of hyperhomocysteinemia due to marginal deficiency of cobalamin or folate in chronic myeloproliferative disorders. Am. J. Hematol. 2000, 65, 136–140. [Google Scholar] [CrossRef]
- Hultberg, B.; Agardh, C.D.; Agardh, E.; Lovestam-Adrian, M. Poor metabolic control, early age at onset, and marginal folate deficiency are associated with increasing levels of plasma homocysteine in insulin-dependent diabetes mellitus. A five-year follow-up study. Scand. J. Clin. Lab. Investig. 1997, 57, 595–600. [Google Scholar] [CrossRef]
- Hultberg, B.; Andersson, A.; Lindgren, A. Marginal folate deficiency as a possible cause of hyperhomocystinaemia in stroke patients. Eur. J. Clin. Chem. Clin. Biochem. 1997, 35, 25–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.W.; Ting, A.C.; Wong, J. Fasting total plasma homocysteine and atherosclerotic peripheral vascular disease. Ann. Vasc. Surg. 1997, 11, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Beaumont, V.; Malinow, M.R.; Sexton, G.; Wilson, D.; Lemort, N.; Upson, B.; Beaumont, J.L. Hyperhomocyst(e)inemia, anti-estrogen antibodies and other risk factors for thrombosis in women on oral contraceptives. Atherosclerosis 1992, 94, 147–152. [Google Scholar] [CrossRef]
- Urnov, F.D. Methylation and the genome: The power of a small amendment. J. Nutr. 2002, 132, 2450S–2456S. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.T. On the mechanism of homocysteine pathophysiology and pathogenesis: A unifying hypothesis. Histol. Histopathol. 2002, 17, 1283–1291. [Google Scholar] [CrossRef]
- Bautista, L.E.; Arenas, I.A.; Peñuela, A.; Martínez, L.X. Total plasma homocysteine level and risk of cardiovascular disease: A meta-analysis of prospective cohort studies. J. Clin. Epidemiol. 2002, 55, 882–887. [Google Scholar] [CrossRef]
- Khare, A.; Lopez, M.; Gogtay, J. Homocysteine, B vitamins, and cardiovascular disease. N. Engl. J. Med. 2006, 355, 206. [Google Scholar]
- Celik, N.; Vurmaz, A.; Kahraman, A. Protective effect of quercetin on homocysteine-induced oxidative stress. Nutrition 2017, 33, 291–296. [Google Scholar] [CrossRef]
- Selhub, J. Homocysteine metabolism. Annu. Rev. Nutr. 1999, 19, 217–246. [Google Scholar] [CrossRef] [Green Version]
- Rizzo, A.; Sciorsci, R.L. Role of homocysteine metabolism in animal reproduction: A review. Res. Vet. Sci. 2019, 122, 29–35. [Google Scholar] [CrossRef]
- Rehman, T.; Shabbir, M.A.; Inam-Ur-Raheem, M.; Manzoor, M.F.; Ahmad, N.; Liu, Z.W.; Ahmad, M.H.; Siddeeg, A.; Abid, M.; Aadil, R.M. Cysteine and homocysteine as biomarker of various diseases. Food Sci. Nutr. 2020, 8, 4696–4707. [Google Scholar] [CrossRef] [PubMed]
- Stead, L.M.; Brosnan, J.T.; Brosnan, M.E.; Vance, D.E.; Jacobs, R.L. Is it time to reevaluate methyl balance in humans? Am. J. Clin. Nutr. 2006, 83, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Carmel, R.; Gott, P.S.; Waters, C.H.; Cairo, K.; Green, R.; Bondareff, W.; DeGiorgio, C.M.; Cummings, J.L.; Jacobsen, D.W.; Buckwalter, G.; et al. The frequently low cobalamin levels in dementia usually signify treatable metabolic, neurologic and electrophysiologic abnormalities. Eur. J. Haematol. 1995, 54, 245–253. [Google Scholar] [CrossRef]
- Allen, R.H.; Stabler, S.P.; Savage, D.G.; Lindenbaum, J. Metabolic abnormalities in cobalamin (vitamin B12) and folate deficiency. FASEB J. 1993, 7, 1344–1353. [Google Scholar] [CrossRef] [PubMed]
- Galbiatti, A.L.; Ruiz, M.T.; Rezende Pinto, D.; Raposo, L.S.; Maniglia, J.V.; Pavarino-Bertelli, E.C.; Goloni-Bertollo, E.M. A80G polymorphism of reduced folate carrier 1 (RFC1) gene and head and neck squamous cell carcinoma etiology in Brazilian population. Mol. Biol. Rep. 2011, 38, 1071–1078. [Google Scholar] [CrossRef]
- Biselli, J.M.; Brumati, D.; Frigeri, V.F.; Zampieri, B.L.; Goloni-Bertollo, E.M.; Pavarino-Bertelli, E.C. A80G polymorphism of reduced folate carrier 1 (RFC1) and C776G polymorphism of transcobalamin 2 (TC2) genes in Down’s syndrome etiology. Sao Paulo Med. J. 2008, 126, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Desouza, C.; Keebler, M.; McNamara, D.B.; Fonseca, V. Drugs affecting homocysteine metabolism: Impact on cardiovascular risk. Drugs 2002, 62, 605–616. [Google Scholar] [CrossRef]
- Deedwania, P.C. New oral anticoagulants in elderly patients with atrial fibrillation. Am. J. Med. 2013, 126, 289–296. [Google Scholar] [CrossRef]
- Miller, D.J.; Simpson, J.R.; Silver, B. Safety of thrombolysis in acute ischemic stroke: A review of complications, risk factors, and newer technologies. Neurohospitalist 2011, 1, 138–147. [Google Scholar] [CrossRef] [Green Version]
- Sacco, R.L. Newer risk factors for stroke. Neurology 2001, 57, S31–S34. [Google Scholar] [CrossRef]
- Cotter, A.M.; Molloy, A.M.; Scott, J.M.; Daly, S.F. Elevated plasma homocysteine in early pregnancy: A risk factor for the development of nonsevere preeclampsia. Am. J. Obstet. Gynecol. 2003, 189, 391–394. [Google Scholar] [CrossRef]
- Ramlau-Hansen, C.H.; Moller, U.K.; Moller, J.; Thulstrup, A.M. Lactation—A risk factor for elevated plasma homocysteine? Ugeskr. Laeger 2003, 165, 2819–2823. [Google Scholar] [PubMed]
- Hultberg, B. Modulation of extracellular homocysteine concentration in human cell lines. Clin. Chim. Acta 2003, 330, 151–159. [Google Scholar] [CrossRef]
- Sakamoto, A.; Nishimura, Y.; Ono, H.; Sakura, N. Betaine and homocysteine concentrations in foods. Pediatr. Int. 2002, 44, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Chen, M.; Gao, J.; Ji, X.; He, J.; Zhang, J.; Zhao, W. A series of BODIPY-based probes for the detection of cysteine and homocysteine in living cells. Talanta 2019, 195, 281–289. [Google Scholar] [CrossRef]
- Kostic, S.; Micovic, Z.; Andrejevic, L.; Cvetkovic, S.; Stamenkovic, A.; Stankovic, S.; Obrenovic, R.; Labudovic-Borovic, M.; Hrncic, D.; Jakovljevic, V.; et al. The effects of L-cysteine and N-acetyl-L-cysteine on homocysteine metabolism and haemostatic markers, and on cardiac and aortic histology in subchronically methionine-treated Wistar male rats. Mol. Cell. Biochem. 2019, 451, 43–54. [Google Scholar] [CrossRef]
- Hannibal, L.; Blom, H.J. Homocysteine and disease: Causal associations or epiphenomenons? Mol. Asp. Med. 2017, 53, 36–42. [Google Scholar] [CrossRef]
- Kolling, J.; Scherer, E.B.; da Cunha, A.A.; da Cunha, M.J.; Wyse, A.T. Homocysteine induces oxidative-nitrative stress in heart of rats: Prevention by folic acid. Cardiovasc. Toxicol. 2011, 11, 67–73. [Google Scholar] [CrossRef]
- Scherer, E.B.; da Cunha, A.A.; Kolling, J.; da Cunha, M.J.; Schmitz, F.; Sitta, A.; Lima, D.D.; Delwing, D.; Vargas, C.R.; Wyse, A.T. Development of an animal model for chronic mild hyperhomocysteinemia and its response to oxidative damage. Int. J. Dev. Neurosci. 2011, 29, 693–699. [Google Scholar] [CrossRef]
- Kaplan, P.; Tatarkova, Z.; Sivonova, M.K.; Racay, P.; Lehotsky, J. Homocysteine and Mitochondria in Cardiovascular and Cerebrovascular Systems. Int. J. Mol. Sci. 2020, 21, 7698. [Google Scholar] [CrossRef]
- Perna, A.F.; Ingrosso, D.; Lombardi, C.; Acanfora, F.; Satta, E.; Cesare, C.M.; Violetti, E.; Romano, M.M.; De Santo, N.G. Possible mechanisms of homocysteine toxicity. Kidney Int. Suppl. 2003, S137–S140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esse, R.; Barroso, M.; Tavares de Almeida, I.; Castro, R. The Contribution of Homocysteine Metabolism Disruption to Endothelial Dysfunction: State-of-the-Art. Int. J. Mol. Sci. 2019, 20, 867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrakhovitch, E.A.; Tabibzadeh, S. Homocysteine and age-associated disorders. Ageing Res. Rev. 2019, 49, 144–164. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef] [PubMed]
- Lash, L.H.; Anders, M.W. Mechanism of S-(1,2-dichlorovinyl)-L-cysteine- and S-(1,2-dichlorovinyl)-L-homocysteine-induced renal mitochondrial toxicity. Mol. Pharmacol. 1987, 32, 549–556. [Google Scholar]
- Dos Santos, T.M.; Siebert, C.; de Oliveira, M.F.; Manfredini, V.; Wyse, A.T.S. Chronic mild Hyperhomocysteinemia impairs energy metabolism, promotes DNA damage and induces a Nrf2 response to oxidative stress in rats brain. Cell. Mol. Neurobiol. 2019, 39, 687–700. [Google Scholar] [CrossRef]
- Wyse, A.T.S.; Sanches, E.F.; Dos Santos, T.M.; Siebert, C.; Kolling, J.; Netto, C.A. Chronic mild hyperhomocysteinemia induces anxiety-like symptoms, aversive memory deficits and hippocampus atrophy in adult rats: New insights into physiopathological mechanisms. Brain Res. 2020, 1728, 146592. [Google Scholar] [CrossRef]
- Kumar, M.; Sandhir, R. Hydrogen sulfide attenuates hyperhomocysteinemia-induced mitochondrial dysfunctions in brain. Mitochondrion 2020, 50, 158–169. [Google Scholar] [CrossRef]
- Folbergrova, J.; Jesina, P.; Drahota, Z.; Lisy, V.; Haugvicova, R.; Vojtiskova, A.; Houstek, J. Mitochondrial complex I inhibition in cerebral cortex of immature rats following homocysteic acid-induced seizures. Exp. Neurol. 2007, 204, 597–609. [Google Scholar] [CrossRef]
- Bhattacharjee, N.; Borah, A. Oxidative stress and mitochondrial dysfunction are the underlying events of dopaminergic neurodegeneration in homocysteine rat model of Parkinson’s disease. Neurochem. Int. 2016, 101, 48–55. [Google Scholar] [CrossRef] [PubMed]
- Kolling, J.; Scherer, E.B.; Siebert, C.; Longoni, A.; Loureiro, S.; Weis, S.; Petenuzzo, L.; Wyse, A.T. Severe Hyperhomocysteinemia Decreases Respiratory Enzyme and Na(+)-K(+) ATPase Activities, and Leads to Mitochondrial Alterations in Rat Amygdala. Neurotox. Res. 2016, 29, 408–418. [Google Scholar] [CrossRef] [PubMed]
- Folbergrova, J.; Jesina, P.; Haugvicova, R.; Lisy, V.; Houstek, J. Sustained deficiency of mitochondrial complex I activity during long periods of survival after seizures induced in immature rats by homocysteic acid. Neurochem. Int. 2010, 56, 394–403. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharjee, N.; Paul, R.; Giri, A.; Borah, A. Chronic exposure of homocysteine in mice contributes to dopamine loss by enhancing oxidative stress in nigrostriatum and produces behavioral phenotypes of Parkinson’s disease. Biochem. Biophys. Rep. 2016, 6, 47–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mishra, D.K.; Gautam, S.; Goyal, B.K.; Kawar, R. Hyperhomocysteinemia as a cause of left main artery thrombosis manifesting as extensive anterior wall MI in a 10 year old girl. J. Assoc. Physicians India 2013, 61, 829–831. [Google Scholar] [PubMed]
- Mishra, P.K.; Tyagi, N.; Sen, U.; Joshua, I.G.; Tyagi, S.C. Synergism in hyperhomocysteinemia and diabetes: Role of PPAR gamma and tempol. Cardiovasc. Diabetol. 2010, 9, 49. [Google Scholar] [CrossRef] [Green Version]
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef]
- Frosst, P.; Blom, H.J.; Milos, R.; Goyette, P.; Sheppard, C.A.; Matthews, R.G.; Boers, G.J.; den Heijer, M.; Kluijtmans, L.A.; van den Heuvel, L.P.; et al. A candidate genetic risk factor for vascular disease: A common mutation in methylenetetrahydrofolate reductase. Nat. Genet. 1995, 10, 111–113. [Google Scholar] [CrossRef]
- Bailey, L.B.; Gregory, J.F., 3rd. Polymorphisms of methylenetetrahydrofolate reductase and other enzymes: Metabolic significance, risks and impact on folate requirement. J. Nutr. 1999, 129, 919–922. [Google Scholar] [CrossRef]
- Refsum, H.; Ueland, P.M.; Nygard, O.; Vollset, S.E. Homocysteine and cardiovascular disease. Annu. Rev. Med. 1998, 49, 31–62. [Google Scholar] [CrossRef]
- Warren, C.J. Emergent cardiovascular risk factor: Homocysteine. Prog. Cardiovasc. Nurs. 2002, 17, 35–41. [Google Scholar] [CrossRef]
- Medina, M.A.; Amores-Sanchez, M.I. Homocysteine: An emergent cardiovascular risk factor? Eur. J. Clin. Invest. 2000, 30, 754–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, S.; Pan, M.; Wu, S.; Venners, S.A.; Zhong, G.; Hsu, Y.H.; Weinstock, J.; Wang, B.; Tang, G.; Liu, D.; et al. Elevation in Total Homocysteine Levels in Chinese Patients with Essential Hypertension Treated with Antihypertensive Benazepril. Clin. Appl. Thromb. Hemost. 2016, 22, 191–198. [Google Scholar] [CrossRef] [PubMed]
- Muller, T.; Jugel, C.; Ehret, R.; Ebersbach, G.; Bengel, G.; Muhlack, S.; Klostermann, F. Elevation of total homocysteine levels in patients with Parkinson’s disease treated with duodenal levodopa/carbidopa gel. J. Neural Transm. (Vienna) 2011, 118, 1329–1333. [Google Scholar] [CrossRef]
- Stabler, S.P.; Marcell, P.D.; Podell, E.R.; Allen, R.H.; Savage, D.G.; Lindenbaum, J. Elevation of total homocysteine in the serum of patients with cobalamin or folate deficiency detected by capillary gas chromatography-mass spectrometry. J. Clin. Invest. 1988, 81, 466–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Selhub, J.; Miller, J.W. The pathogenesis of homocysteinemia: Interruption of the coordinate regulation by S-adenosylmethionine of the remethylation and transsulfuration of homocysteine. Am. J. Clin. Nutr. 1992, 55, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Kong, H.; Pang, W.; Yang, H.; Lu, H.; Huang, C.; Jiang, Y. B vitamin supplementation improves cognitive function in the middle aged and elderly with hyperhomocysteinemia. Nutr. Neurosci. 2016, 19, 461–466. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R. Homocysteine, B vitamins, and the risk of cardiovascular disease. Clin. Chem. 2011, 57, 1201–1202. [Google Scholar] [CrossRef] [Green Version]
- Blacher, J.; Czernichow, S.; Horrellou, M.H.; Conad, J.; David, P.; Chadefaux-Vekemans, B.; Ankria, A.; Galan, P.; Hercberg, S.; Ducimetiere, P. Homocysteine, folic acid, group B vitamins and cardiovascular risk. Arch. Mal. Coeur Vaiss. 2005, 98, 145–152. [Google Scholar]
- Zhuo, J.M.; Wang, H.; Pratico, D. Is hyperhomocysteinemia an Alzheimer’s disease (AD) risk factor, an AD marker, or neither? Trends Pharmacol. Sci. 2011, 32, 562–571. [Google Scholar] [CrossRef] [Green Version]
- Carmel, R.; Green, R.; Rosenblatt, D.S.; Watkins, D. Update on cobalamin, folate, and homocysteine. Hematology 2003, 2003, 62–81. [Google Scholar] [CrossRef] [Green Version]
- Ansari, R.; Mahta, A.; Mallack, E.; Luo, J.J. Hyperhomocysteinemia and neurologic disorders: A review. J. Clin. Neurol. 2014, 10, 281–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choe, Y.M.; Sohn, B.K.; Choi, H.J.; Byun, M.S.; Seo, E.H.; Han, J.Y.; Kim, Y.K.; Yoon, E.J.; Lee, J.M.; Park, J.; et al. Association of homocysteine with hippocampal volume independent of cerebral amyloid and vascular burden. Neurobiol. Aging 2014, 35, 1519–1525. [Google Scholar] [CrossRef]
- Skovierova, H.; Vidomanova, E.; Mahmood, S.; Sopkova, J.; Drgova, A.; Cervenova, T.; Halasova, E.; Lehotsky, J. The Molecular and Cellular Effect of Homocysteine Metabolism Imbalance on Human Health. Int. J. Mol. Sci. 2016, 17, 1733. [Google Scholar] [CrossRef]
- Kamat, P.K.; Vacek, J.C.; Kalani, A.; Tyagi, N. Homocysteine Induced Cerebrovascular Dysfunction: A Link to Alzheimer’s Disease Etiology. Open Neurol. J. 2015, 9, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Gillespie, W.; Vacek, J.C.; Sen, U.; Tyagi, S.C.; Lominadze, D. Activation of GABA-A receptor ameliorates homocysteine-induced MMP-9 activation by ERK pathway. J. Cell. Physiol. 2009, 220, 257–266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.E.; Tian, Q.; Wei, W.; Peng, J.H.; Liu, G.P.; Zhou, X.W.; Wang, Q.; Wang, D.W.; Wang, J.Z. Homocysteine induces tau phosphorylation by inactivating protein phosphatase 2A in rat hippocampus. Neurobiol. Aging 2008, 29, 1654–1665. [Google Scholar] [CrossRef]
- Chen, S.; Dong, Z.; Cheng, M.; Zhao, Y.; Wang, M.; Sai, N.; Wang, X.; Liu, H.; Huang, G.; Zhang, X. Homocysteine exaggerates microglia activation and neuroinflammation through microglia localized STAT3 overactivation following ischemic stroke. J. Neuroinflamm. 2017, 14, 187. [Google Scholar] [CrossRef] [Green Version]
- Fan, X.; Zhang, L.; Li, H.; Chen, G.; Qi, G.; Ma, X.; Jin, Y. Role of homocysteine in the development and progression of Parkinson’s disease. Ann. Clin. Transl. Neurol. 2020. [Google Scholar] [CrossRef]
- Zieminska, E.; Lazarewicz, J.W. Excitotoxic neuronal injury in chronic homocysteine neurotoxicity studied in vitro: The role of NMDA and group I metabotropic glutamate receptors. Acta Neurobiol. Exp. (Wars) 2006, 66, 301–309. [Google Scholar]
- Valkovic, P.; Benetin, J.; Blazicek, P.; Valkovicova, L.; Gmitterova, K.; Kukumberg, P. Reduced plasma homocysteine levels in levodopa/entacapone treated Parkinson patients. Parkinsonism Relat. Disord. 2005, 11, 253–256. [Google Scholar] [CrossRef]
- Racek, J.; Rusnakova, H.; Trefil, L.; Siala, K.K. The influence of folate and antioxidants on homocysteine levels and oxidative stress in patients with hyperlipidemia and hyperhomocysteinemia. Physiol. Res. 2005, 54, 87–95. [Google Scholar] [PubMed]
- Lee, S.H.; Kim, M.J.; Kim, B.J.; Kim, S.R.; Chun, S.; Ryu, J.S.; Kim, G.S.; Lee, M.C.; Koh, J.M.; Chung, S.J. Homocysteine-lowering therapy or antioxidant therapy for bone loss in Parkinson’s disease. Mov. Disord. 2010, 25, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Johnson, N.L.; Burkett, K.; Reinhold, J.; Bultas, M.W. Translating Research to Practice for Children with Autism Spectrum Disorder: Part I: Definition, Associated Behaviors, Prevalence, Diagnostic Process, and Interventions. J. Pediatr. Health Care 2016, 30, 15–26. [Google Scholar] [CrossRef] [PubMed]
- Yoo, H. Genetics of Autism Spectrum Disorder: Current Status and Possible Clinical Applications. Exp. Neurobiol. 2015, 24, 257–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Subramanian, M.; Timmerman, C.K.; Schwartz, J.L.; Pham, D.L.; Meffert, M.K. Characterizing autism spectrum disorders by key biochemical pathways. Front. Neurosci. 2015, 9, 313. [Google Scholar] [CrossRef] [Green Version]
- Kiykim, E.; Zeybek, C.A.; Zubarioglu, T.; Cansever, S.; Yalcinkaya, C.; Soyucen, E.; Aydin, A. Inherited metabolic disorders in Turkish patients with autism spectrum disorders. Autism Res. 2016, 9, 217–223. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Cutler, P.; Melnyk, S.; Jernigan, S.; Janak, L.; Gaylor, D.W.; Neubrander, J.A. Metabolic biomarkers of increased oxidative stress and impaired methylation capacity in children with autism. Am. J. Clin. Nutr. 2004, 80, 1611–1617. [Google Scholar] [CrossRef] [Green Version]
- Tu, W.J.; Yin, C.H.; Guo, Y.Q.; Li, S.O.; Chen, H.; Zhang, Y.; Feng, Y.L.; Long, B.H. Serum homocysteine concentrations in Chinese children with autism. Clin. Chem. Lab. Med. 2013, 51, e19–e22. [Google Scholar] [CrossRef] [PubMed]
- Ali, A.; Waly, M.I.; Al-Farsi, Y.M.; Essa, M.M.; Al-Sharbati, M.M.; Deth, R.C. Hyperhomocysteinemia among Omani autistic children: A case-control study. Acta Biochim. Pol. 2011, 58, 547–551. [Google Scholar] [CrossRef]
- Kaluzna-Czaplinska, J.; Michalska, M.; Rynkowski, J. Homocysteine level in urine of autistic and healthy children. Acta Biochim. Pol. 2011, 58, 31–34. [Google Scholar] [CrossRef] [PubMed]
- Ghanizadeh, A. Increased glutamate and homocysteine and decreased glutamine levels in autism: A review and strategies for future studies of amino acids in autism. Dis. Markers 2013, 35, 281–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kral, T.V.; Eriksen, W.T.; Souders, M.C.; Pinto-Martin, J.A. Eating behaviors, diet quality, and gastrointestinal symptoms in children with autism spectrum disorders: A brief review. J. Pediatr. Nurs. 2013, 28, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Ranjan, S.; Nasser, J.A. Nutritional status of individuals with autism spectrum disorders: Do we know enough? Adv. Nutr. 2015, 6, 397–407. [Google Scholar] [CrossRef] [PubMed]
- McGrath, J.; Saha, S.; Welham, J.; El Saadi, O.; MacCauley, C.; Chant, D. A systematic review of the incidence of schizophrenia: The distribution of rates and the influence of sex, urbanicity, migrant status and methodology. BMC Med. 2004, 2, 13. [Google Scholar] [CrossRef]
- Arroll, M.A.; Wilder, L.; Neil, J. Nutritional interventions for the adjunctive treatment of schizophrenia: A brief review. Nutr. J. 2014, 13, 91. [Google Scholar] [CrossRef] [Green Version]
- Vita, A.; Barlati, S.; De Peri, L.; Deste, G.; Sacchetti, E. Schizophrenia. Lancet 2016, 388, 1280. [Google Scholar] [CrossRef]
- Regland, B.; Johansson, B.V.; Grenfeldt, B.; Hjelmgren, L.T.; Medhus, M. Homocysteinemia is a common feature of schizophrenia. J. Neural Transm. Gen. Sect. 1995, 100, 165–169. [Google Scholar] [CrossRef]
- Regland, B.; Johansson, B.V.; Gottfries, C.G. Homocysteinemia and schizophrenia as a case of methylation deficiency. J. Neural Transm. Gen. Sect. 1994, 98, 143–152. [Google Scholar] [CrossRef]
- Muntjewerff, J.W.; Kahn, R.S.; Blom, H.J.; den Heijer, M. Homocysteine, methylenetetrahydrofolate reductase and risk of schizophrenia: A meta-analysis. Mol. Psychiatry 2006, 11, 143–149. [Google Scholar] [CrossRef] [Green Version]
- Kim, T.H.; Moon, S.W. Serum homocysteine and folate levels in korean schizophrenic patients. Psychiatry Investig. 2011, 8, 134–140. [Google Scholar] [CrossRef] [Green Version]
- Di Lorenzo, R.; Amoretti, A.; Baldini, S.; Soli, M.; Landi, G.; Pollutri, G.; Corradini, R.; Ferri, P. Homocysteine levels in schizophrenia patients newly admitted to an acute psychiatric ward. Acta Neuropsychiatr. 2015, 27, 336–344. [Google Scholar] [CrossRef]
- Ayesa-Arriola, R.; Perez-Iglesias, R.; Rodriguez-Sanchez, J.M.; Mata, I.; Gomez-Ruiz, E.; Garcia-Unzueta, M.; Martinez-Garcia, O.; Tabares-Seisdedos, R.; Vazquez-Barquero, J.L.; Crespo-Facorro, B. Homocysteine and cognition in first-episode psychosis patients. Eur. Arch. Psychiatry Clin. Neurosci. 2012, 262, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, M.; Numata, S.; Tajima, A.; Nishi, A.; Muraki, S.; Tsuchiya, A.; Umehara, H.; Watanabe, S.Y.; Imoto, I.; Ohmori, T. Cumulative effect of the plasma total homocysteine-related genetic variants on schizophrenia risk. Psychiatry Res. 2016, 246, 833–837. [Google Scholar] [CrossRef]
- Numata, S.; Kinoshita, M.; Tajima, A.; Nishi, A.; Imoto, I.; Ohmori, T. Evaluation of an association between plasma total homocysteine and schizophrenia by a Mendelian randomization analysis. BMC Med. Genet. 2015, 16, 54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kinoshita, M.; Numata, S.; Tajima, A.; Shimodera, S.; Imoto, I.; Ohmori, T. Plasma total homocysteine is associated with DNA methylation in patients with schizophrenia. Epigenetics 2013, 8, 584–590. [Google Scholar] [CrossRef] [PubMed]
- Chia, S.C.; Henry, J.; Mok, Y.M.; Honer, W.G.; Sim, K. Fatty acid and vitamin interventions in adults with schizophrenia: A systematic review of the current evidence. J. Neural Transm. (Vienna) 2015, 122, 1721–1732. [Google Scholar] [CrossRef] [PubMed]
- Roffman, J.L.; Lamberti, J.S.; Achtyes, E.; Macklin, E.A.; Galendez, G.C.; Raeke, L.H.; Silverstein, N.J.; Smoller, J.W.; Hill, M.; Goff, D.C. Randomized multicenter investigation of folate plus vitamin B12 supplementation in schizophrenia. JAMA Psychiatry 2013, 70, 481–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacerda, A.L.; Keshavan, M.S.; Hardan, A.Y.; Yorbik, O.; Brambilla, P.; Sassi, R.B.; Nicoletti, M.; Mallinger, A.G.; Frank, E.; Kupfer, D.J.; et al. Anatomic evaluation of the orbitofrontal cortex in major depressive disorder. Biol. Psychiatry 2004, 55, 353–358. [Google Scholar] [CrossRef]
- Hasler, G. Pathophysiology of depression: Do we have any solid evidence of interest to clinicians? World Psychiatry 2010, 9, 155–161. [Google Scholar] [CrossRef] [Green Version]
- Copeland, W.E.; Adair, C.E.; Smetanin, P.; Stiff, D.; Briante, C.; Colman, I.; Fergusson, D.; Horwood, J.; Poulton, R.; Costello, E.J.; et al. Diagnostic transitions from childhood to adolescence to early adulthood. J. Child Psychol. Psychiatry 2013, 54, 791–799. [Google Scholar] [CrossRef]
- Djernes, J.K. Prevalence and predictors of depression in populations of elderly: A review. Acta Psychiatr. Scand. 2006, 113, 372–387. [Google Scholar] [CrossRef] [PubMed]
- Bottiglieri, T.; Laundy, M.; Crellin, R.; Toone, B.K.; Carney, M.W.; Reynolds, E.H. Homocysteine, folate, methylation, and monoamine metabolism in depression. J. Neurol. Neurosurg. Psychiatry 2000, 69, 228–232. [Google Scholar] [CrossRef]
- Cosar, A.; Ipcioglu, O.M.; Ozcan, O.; Gultepe, M. Folate and homocysteine metabolisms and their roles in the biochemical basis of neuropsychiatry. Turk. J. Med. Sci. 2014, 44, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Baek, J.H.; Bernstein, E.E.; Nierenberg, A.A. One-carbon metabolism and bipolar disorder. Aust. N. Z. J. Psychiatry 2013, 47, 1013–1018. [Google Scholar] [CrossRef]
- Coppen, A.; Bolander-Gouaille, C. Treatment of depression: Time to consider folic acid and vitamin B12. J. Psychopharmacol. 2005, 19, 59–65. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.S.Y.; Chen, H.T.; Soliman, K.F.A.; Charlton, C.G. Effects of homocysteine on the dopaminergic system and behavior in rodents. Neurotoxicology 2005, 26, 361–371. [Google Scholar] [CrossRef]
- Berk, M.; Dodd, S.; Kauer-Sant’Anna, M.; Malhi, G.S.; Bourin, M.; Kapczinski, F.; Norman, T. Dopamine dysregulation syndrome: Implications for a dopamine hypothesis of bipolar disorder. Acta Psychiatr. Scand. 2007, 116, 41–49. [Google Scholar] [CrossRef]
- Ghanizadeh, A.; Singh, A.B.; Berk, M.; Torabi-Nami, M. Homocysteine as a potential biomarker in bipolar disorders: A critical review and suggestions for improved studies. Expert Opin. Ther. Targets 2015, 19, 927–939. [Google Scholar] [CrossRef]
- Permoda-Osip, A.; Dorszewska, J.; Skibinska, M.; Chlopocka-Wozniak, M.; Rybakowski, J.K. Hyperhomocysteinemia in Bipolar Depression: Clinical and Biochemical Correlates. Neuropsychobiology 2013, 68, 193–196. [Google Scholar] [CrossRef]
- Moustafa, A.A.; Hewedi, D.H.; Eissa, A.M.; Frydecka, D.; Misiak, B. Homocysteine levels in schizophrenia and affective disorders—focus on cognition. Front. Behav. Neurosci. 2014, 8, 343. [Google Scholar] [CrossRef] [Green Version]
- Ezzaher, A.; Mouhamed, D.H.; Mechri, A.; Omezzine, A.; Neffati, F.; Douki, W.; Bouslama, A.; Gaha, L.; Najjar, M.F. Hyperhomocysteinemia in Tunisian bipolar I patients. Psychiatry Clin. Neurosci. 2011, 65, 664–671. [Google Scholar] [CrossRef] [PubMed]
- Rodrigo, C.; De Silva, N.L.; Gunaratne, R.; Rajapakse, S.; De Silva, V.A.; Hanwella, R. Lower Estimated Glomerular Filtration Rates in Patients on Long Term Lithium; a Comparative Study and a Meta-analysis of Literature. Eur. Psychiatry 2015, 30. [Google Scholar] [CrossRef] [Green Version]
- Enderle, J.; Klink, U.; di Giuseppe, R.; Koch, M.; Seidel, U.; Weber, K.; Birringer, M.; Ratjen, I.; Rimbach, G.; Lieb, W. Plasma Lithium Levels in the General Population: A Cross-Sectional Analysis of Metabolic and Dietary Correlates. Nutrients 2020, 12, 2489. [Google Scholar] [CrossRef] [PubMed]
- Ubeda, N.; Alonso-Aperte, E.; Varela-Moreiras, G. Acute valproate administration impairs methionine metabolism in rats. J. Nutr. 2002, 132, 2737–2742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lowenstein, C.J.; Dinerman, J.L.; Snyder, S.H. Nitric oxide: A physiologic messenger. Ann. Intern. Med. 1994, 120, 227–237. [Google Scholar] [CrossRef]
- Narayan, S.K.; Verman, A.; Kattimani, S.; Ananthanarayanan, P.H.; Adithan, C. Plasma homocysteine levels in depression and schizophrenia in South Indian Tamilian population. Indian J. Psychiatry 2014, 56, 46–53. [Google Scholar] [CrossRef]
- Misiak, B.; Frydecka, D.; Slezak, R.; Piotrowski, P.; Kiejna, A. Elevated homocysteine level in first-episode schizophrenia patients—The relevance of family history of schizophrenia and lifetime diagnosis of cannabis abuse. Metab. Brain Dis. 2014, 29, 661–670. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.Y.; Shek, C.C.; Wong, M.C.; Yip, K.C.; Ng, R.M.; Nguyen, D.G.; Poon, T.K. Homocysteine level in schizophrenia patients. Aust. N. Z. J. Psychiatry 2009, 43, 760–765. [Google Scholar] [CrossRef]
- Nishi, A.; Numata, S.; Tajima, A.; Kinoshita, M.; Kikuchi, K.; Shimodera, S.; Tomotake, M.; Ohi, K.; Hashimoto, R.; Imoto, I.; et al. Meta-analyses of blood homocysteine levels for gender and genetic association studies of the MTHFR C677T polymorphism in schizophrenia. Schizophr. Bull. 2014, 40, 1154–1163. [Google Scholar] [CrossRef] [Green Version]
- Kevere, L.; Purvina, S.; Bauze, D.; Zeibarts, M.; Andrezina, R.; Rizevs, A.; Jelisejevs, S.; Piekuse, L.; Kreile, M.; Purvins, I. Elevated serum levels of homocysteine as an early prognostic factor of psychiatric disorders in children and adolescents. Schizophr. Res. Treat. 2012, 2012, 373261. [Google Scholar] [CrossRef] [Green Version]
- Li, J.B.; Cheng, Y.C.; Shi, M.; Tang, J.R.; Dai, Q.; Zhang, Y.; Chen, J.W.; Wang, H.X. Association of homocysteine with peripheral neuropathy in Chinese patients with type 2 diabetes. Diabetes Res. Clin. Pract. 2011, 93, 38–42. [Google Scholar] [CrossRef]
- Belardo, A.; Gevi, F.; Zolla, L. The concomitant lower concentrations of vitamins B6, B9 and B12 may cause methylation deficiency in autistic children. J. Nutr. Biochem. 2019, 70, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Weir, D.G.; Keating, S.; Molloy, A.; Mcpartlin, J.; Kennedy, S.; Blanchflower, J.; Kennedy, D.G.; Rice, D.; Scott, J.M. Methylation Deficiency Causes Vitamin-B12-Associated Neuropathy in the Pig. J. Neurochem. 1988, 51, 1949–1952. [Google Scholar] [CrossRef] [PubMed]
- Varshney, K.K.; Gupta, J.K.; Mujwar, S. Homocysteine Induced Neurological Dysfunctions: A Link to Neurodegenerative Disorders. Int. J. Med. Res. Health Sci. 2019, 8, 135–146. [Google Scholar]
- Faverzani, J.L.; Hammerschmidt, T.G.; Sitta, A.; Deon, M.; Wajner, M.; Vargas, C.R. Oxidative Stress in Homocystinuria Due to Cystathionine -Synthase Deficiency: Findings in Patients and in Animal Models. Cell. Mol. Neurobiol. 2017, 37, 1477–1485. [Google Scholar] [CrossRef]
- Holmes, M.V.; Newcombe, P.; Hubacek, J.A.; Sofat, R.; Ricketts, S.L.; Cooper, J.; Breteler, M.M.B.; Bautista, L.E.; Sharma, P.; Whittaker, J.C.; et al. Effect modification by population dietary folate on the association between MTHFR genotype, homocysteine, and stroke risk: A meta-analysis of genetic studies and randomised trials. Lancet 2011, 378, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Casas, J.P.; Bautista, L.E.; Smeeth, L.; Sharma, P.; Hingorani, A.D. Homocysteine and stroke: Evidence on a causal link from mendelian randomisation. Lancet 2005, 365, 224–232. [Google Scholar] [CrossRef]
- Casas, J.P.; Bautista, L.E.; Hingorani, A.D.; Sharma, P. Plasma homocysteine, and ischaemic stroke: “Mendelian randomization” provides further evidence of causal link. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1218. [Google Scholar]
- Lehotsky, J.; Tothova, B.; Kovalska, M.; Dobrota, D.; Benova, A.; Kalenska, D.; Kaplan, P. Role of Homocysteine in the Ischemic Stroke and Development of Ischemic Tolerance. Front. Neurosci. 2016, 10. [Google Scholar] [CrossRef] [Green Version]
- Poddar, R.; Paul, S. Homocysteine-NMDA receptor-mediated activation of extracellular signal-regulated kinase leads to neuronal cell death. J. Neurochem. 2009, 110, 1095–1106. [Google Scholar] [CrossRef] [Green Version]
- Gorgone, G.; Caccamo, D.; Pisani, L.R.; Curro, M.; Parisi, G.; Oteri, G.; Ientile, R.; Rossini, P.M.; Pisani, F. Hyperhomocysteinemia in patients with epilepsy: Does it play a role in the pathogenesis of brain atrophy? A preliminary report. Epilepsia 2009, 50, 33–36. [Google Scholar] [CrossRef] [PubMed]
- Caccamo, D.; Condello, S.; Gorgone, G.; Crisafulli, G.; Belcastro, V.; Gennaro, S.; Striano, P.; Pisani, F.; Ientile, R. Screening for C677T and A1298C MTHFR polymorphisms in patients with epilepsy and risk of hyperhomocysteinemia. Neuromol. Med. 2004, 6, 117–126. [Google Scholar] [CrossRef]
- Folbergrova, J. Anticonvulsant action of both NMDA and Non-NMDA receptor antagonists against seizures induced by homocysteine in immature rats. Exp. Neurol. 1997, 145, 442–450. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Arrastia, R. Homocysteine and neurologic disease. Arch. Neurol. 2000, 57, 1422–1427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamed, S.A. The vascular risk associations with migraine: Relation to migraine susceptibility and progression. Atherosclerosis 2009, 205, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Kara, I.; Sazci, A.; Ergul, E.; Kaya, G.; Kilic, G. Association of the C677T and A1298C polymorphisms in the 5,10 methylenetetrahydrofolate reductase gene in patients with migraine risk. Mol. Brain Res. 2003, 111, 84–90. [Google Scholar] [CrossRef]
- Cacciapuoti, F. Migraine homocysteine-related: Old and new mechanisms. Neurol. Clin. Neurosci. 2017, 5, 137–140. [Google Scholar] [CrossRef]
- Al-Qasmi, M.M.; Athanas, K.; Dafer, R.M.; Tietjen, G.E. Von Willebrand Factor is elevated in migraineurs with aura, transient ischemic attacks, and stroke: A retrospective analysis. Neurology 2000, 54, A405. [Google Scholar]
- Hering-Hanit, R.; Friedman, Z.; Schlesinger, I.; Ellis, M. Evidence for activation of the coagulation system in migraine with aura. Cephalalgia 2001, 21, 137–139. [Google Scholar] [CrossRef]
- Scher, A.I.; Terwindt, G.M.; Verschuren, W.M.M.; Kruit, M.C.; Blom, H.J.; Kowa, H.; Frants, R.R.; Maagdenberg, A.M.J.M.V.D.; A Van Buchem, M.; Ferrari, M.D.; et al. Migraine and MTHFR C677T genotype in a population-based sample. Ann. Neurol. Off. J. Am. Neurol. Assoc. Child Neurol. Soc. 2006, 59, 372–375. [Google Scholar] [CrossRef]
- Hering-Hanit, R.; Friedman, Z.; Schlesinger, I.; Ellis, M. Activation of the coagulation system in migraine with aura. Cephalalgia 1999, 19, 342. [Google Scholar] [CrossRef] [PubMed]
- Edvinsson, L.; Uddman, R. Neurobiology in primary headaches. Brain Res. Rev. 2005, 48, 438–456. [Google Scholar] [CrossRef] [Green Version]
- Liampas, I.; Siokas, V.; Mentis, A.A.; Aloizou, A.; Dastamani, M.; Tsouris, Z.; Aslanidou, P.; Brotis, A.; Dardiotis, E. Serum Homocysteine, Pyridoxine, Folate, and Vitamin B12 Levels in Migraine: Systematic Review and Meta-Analysis. Headache J. Head Face Pain 2020, 60, 1508–1534. [Google Scholar] [CrossRef] [PubMed]
- Goadsby, P.J. Migraine and other primary headache disorders. In Pain: The Person, the Science, the Clinical Interface; BPA Print Group: Melbourne, Australia, 2015; Chapter 8; p. 107. [Google Scholar]
- Smith, A.D.; Kim, Y.I.; Refsum, H. Is folic acid good for everyone? Am. J. Clin. Nutr. 2008, 87, 517–533. [Google Scholar] [CrossRef] [Green Version]
- Goodwin, G.M.; Consensus Group of the British Association for Psychopharmacology. Evidence-based guidelines for treating bipolar disorder: Revised second edition—recommendations from the British Association for Psychopharmacology. J. Psychopharmacol. 2009, 23, 346–388. [Google Scholar] [CrossRef] [PubMed]
- Merikangas, K.R.; Akiskal, H.S.; Angst, J.; Greenberg, P.E.; Hirschfeld, R.M.A.; Petukhova, M.; Kessler, R.C. Lifetime and 12-month prevalence of bipolar spectrum disorder in the national comorbidity survey replication. Arch. Gen. Psychiatry 2007, 64, 543–552. [Google Scholar] [CrossRef]
- Merikangas, K.R.; Jin, R.; He, J.P.; Kessler, R.C.; Lee, S.; Sampson, N.A.; Viana, M.C.; Andrade, L.H.; Hu, C.Y.; Karam, E.G.; et al. Prevalence and Correlates of Bipolar Spectrum Disorder in the World Mental Health Survey Initiative. Arch. Gen. Psychiatry 2011, 68, 241–251. [Google Scholar] [CrossRef]
- Blaise, S.A.; Nedelec, E.; Alberto, J.M.; Schroeder, H.; Audonnet, S.; Bossenmeyer-Pourie, C.; Gueant, J.L.; Daval, J.L. Short hypoxia could attenuate the adverse effects of hyperhomocysteinemia on the developing rat brain by inducing neurogenesis. Exp. Neurol. 2009, 216, 231–238. [Google Scholar] [CrossRef]
- Hrncic, D.; Rasic-Markovic, A.; Lekovic, J.; Krstic, D.; Colovic, M.; Macut, D.; Susic, V.; Djuric, D.; Stanojlovic, O. Exercise Decreases Susceptibility to Homocysteine Seizures: The Role of Oxidative Stress. Int. J. Sports Med. 2014, 35, 544–550. [Google Scholar] [CrossRef]
- Oterino, A.; Valle, N.; Bravo, Y.; Munoz, P.; Sanchez-Velasco, P.; Ruiz-Alegria, C.; Castillo, J.; Leyva-Cobian, F.; Vadillo, A.; Pascual, J. MTHFR T677 homozygosis influences the presence of aura in migraineurs. Cephalalgia 2004, 24, 491–494. [Google Scholar] [CrossRef]
- Cacciapuoti, F. Lowering homocysteine levels may prevent cardiovascular impairments? Possible therapeutic behaviors. Blood Coagul. Fibrin. 2012, 23, 677–679. [Google Scholar] [CrossRef] [PubMed]
- Lippi, G.; Mattiuzzi, C.; Meschi, T.; Cervellin, G.; Borghi, L. Homocysteine and migraine. A narrative review. Clin. Chim. Acta 2014, 433, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Refsum, H.; Smith, A.D.; Ueland, P.M.; Nexo, E.; Clarke, R.; McPartlin, J.; Johnston, C.; Engbaek, F.; Schneede, J.; McPartlin, C.; et al. Facts and recommendations about total homocysteine determinations: An expert opinion. Clin. Chem. 2004, 50, 3–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Lim, I.K.; Park, G.H.; Paik, W.K. Biological methylation of myelin basic protein: Enzymology and biological significance. Int. J. Biochem. Cell B 1997, 29, 743–751. [Google Scholar] [CrossRef]
- Reynolds, E. Vitamin B12, folic acid, and the nervous system. Lancet Neurol. 2006, 5, 949–960. [Google Scholar] [CrossRef]
- Kruman, I.I.; Culmsee, C.; Chan, S.L.; Kruman, Y.; Guo, Z.H.; Penix, L.; Mattson, M.P. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J. Neurosci. 2000, 20, 6920–6926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sengupta, S.; Wehbe, C.; Majors, A.K.; Ketterer, M.E.; DiBello, P.M.; Jacobsen, D.W. Relative roles of albumin and ceruloplasmin in the formation of homocystine, homocysteine-cysteine-mixed disulfide, and cystine in circulation. J. Biol. Chem. 2001, 276, 46896–46904. [Google Scholar] [CrossRef] [Green Version]
- Au-Yeung, K.K.W.; Yip, J.C.W.; Siow, Y.L.; Karmin, O. Folic acid inhibits homocysteine-induced superoxide anion production and nuclear factor kappa B activation in macrophages. Can. J. Physiol. Pharm. 2006, 84, 141–147. [Google Scholar] [CrossRef]
- Tajouri, L.; Martin, V.; Gasparini, C.; Ovcaric, M.; Curtain, R.; Lea, R.A.; Haupt, L.M.; Csurhes, P.; Pender, M.P.; Griffiths, L.R. Genetic investigation of methylenetetrahydrofolate reductase (MTHFR) and catechol-O-methyl transferase (COMT) in multiple sclerosis. Brain Res. Bull. 2006, 69, 327–331. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.S.; Zhou, J.; Wong, P.W.K.; Kowalisyn, J.; Strokosch, G. Intermediate Homocysteinemia—A Thermolabile Variant of Methylenetetrahydrofolate Reductase. Am. J. Hum. Genet. 1988, 43, 414–421. [Google Scholar]
- Szvetko, A.L.; Fowdar, J.; Nelson, J.; Colson, N.; Tajouri, L.; Csurhes, P.A.; Pender, M.P.; Griffiths, L.R. No association between MTHFR A1298C and MTRR A66G polymorphisms, and MS in an Australian cohort. J. Neurol. Sci. 2007, 252, 49–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, S.R.; Flauzino, T.; Sabino, B.S.; Kallaur, A.P.; Alfieri, D.F.; Kaimen-Maciel, D.R.; Morimoto, H.K.; de Almeida, E.R.D.; Lozovoy, M.A.B.; Reiche, E.M.V.; et al. Elevated plasma homocysteine levels are associated with disability progression in patients with multiple sclerosis. Metab. Brain Dis. 2018, 33, 1393–1399. [Google Scholar] [CrossRef] [PubMed]
- Kararizou, E.; Paraskevas, G.; Triantafyllou, N.; Koutsis, G.; Evangelopoulos, M.E.; Mandellos, D.; Sfagos, C.; Kapaki, E. Plasma homocysteine levels in patients with multiple sclerosis in the Greek population. J. Chin. Med Assoc. 2013, 76, 611–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zoccolella, S.; Tortorella, C.; Iaffaldano, P.; Direnzo, V.; D’Onghia, M.; Paolicelli, D.; Livrea, P.; Trojano, M. Elevated plasma homocysteine levels in patients with multiple sclerosis are associated with male gender. J. Neurol. 2012, 259, 2105–2110. [Google Scholar] [CrossRef] [PubMed]
- Akanji, A.O.; Al-Shammri, S. Plasma homocysteine and C-reactive protein levels in relation to disease progression in multiple sclerosis. Clin. Chem. 2008, 54, A76. [Google Scholar]
- Sahin, S.; Aksungar, F.; Topkaya, A.; Yildiz, Z.; Boru, U.; Ayalpl, S.; Karsidag, S. Increased plasma homocysteine levels in multiple sclerosis. Mult. Scler. J. 2007, 13, 945–946. [Google Scholar] [CrossRef]
- Ramsaransing, G.M.; Fokkema, M.R.; Teelken, A.; Arutiunyan, A.V.; Koch, M.; De Keyser, J. Plasma homocysteine levels in multiple sclerosis. J. Neurol. Neurosur. Psychiatry 2006, 77, 189–192. [Google Scholar] [CrossRef] [Green Version]
- Besler, H.T.; Comoglu, S. Lipoprotein oxidation, plasma total antioxidant capacity and homocysteine level in patients with multiple sclerosis. Nutr. Neurosci. 2003, 6, 189–196. [Google Scholar] [CrossRef]
- Vrethem, M.; Mattsson, E.; Hebelka, H.; Leerbeck, K.; Osterberg, A.; Landtblom, A.M.; Balla, B.; Nilsson, H.; Hultgren, M.; Brattstrom, L.; et al. Increased plasma homocysteine levels without signs of vitamin B-12 deficiency in patients with multiple sclerosis assessed by blood and cerebrospinal fluid homocysteine and methylmalonic acid. Mult. Scler. 2003, 9, 239–245. [Google Scholar] [CrossRef]
- Li, X.T.; Yuan, J.L.; Han, J.M.; Hu, W.L. Serum levels of Homocysteine, Vitamin B12 and Folate in Patients with Multiple Sclerosis: An Updated Meta-Analysis. Int. J. Med Sci. 2020, 17, 751–761. [Google Scholar] [CrossRef] [Green Version]
- Onar, M.K.; Yazici, T. Serum Homocysteine Levels and Cognitive Dysfunction in Multiple Sclerosis Patients. Mult. Scler. J. 2018, 24, 391. [Google Scholar]
- Fahmy, E.M.; Elfayoumy, N.M.; Abdelalim, A.M.; Sharaf, S.A.A.; Ismail, R.S.; Elshebawy, H. Relation of serum levels of homocysteine, vitamin B12 and folate to cognitive functions in multiple sclerosis patients. Int. J. Neurosci. 2018, 128, 835–841. [Google Scholar] [CrossRef] [PubMed]
- Kocer, B.; Engur, S.; Ak, F.; Yilmaz, M. Serum vitamin B12, folate, and homocysteine levels and their association with clinical and electrophysiological parameters in multiple sclerosis. J. Clin. Neurosci. 2009, 16, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Teunissen, C.E.; Killestein, J.; Kragt, J.J.; Polman, C.H.; Dijkstra, C.D.; Blom, H.J. Serum homocysteine levels in relation to clinical progression in multiple sclerosis. J. Neurol. Neurosur. Psychiatry 2008, 79, 1349–1353. [Google Scholar] [CrossRef]
- Ashtari, F.; Shaygannejad, V. Serum homocysteine level in patients with multiple sclerosis. Eur. J. Neurol. 2006, 13, 236. [Google Scholar]
- Rio, J.; Montalban, J.; Tintore, M.; Codina, A.; Malinow, M.R. Serum Homocysteine Levels in Multiple-Sclerosis. Arch. Neurol. Chic. 1994, 51, 1181. [Google Scholar] [CrossRef]
- Stabler, S.P.; Estacio, R.; Jeffers, B.W.; Cohen, J.A.; Allen, R.H.; Schrier, R.W. Total homocysteine is associated with nephropathy in non-insulin-dependent diabetes mellitus. Metabolism 1999, 48, 1096–1101. [Google Scholar] [CrossRef]
- Cronin, C.C.; McPartlin, J.M.; Barry, D.G.; Ferriss, J.B.; Scott, J.M.; Weir, D.G. Plasma homocysteine concentrations in patients with type 1 diabetes. Diabetes Care 1998, 21, 1843–1847. [Google Scholar] [CrossRef]
- Ambrosch, A.; Dierkes, J.; Lobmann, R.; Kuhne, W.; Konig, W.; Luley, C.; Lehnert, H. Relation between homocysteinaemia and diabetic neuropathy in patients with Type 2 diabetes mellitus. Diabet. Med. 2001, 18, 185–192. [Google Scholar] [CrossRef]
- Buysschaert, M.; Dramais, A.S.; Wallemacq, P.E.; Hermans, M.P. Hyperhomocysteinemia in type 2 diabetes: Relationship to macroangiopathy, nephropathy, and insulin resistance. Diabetes Care 2000, 23, 1816–1822. [Google Scholar] [CrossRef] [Green Version]
- Chico, A.; Perez, A.; Cordoba, A.; Arcelus, R.; Carreras, G.; de Leiva, A.; Gonzalez-Sastre, F.; Blanco-Vaca, F. Plasma homocysteine is related to albumin excretion rate in patients with diabetes mellitus: A new link between diabetic nephropathy and cardiovascular disease? Diabetologia 1998, 41, 684–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogeveen, E.K.; Kostense, P.J.; Valk, G.D.; Bertelsmann, F.W.; Jakobs, C.; Dekker, J.M.; Nijpels, G.; Heine, R.J.; Bouter, L.M.; Stehouwer, C.D. Hyperhomocysteinaemia is not related to risk of distal somatic polyneuropathy: The Hoorn Study. J. Intern. Med. 1999, 246, 561–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganapathy, P.S.; White, R.E.; Ha, Y.; Bozard, B.R.; McNeil, P.L.; Caldwell, R.W.; Kumar, S.; Black, S.M.; Smith, S.B. The role of N-methyl-D-aspartate receptor activation in homocysteine-induced death of retinal ganglion cells. Invest. Ophthalmol. Vis. Sci. 2011, 52, 5515–5524. [Google Scholar] [CrossRef] [PubMed]
- Lipton, S.A.; Kim, W.K.; Choi, Y.B.; Kumar, S.; D’Emilia, D.M.; Rayudu, P.V.; Arnelle, D.R.; Stamler, J.S. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1997, 94, 5923–5928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chmiel-Perzynska, I.; Perzynski, A.; Wielosz, M.; Urbanska, E.M. Hyperglycemia enhances the inhibitory effect of mitochondrial toxins and D,L-homocysteine on the brain production of kynurenic acid. Pharmacol. Rep. 2007, 59, 268–273. [Google Scholar] [PubMed]
- Munipally, P.K.; Agraharm, S.G.; Valavala, V.K.; Gundae, S.; Turlapati, N.R. Evaluation of indoleamine 2,3-dioxygenase expression and kynurenine pathway metabolites levels in serum samples of diabetic retinopathy patients. Arch. Physiol. Biochem. 2011, 117, 254–258. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathology | Reference |
|---|---|
| Alzheimer’s Disease | [79,80,81,82,83,84,85,86] |
| Parkinson’s Disease | [81,87,88,89,90,91,92] |
| Autism | [93,94,95,96,97,98,99,100,101,102,103] |
| Schizophrenia | [104,105,106,107,108,109,110,111,112,113,114,115,116,117] |
| Major Depressive and Bipolar Disorder | [118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134] |
| Vascular Dementia | [110,111,112,114,135,136,137,138,139,140] |
| Peripheral Neuritis | [80,81,141,142,143,144] |
| Stroke | [80,81,145,146,147,148,149] |
| Epilepsy | [150,151,152,153,154] |
| Headache | [155,156,157,158,159,160,161,162,163,164] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cordaro, M.; Siracusa, R.; Fusco, R.; Cuzzocrea, S.; Di Paola, R.; Impellizzeri, D. Involvements of Hyperhomocysteinemia in Neurological Disorders. Metabolites 2021, 11, 37. https://doi.org/10.3390/metabo11010037
Cordaro M, Siracusa R, Fusco R, Cuzzocrea S, Di Paola R, Impellizzeri D. Involvements of Hyperhomocysteinemia in Neurological Disorders. Metabolites. 2021; 11(1):37. https://doi.org/10.3390/metabo11010037
Chicago/Turabian StyleCordaro, Marika, Rosalba Siracusa, Roberta Fusco, Salvatore Cuzzocrea, Rosanna Di Paola, and Daniela Impellizzeri. 2021. "Involvements of Hyperhomocysteinemia in Neurological Disorders" Metabolites 11, no. 1: 37. https://doi.org/10.3390/metabo11010037
APA StyleCordaro, M., Siracusa, R., Fusco, R., Cuzzocrea, S., Di Paola, R., & Impellizzeri, D. (2021). Involvements of Hyperhomocysteinemia in Neurological Disorders. Metabolites, 11(1), 37. https://doi.org/10.3390/metabo11010037