Bioactive Ether Lipids: Primordial Modulators of Cellular Signaling




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Ether Lipid Biosynthesis
1.2. Ether Lipid Physical Characteristics
1.3. Signaling Ether Lipids in Bacteria
1.4. Mammalian Ether Lipid Signaling: PAF
PAF Biosynthesis and Signaling
1.5. PAF-Like Signaling Lipids
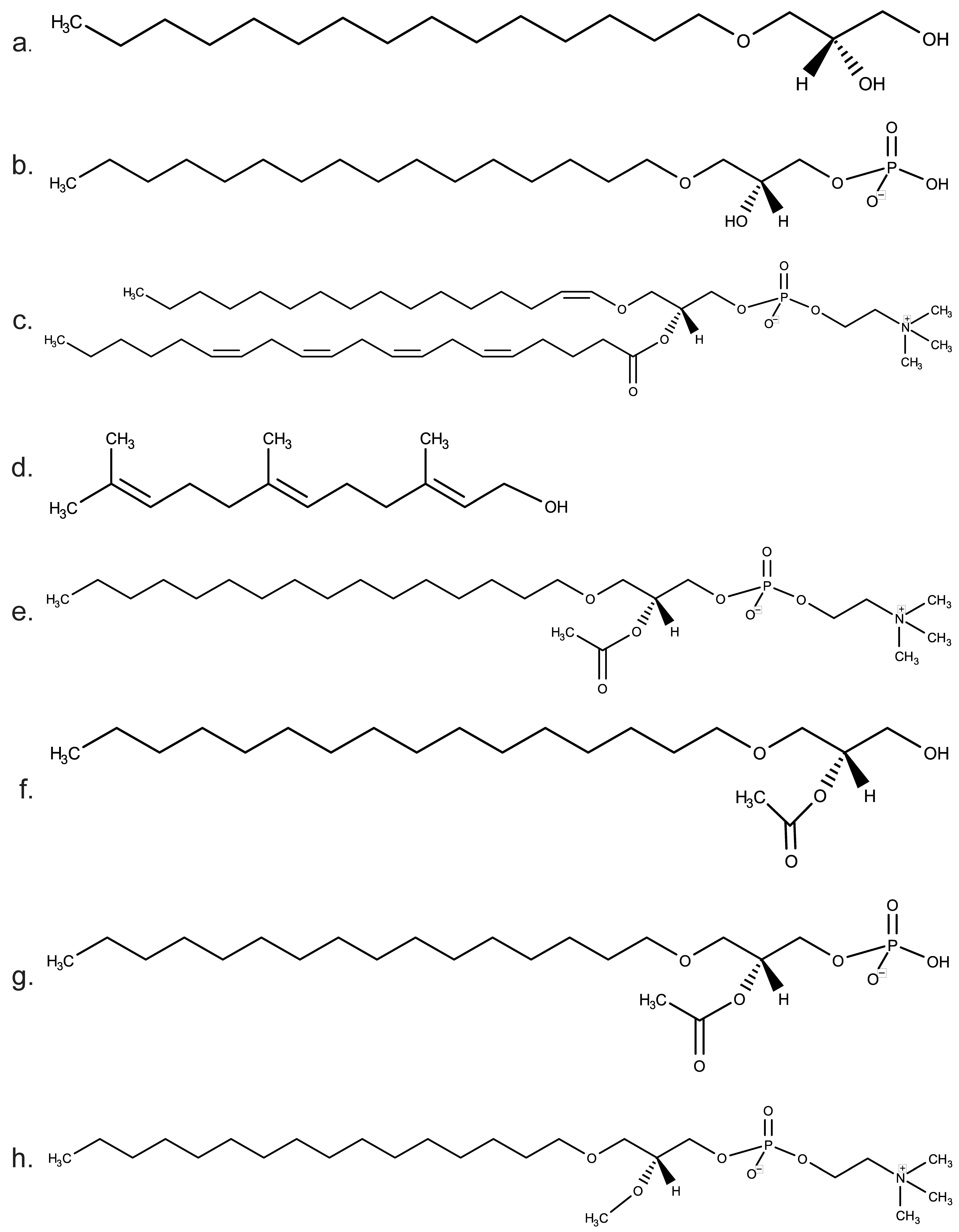
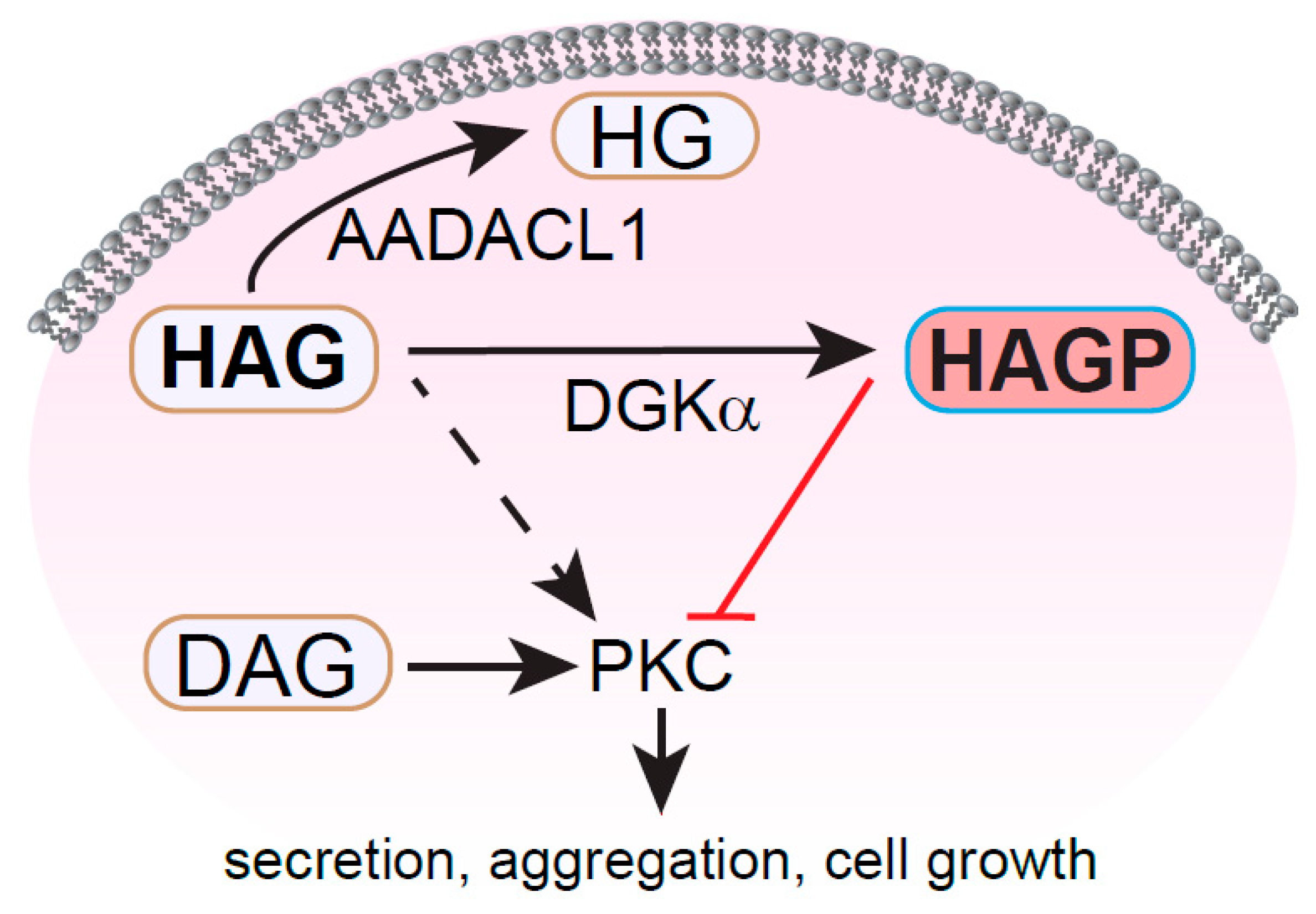
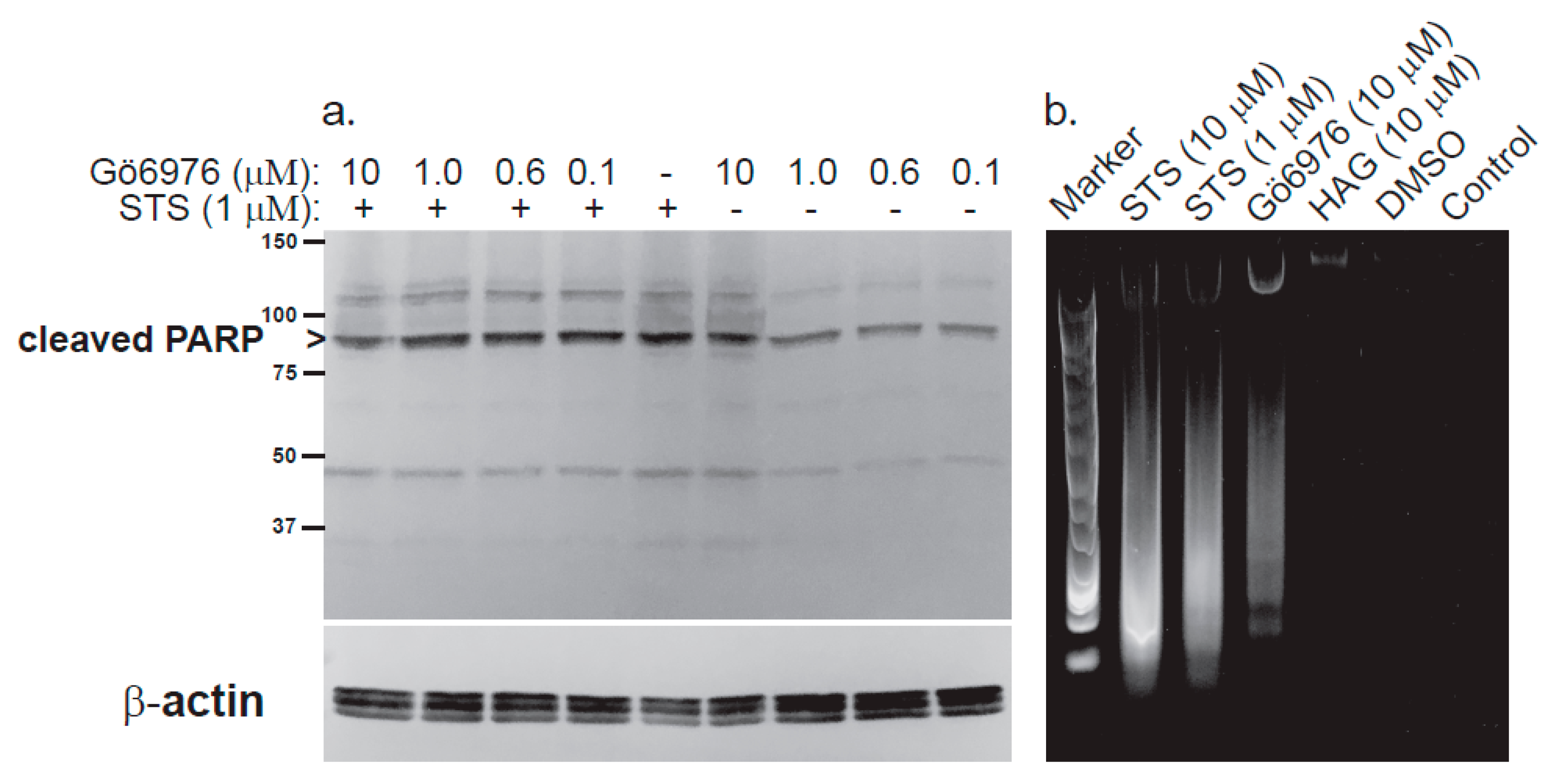
1.6. HAG and HG
1.7. HAGP
1.8. Ether Lipids and Cancer
1.8.1. Alkyl-LPA
1.8.2. Synthetic Anticancer Ether Lipids
1.8.3. Edelfosine
1.8.4. Perifosine
1.9. Ether Lipid Metabolism and Other Lipid Pathways
1.10. Plasmalogens
2. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lopez-Lara, I.M.; Geiger, O. Bacterial lipid diversity. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2017, 1862, 1287–1299. [Google Scholar] [CrossRef] [PubMed]
- Fahy, E.; Subramaniam, S.; Brown, H.A.; Glass, C.K.; Merrill, A.H., Jr.; Murphy, R.C.; Raetz, C.R.; Russell, D.W.; Seyama, Y.; Shaw, W.; et al. A comprehensive classification system for lipids. J. Lipid Res. 2005, 46, 839–861. [Google Scholar] [CrossRef] [Green Version]
- Magnusson, C.D.; Haraldsson, G.G. Ether lipids. Chem. Phys. Lipids 2011, 164, 315–340. [Google Scholar] [CrossRef]
- von Euler, U.S. On the specific vaso-dilating and plain muscle stimulating substances from accessory genital glands in man and certain animals (prostaglandin and vesiglandin). J. Physiol. 1936, 88, 213–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, V.B.; Aldrovandi, M.; Murphy, R.C.; Kronke, G. Enzymatically oxidized phospholipids assume center stage as essential regulators of innate immunity and cell death. Sci. Signal. 2019, 12, eaau2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Signini, E.F.; Nieman, D.C.; Silva, C.D.; Sakaguchi, C.A.; Catai, A.M. Oxylipin Response to Acute and Chronic Exercise: A Systematic Review. Metabolites 2020, 10, 264. [Google Scholar] [CrossRef]
- Yeung, J.; Hawley, M.; Holinstat, M. The expansive role of oxylipins on platelet biology. J. Mol. Med. 2017, 95, 575–588. [Google Scholar] [CrossRef] [Green Version]
- Bhat, S.; Ahrendt, T.; Dauth, C.; Bode, H.B.; Shimkets, L.J. Two lipid signals guide fruiting body development of Myxococcus xanthus. mBio 2014, 5, e00939-13. [Google Scholar] [CrossRef] [Green Version]
- Hoiczyk, E.; Ring, M.W.; McHugh, C.A.; Schwar, G.; Bode, E.; Krug, D.; Altmeyer, M.O.; Lu, J.Z.; Bode, H.B. Lipid body formation plays a central role in cell fate determination during developmental differentiation of Myxococcus xanthus. Mol. Microbiol. 2009, 74, 497–517. [Google Scholar] [CrossRef] [Green Version]
- Lorenzen, W.; Ahrendt, T.; Bozhuyuk, K.A.; Bode, H.B. A multifunctional enzyme is involved in bacterial ether lipid biosynthesis. Nat. Chem. Biol. 2014, 10, 425–427. [Google Scholar] [CrossRef]
- Ring, M.W.; Schwar, G.; Thiel, V.; Dickschat, J.S.; Kroppenstedt, R.M.; Schulz, S.; Bode, H.B. Novel iso-branched ether lipids as specific markers of developmental sporulation in the myxobacterium Myxococcus xanthus. J. Biol. Chem. 2006, 281, 36691–36700. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dean, J.M.; Lodhi, I.J. Structural and functional roles of ether lipids. Protein Cell 2018, 9, 196–206. [Google Scholar] [CrossRef] [PubMed]
- Dorninger, F.; Forss-Petter, S.; Wimmer, I.; Berger, J. Plasmalogens, platelet-activating factor and beyond—Ether lipids in signaling and neurodegeneration. Neurobiol. Dis. 2020, 145, 105061. [Google Scholar] [CrossRef]
- Jimenez-Rojo, N.; Riezman, H. On the road to unraveling the molecular functions of ether lipids. FEBS Lett. 2019, 593, 2378–2389. [Google Scholar] [CrossRef] [Green Version]
- Braverman, N.E.; Moser, A.B. Functions of plasmalogen lipids in health and disease. Biochim. Biophys. Acta 2012, 1822, 1442–1452. [Google Scholar] [CrossRef] [Green Version]
- Brites, P.; Waterham, H.R.; Wanders, R.J. Functions and biosynthesis of plasmalogens in health and disease. Biochim. Biophys. Acta 2004, 1636, 219–231. [Google Scholar] [CrossRef]
- Hillebrand, M.; Gersting, S.W.; Lotz-Havla, A.S.; Schafer, A.; Rosewich, H.; Valerius, O.; Muntau, A.C.; Gartner, J. Identification of a new fatty acid synthesis-transport machinery at the peroxisomal membrane. J. Biol. Chem. 2012, 287, 210–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benjamin, D.I.; Cozzo, A.; Ji, X.; Roberts, L.S.; Louie, S.M.; Mulvihill, M.M.; Luo, K.; Nomura, D.K. Ether lipid generating enzyme AGPS alters the balance of structural and signaling lipids to fuel cancer pathogenicity. Proc. Natl. Acad. Sci. USA 2013, 110, 14912–14917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorninger, F.; Konig, T.; Scholze, P.; Berger, M.L.; Zeitler, G.; Wiesinger, C.; Gundacker, A.; Pollak, D.D.; Huck, S.; Just, W.W.; et al. Disturbed neurotransmitter homeostasis in ether lipid deficiency. Hum. Mol. Genet. 2019, 28, 2046–2061. [Google Scholar] [CrossRef] [PubMed]
- Lodhi, I.J.; Yin, L.; Jensen-Urstad, A.P.; Funai, K.; Coleman, T.; Baird, J.H.; El Ramahi, M.K.; Razani, B.; Song, H.; Fu-Hsu, F.; et al. Inhibiting adipose tissue lipogenesis reprograms thermogenesis and PPARgamma activation to decrease diet-induced obesity. Cell Metab. 2012, 16, 189–201. [Google Scholar] [CrossRef] [Green Version]
- Mueller, P.; Ye, S.; Morris, A.; Smyth, S.S. Lysophospholipid mediators in the vasculature. Exp. Cell Res. 2015, 333, 190–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magkrioti, C.; Galaris, A.; Kanellopoulou, P.; Stylianaki, E.A.; Kaffe, E.; Aidinis, V. Autotaxin and chronic inflammatory diseases. J. Autoimmun. 2019, 104, 102327. [Google Scholar] [CrossRef] [PubMed]
- Horibata, Y.; Elpeleg, O.; Eran, A.; Hirabayashi, Y.; Savitzki, D.; Tal, G.; Mandel, H.; Sugimoto, H. EPT1 (selenoprotein I) is critical for the neural development and maintenance of plasmalogen in humans. J. Lipid Res. 2018, 59, 1015–1026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, M.Y.; Al-Khayat, A.; Al-Murshedi, F.; Al-Futaisi, A.; Chioza, B.A.; Pedro Fernandez-Murray, J.; Self, J.E.; Salter, C.G.; Harlalka, G.V.; Rawlins, L.E.; et al. A mutation of EPT1 (SELENOI) underlies a new disorder of Kennedy pathway phospholipid biosynthesis. Brain 2017, 140, 547–554. [Google Scholar]
- Gallego-Garcia, A.; Monera-Girona, A.J.; Pajares-Martinez, E.; Bastida-Martinez, E.; Perez-Castano, R.; Iniesta, A.A.; Fontes, M.; Padmanabhan, S.; Elias-Arnanz, M. A bacterial light response reveals an orphan desaturase for human plasmalogen synthesis. Science 2019, 366, 128–132. [Google Scholar] [CrossRef]
- Werner, E.R.; Keller, M.A.; Sailer, S.; Lackner, K.; Koch, J.; Hermann, M.; Coassin, S.; Golderer, G.; Werner-Felmayer, G.; Zoeller, R.A.; et al. The TMEM189 gene encodes plasmanylethanolamine desaturase which introduces the characteristic vinyl ether double bond into plasmalogens. Proc. Natl. Acad. Sci. USA 2020, 117, 7792–7798. [Google Scholar] [CrossRef] [Green Version]
- Horibata, Y.; Ando, H.; Sugimoto, H. Locations and contributions of the phosphotransferases EPT1 and CEPT1 to the biosynthesis of ethanolamine phospholipids. J. Lipid Res. 2020, 61, 1221–1231. [Google Scholar] [CrossRef]
- Grossi, V.; Mollex, D.; Vincon-Laugier, A.; Hakil, F.; Pacton, M.; Cravo-Laureau, C. Mono- and dialkyl glycerol ether lipids in anaerobic bacteria: Biosynthetic insights from the mesophilic sulfate reducer Desulfatibacillum alkenivorans PF2803T. Appl. Environ. Microbiol. 2015, 81, 3157–3168. [Google Scholar] [CrossRef] [Green Version]
- Valentine, D.L. Adaptations to energy stress dictate the ecology and evolution of the Archaea. Nat. Rev. Microbiol. 2007, 5, 316–323. [Google Scholar] [CrossRef]
- Bergan, J.; Skotland, T.; Lingelem, A.B.; Simm, R.; Spilsberg, B.; Lindback, T.; Sylvanne, T.; Simolin, H.; Ekroos, K.; Sandvig, K. The ether lipid precursor hexadecylglycerol protects against Shiga toxins. Cell Mol. Life Sci. 2014, 71, 4285–4300. [Google Scholar] [CrossRef]
- Ahrendt, T.; Wolff, H.; Bode, H.B. Neutral and Phospholipids of the Myxococcus xanthus Lipodome during Fruiting Body Formation and Germination. Appl. Environ. Microbiol. 2015, 81, 6538–6547. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benveniste, J.; Henson, P.M.; Cochrane, C.G. Leukocyte-dependent histamine release from rabbit platelets. The role of IgE, basophils, and a platelet-activating factor. J. Exp. Med. 1972, 136, 1356–1377. [Google Scholar] [CrossRef] [PubMed]
- Blank, M.L.; Snyder, F.; Byers, L.W.; Brooks, B.; Muirhead, E.E. Antihypertensive activity of an alkyl ether analog of phosphatidylcholine. Biochem. Biophys. Res. Commun. 1979, 90, 1194–1200. [Google Scholar] [CrossRef]
- Muirhead, E.E.; Byers, L.W.; Pitcock, J.A.; Desiderio, D.M.; Brooks, B.; Brown, P.; Brosius, W.L. Derivation of neutral antihypertensive lipid from renal venous effluent in rats. Clin. Sci. 1981, 61 (Suppl. 7), 331s–333s. [Google Scholar] [CrossRef]
- Prewitt, R.L.; Leach, B.E.; Byers, L.W.; Brooks, B.; Lands, W.E.; Muirhead, E.E. Antihypertensive polar renomedullary lipid, a semisynthetic vasodilator. Hypertension 1979, 1, 299–308. [Google Scholar] [CrossRef] [Green Version]
- Lordan, R.; Tsoupras, A.; Zabetakis, I.; Demopoulos, C.A. Forty Years Since the Structural Elucidation of Platelet-Activating Factor (PAF): Historical, Current, and Future Research Perspectives. Molecules 2019, 24, 4414. [Google Scholar] [CrossRef] [Green Version]
- Marathe, G.K.; Pandit, C.; Lakshmikanth, C.L.; Chaithra, V.H.; Jacob, S.P.; D’Souza, C.J. To hydrolyze or not to hydrolyze: The dilemma of platelet-activating factor acetylhydrolase. J. Lipid Res. 2014, 55, 1847–1854. [Google Scholar] [CrossRef] [Green Version]
- Prescott, S.M.; Zimmerman, G.A.; Stafforini, D.M.; McIntyre, T.M. Platelet-activating factor and related lipid mediators. Annu. Rev. Biochem. 2000, 69, 419–445. [Google Scholar] [CrossRef]
- Snyder, F. The ether lipid trail: A historical perspective. Biochim. Biophys. Acta 1999, 1436, 265–278. [Google Scholar] [CrossRef]
- Lee, T.C.; Malone, B.; Blank, M.L.; Fitzgerald, V.; Snyder, F. Regulation of the synthesis of platelet-activating factor and its inactive storage precursor (1-alkyl-2-acyl-sn-glycero-3-phosphocholine) from 1-alkyl-2-acetyl-sn-glycerol by rabbit platelets. J. Biol. Chem. 1990, 265, 9181–9187. [Google Scholar] [CrossRef]
- Bonventre, J.V.; Huang, Z.; Taheri, M.R.; O’Leary, E.; Li, E.; Moskowitz, M.A.; Sapirstein, A. Reduced fertility and postischaemic brain injury in mice deficient in cytosolic phospholipase A2. Nature 1997, 390, 622–625. [Google Scholar] [CrossRef] [PubMed]
- Uozumi, N.; Kume, K.; Nagase, T.; Nakatani, N.; Ishii, S.; Tashiro, F.; Komagata, Y.; Maki, K.; Ikuta, K.; Ouchi, Y.; et al. Role of cytosolic phospholipase A2 in allergic response and parturition. Nature 1997, 390, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Renooij, W.; Snyder, F. Biosynthesis of 1-alkyl-2-acetyl-sn-glycero-3-phosphocholine (platelet activating factor and a hypotensive lipid) by cholinephosphotransferase in various rat tissues. Biochim. Biophys. Acta 1981, 663, 545–556. [Google Scholar] [CrossRef]
- Tsoupras, A.B.; Fragopoulou, E.; Nomikos, T.; Iatrou, C.; Antonopoulou, S.; Demopoulos, C.A. Characterization of the de novo biosynthetic enzyme of platelet activating factor, DDT-insensitive cholinephosphotransferase, of human mesangial cells. Mediat. Inflamm. 2007, 2007, 27683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henneberry, A.L.; Wistow, G.; McMaster, C.R. Cloning, genomic organization, and characterization of a human cholinephosphotransferase. J. Biol. Chem. 2000, 275, 29808–29815. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Detopoulou, P.; Nomikos, T.; Fragopoulou, E.; Antonopoulou, S.; Kotroyiannis, I.; Vassiliadou, C.; Panagiotakos, D.B.; Chrysohoou, C.; Pitsavos, C.; Stefanadis, C. Platelet activating factor (PAF) and activity of its biosynthetic and catabolic enzymes in blood and leukocytes of male patients with newly diagnosed heart failure. Clin. Biochem. 2009, 42, 44–49. [Google Scholar] [CrossRef]
- Lynch, J.M.; Henson, P.M. The intracellular retention of newly synthesized platelet-activating factor. J. Immunol. 1986, 137, 2653–2661. [Google Scholar]
- McIntyre, T.M.; Zimmerman, G.A.; Satoh, K.; Prescott, S.M. Cultured endothelial cells synthesize both platelet-activating factor and prostacyclin in response to histamine, bradykinin, and adenosine triphosphate. J. Clin. Investig. 1985, 76, 271–280. [Google Scholar] [CrossRef]
- Valone, F.H. Isolation of a platelet membrane protein which binds the platelet-activating factor 1-0-hexadecyl-2-acetyl-SN-glycero-3-phosphorylcholine. Immunology 1984, 52, 169–174. [Google Scholar]
- Vercellotti, G.M.; Yin, H.Q.; Gustafson, K.S.; Nelson, R.D.; Jacob, H.S. Platelet-activating factor primes neutrophil responses to agonists: Role in promoting neutrophil-mediated endothelial damage. Blood 1988, 71, 1100–1107. [Google Scholar] [CrossRef]
- Elstad, M.R.; Prescott, S.M.; McIntyre, T.M.; Zimmerman, G.A. Synthesis and release of platelet-activating factor by stimulated human mononuclear phagocytes. J. Immunol. 1988, 140, 1618–1624. [Google Scholar] [PubMed]
- Heller, R.; Bussolino, F.; Ghigo, D.; Garbarino, G.; Pescarmona, G.; Till, U.; Bosia, A. Human endothelial cells are target for platelet-activating factor. II. Platelet-activating factor induces platelet-activating factor synthesis in human umbilical vein endothelial cells. J. Immunol. 1992, 149, 3682–3688. [Google Scholar] [PubMed]
- Ostrovsky, L.; King, A.J.; Bond, S.; Mitchell, D.; Lorant, D.E.; Zimmerman, G.A.; Larsen, R.; Niu, X.F.; Kubes, P. A juxtacrine mechanism for neutrophil adhesion on platelets involves platelet-activating factor and a selectin-dependent activation process. Blood 1998, 91, 3028–3036. [Google Scholar] [CrossRef] [PubMed]
- Amatruda, T.T., 3rd; Gerard, N.P.; Gerard, C.; Simon, M.I. Specific interactions of chemoattractant factor receptors with G-proteins. J. Biol. Chem. 1993, 268, 10139–10144. [Google Scholar] [CrossRef]
- Brown, S.L.; Jala, V.R.; Raghuwanshi, S.K.; Nasser, M.W.; Haribabu, B.; Richardson, R.M. Activation and regulation of platelet-activating factor receptor: Role of G(i) and G(q) in receptor-mediated chemotactic, cytotoxic, and cross-regulatory signals. J. Immunol. 2006, 177, 3242–3249. [Google Scholar] [CrossRef]
- Shi, L.C.; Wang, H.Y.; Horwitz, J.; Friedman, E. Guanine nucleotide regulatory proteins, Gq and Gi1/2, mediate platelet-activating factor-stimulated phosphoinositide metabolism in immortalized hippocampal cells. J. Neurochem. 1996, 67, 1478–1484. [Google Scholar] [CrossRef]
- Yue, T.L.; Stadel, J.M.; Sarau, H.M.; Friedman, E.; Gu, J.L.; Powers, D.A.; Gleason, M.M.; Feuerstein, G.; Wang, H.Y. Platelet-activating factor stimulates phosphoinositide turnover in neurohybrid NCB-20 cells: Involvement of pertussis toxin-sensitive guanine nucleotide-binding proteins and inhibition by protein kinase C. Mol. Pharm. 1992, 41, 281–289. [Google Scholar]
- McMeekin, S.R.; Dransfield, I.; Rossi, A.G.; Haslett, C.; Walker, T.R. E-selectin permits communication between PAF receptors and TRPC channels in human neutrophils. Blood 2006, 107, 4938–4945. [Google Scholar] [CrossRef] [Green Version]
- Bye, A.P.; Unsworth, A.J.; Gibbins, J.M. Platelet signaling: A complex interplay between inhibitory and activatory networks. J. Thromb. Haemost. 2016, 14, 918–930. [Google Scholar] [CrossRef] [Green Version]
- Stefanini, L.; Paul, D.S.; Robledo, R.F.; Chan, E.R.; Getz, T.M.; Campbell, R.A.; Kechele, D.O.; Casari, C.; Piatt, R.; Caron, K.M.; et al. RASA3 is a critical inhibitor of RAP1-dependent platelet activation. J. Clin. Investig. 2015, 125, 1419–1432. [Google Scholar] [CrossRef] [Green Version]
- Stefanini, L.; Roden, R.C.; Bergmeier, W. CalDAG-GEFI is at the nexus of calcium-dependent platelet activation. Blood 2009, 114, 2506–2514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lagarrigue, F.; Paul, D.S.; Gingras, A.R.; Valadez, A.J.; Sun, H.; Lin, J.; Cuevas, M.N.; Ablack, J.N.; Lopez-Ramirez, M.A.; Bergmeier, W.; et al. Talin-1 is the principal platelet Rap1 effector of integrin activation. Blood 2020, 136, 1180–1190. [Google Scholar] [CrossRef] [PubMed]
- Holly, S.P.; Chang, J.W.; Li, W.; Niessen, S.; Phillips, R.M.; Piatt, R.; Black, J.L.; Smith, M.C.; Boulaftali, Y.; Weyrich, A.S.; et al. Chemoproteomic discovery of AADACL1 as a regulator of human platelet activation. Chem. Biol. 2013, 20, 1125–1134. [Google Scholar] [CrossRef] [Green Version]
- Houck, K.L.; Fox, T.E.; Sandirasegarane, L.; Kester, M. Ether-linked diglycerides inhibit vascular smooth muscle cell growth via decreased MAPK and PI3K/Akt signaling. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H1657–H1668. [Google Scholar] [CrossRef] [PubMed]
- Marigny, K.; Pedrono, F.; Martin-Chouly, C.A.; Youmine, H.; Saiag, B.; Legrand, A.B. Modulation of endothelial permeability by 1-O-alkylglycerols. Acta Physiol. Scand. 2002, 176, 263–268. [Google Scholar] [CrossRef]
- Vallari, D.S.; Record, M.; Snyder, F. Conversion of alkylacetylglycerol to platelet-activating factor in HL-60 cells and subcellular localization of the mediator. Arch. Biochem. Biophys. 1990, 276, 538–545. [Google Scholar] [CrossRef]
- Holly, S.P.; Gera, N.; Wang, P.; Wilson, A.; Guan, Z.; Lin, L.; Cooley, B.; Alfar, H.R.; Patil, R.G.; Piatt, R.; et al. Ether lipid metabolism by AADACL1 regulates platelet function and thrombosis. Blood Adv. 2019, 3, 3818–3828. [Google Scholar] [CrossRef]
- Chiang, K.P.; Niessen, S.; Saghatelian, A.; Cravatt, B.F. An enzyme that regulates ether lipid signaling pathways in cancer annotated by multidimensional profiling. Chem. Biol. 2006, 13, 1041–1050. [Google Scholar] [CrossRef] [Green Version]
- Jessani, N.; Liu, Y.; Humphrey, M.; Cravatt, B.F. Enzyme activity profiles of the secreted and membrane proteome that depict cancer cell invasiveness. Proc. Natl. Acad. Sci. USA 2002, 99, 10335–10340. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Zhang, L.; Chen, X.; Zhang, Q. NCEH1 may be a prognostic biomarker for pancreatic cancer. Int. J. Clin. Exp. Pathol. 2020, 13, 2746–2752. [Google Scholar]
- Xiao, Y.; Xie, J.; Liu, L.; Huang, W.; Han, Q.; Qin, J.; Liu, S.; Jiang, Z. NAD(P)-dependent steroid dehydrogenase-like protein and neutral cholesterol ester hydrolase 1 serve as novel markers for early detection of gastric cancer identified using quantitative proteomics. J. Clin. Lab. Anal. 2020, e23652. [Google Scholar] [CrossRef]
- Chang, J.W.; Nomura, D.K.; Cravatt, B.F. A potent and selective inhibitor of KIAA1363/AADACL1 that impairs prostate cancer pathogenesis. Chem. Biol. 2011, 18, 476–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leonard, T.A.; Rozycki, B.; Saidi, L.F.; Hummer, G.; Hurley, J.H. Crystal structure and allosteric activation of protein kinase C betaII. Cell 2011, 144, 55–66. [Google Scholar] [CrossRef] [Green Version]
- Slater, S.J.; Seiz, J.L.; Stagliano, B.A.; Cook, A.C.; Milano, S.K.; Ho, C.; Stubbs, C.D. Low- and high-affinity phorbol ester and diglyceride interactions with protein kinase C: 1-O-alkyl-2-acyl-sn-glycerol enhances phorbol ester- and diacylglycerol-induced activity but alone does not induce activity. Biochemistry 2001, 40, 6085–6092. [Google Scholar] [CrossRef] [PubMed]
- Daniel, L.W.; Small, G.W.; Schmitt, J.D.; Marasco, C.J.; Ishaq, K.; Piantadosi, C. Alkyl-linked diglycerides inhibit protein kinase C activation by diacylglycerols. Biochem. Biophys. Res. Commun. 1988, 151, 291–297. [Google Scholar] [CrossRef]
- Ford, D.A.; Miyake, R.; Glaser, P.E.; Gross, R.W. Activation of protein kinase C by naturally occurring ether-linked diglycerides. J. Biol. Chem. 1989, 264, 13818–13824. [Google Scholar] [CrossRef]
- Mandal, A.; Wang, Y.; Ernsberger, P.; Kester, M. Interleukin-1-induced ether-linked diglycerides inhibit calcium-insensitive protein kinase C isotypes. Implications for growth senescence. J. Biol. Chem. 1997, 272, 20306–20311. [Google Scholar] [CrossRef] [Green Version]
- Musial, A.; Mandal, A.; Coroneos, E.; Kester, M. Interleukin-1 and endothelin stimulate distinct species of diglycerides that differentially regulate protein kinase C in mesangial cells. J. Biol. Chem. 1995, 270, 21632–21638. [Google Scholar] [CrossRef] [Green Version]
- Tao, J.; Zhang, Y.; Huang, H.; Jiang, X. Activation of corticotropin-releasing factor 2 receptor inhibits Purkinje neuron P-type calcium currents via G(o)alpha-dependent PKC epsilon pathway. Cell Signal. 2009, 21, 1436–1443. [Google Scholar] [CrossRef]
- Cabot, M.C.; Jaken, S. Structural and chemical specificity of diacylglycerols for protein kinase C activation. Biochem. Biophys. Res. Commun. 1984, 125, 163–169. [Google Scholar] [CrossRef]
- Cheeseman, K.L.; Ueyama, T.; Michaud, T.M.; Kashiwagi, K.; Wang, D.; Flax, L.A.; Shirai, Y.; Loegering, D.J.; Saito, N.; Lennartz, M.R. Targeting of protein kinase C-epsilon during Fcgamma receptor-dependent phagocytosis requires the epsilonC1B domain and phospholipase C-gamma1. Mol. Biol. Cell 2006, 17, 799–813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shirai, Y.; Kashiwagi, K.; Sakai, N.; Saito, N. Phospholipase A(2) and its products are involved in the purinergic receptor-mediated translocation of protein kinase C in CHO-K1 cells. J. Cell Sci. 2000, 113 Pt 8, 1335–1343. [Google Scholar]
- Heymans, F.; Da Silva, C.; Marrec, N.; Godfroid, J.J.; Castagna, M. Alkyl analogs of diacylglycerol as activators of protein kinase C. FEBS Lett. 1987, 218, 35–40. [Google Scholar] [CrossRef] [Green Version]
- Stoll, L.L.; Figard, P.H.; Yerram, N.R.; Yorek, M.A.; Spector, A.A. 1-O-alkyl-2-acetyl-sn-glycerol: A platelet-activating factor metabolite with biological activity in vascular smooth muscle cells. Cell Regul. 1989, 1, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gellett, A.M.; Kharel, Y.; Sunkara, M.; Morris, A.J.; Lynch, K.R. Biosynthesis of alkyl lysophosphatidic acid by diacylglycerol kinases. Biochem. Biophys. Res. Commun. 2012, 422, 758–763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Albert, D.H.; Anderson, C.E. Ether-linked glycerolipids in human brain tumors. Lipids 1977, 12, 188–192. [Google Scholar] [CrossRef]
- Snyder, F.; Wood, R. Alkyl and alk-1-enyl ethers of glycerol in lipids from normal and neoplastic human tissues. Cancer Res. 1969, 29, 251–257. [Google Scholar]
- Tokumura, A.; Sinomiya, J.; Kishimoto, S.; Tanaka, T.; Kogure, K.; Sugiura, T.; Satouchi, K.; Waku, K.; Fukuzawa, K. Human platelets respond differentially to lysophosphatidic acids having a highly unsaturated fatty acyl group and alkyl ether-linked lysophosphatidic acids. Biochem. J. 2002, 365, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.R.; Khandoga, A.L.; Goyal, P.; Fells, J.I.; Perygin, D.H.; Siess, W.; Parrill, A.L.; Tigyi, G.; Fujiwara, Y. Unique ligand selectivity of the GPR92/LPA5 lysophosphatidate receptor indicates role in human platelet activation. J. Biol. Chem. 2009, 284, 17304–17319. [Google Scholar] [CrossRef] [Green Version]
- Haseruck, N.; Erl, W.; Pandey, D.; Tigyi, G.; Ohlmann, P.; Ravanat, C.; Gachet, C.; Siess, W. The plaque lipid lysophosphatidic acid stimulates platelet activation and platelet-monocyte aggregate formation in whole blood: Involvement of P2Y1 and P2Y12 receptors. Blood 2004, 103, 2585–2592. [Google Scholar] [CrossRef] [Green Version]
- Rother, E.; Brandl, R.; Baker, D.L.; Goyal, P.; Gebhard, H.; Tigyi, G.; Siess, W. Subtype-selective antagonists of lysophosphatidic Acid receptors inhibit platelet activation triggered by the lipid core of atherosclerotic plaques. Circulation 2003, 108, 741–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, J.; Xiao, Y.J.; Baudhuin, L.M.; Hong, G.; Xu, Y. Role of ether-linked lysophosphatidic acids in ovarian cancer cells. J. Lipid Res. 2002, 43, 463–476. [Google Scholar] [PubMed]
- Jaffres, P.A.; Gajate, C.; Bouchet, A.M.; Couthon-Gourves, H.; Chantome, A.; Potier-Cartereau, M.; Besson, P.; Bougnoux, P.; Mollinedo, F.; Vandier, C. Alkyl ether lipids, ion channels and lipid raft reorganization in cancer therapy. Pharm. Ther. 2016, 165, 114–131. [Google Scholar] [CrossRef] [PubMed]
- van Blitterswijk, W.J.; Verheij, M. Anticancer mechanisms and clinical application of alkylphospholipids. Biochim. Biophys. Acta 2013, 1831, 663–674. [Google Scholar] [CrossRef]
- Diomede, L.; Colotta, F.; Piovani, B.; Re, F.; Modest, E.J.; Salmona, M. Induction of apoptosis in human leukemic cells by the ether lipid 1-octadecyl-2-methyl-rac-glycero-3-phosphocholine. A possible basis for its selective action. Int. J. Cancer 1993, 53, 124–130. [Google Scholar] [CrossRef]
- Mollinedo, F.; Martinez-Dalmau, R.; Modolell, M. Early and selective induction of apoptosis in human leukemic cells by the alkyl-lysophospholipid ET-18-OCH3. Biochem. Biophys. Res. Commun. 1993, 192, 603–609. [Google Scholar] [CrossRef]
- Alonso, M.T.; Gajate, C.; Mollinedo, F.; Modolell, M.; Alvarez, J.; Garcia-Sancho, J. Dissociation of the effects of the antitumour ether lipid ET-18-OCH3 on cytosolic calcium and on apoptosis. Br. J. Pharm. 1997, 121, 1364–1368. [Google Scholar] [CrossRef]
- Gajate, C.; Santos-Beneit, A.M.; Macho, A.; Lazaro, M.; Hernandez-De Rojas, A.; Modolell, M.; Munoz, E.; Mollinedo, F. Involvement of mitochondria and caspase-3 in ET-18-OCH(3)-induced apoptosis of human leukemic cells. Int. J. Cancer 2000, 86, 208–218. [Google Scholar] [CrossRef]
- Diomede, L.; Damia, G.; D’Incalci, M.; Imperatori, L.; Algeri, M.; Modest, E.J.; Salmona, M. In vivo anti-tumor activity of synthetic ether lipids is not enhanced by pharmacological modulation of tumor lipid composition. Int. J. Cancer 1994, 59, 580–581. [Google Scholar] [CrossRef]
- Verdonck, L.F.; van Heugten, H.G. Ether lipids are effective cytotoxic drugs against multidrug-resistant acute leukemia cells and can act by the induction of apoptosis. Leuk. Res. 1997, 21, 37–43. [Google Scholar] [CrossRef]
- Gajate, C.; Fonteriz, R.I.; Cabaner, C.; Alvarez-Noves, G.; Alvarez-Rodriguez, Y.; Modolell, M.; Mollinedo, F. Intracellular triggering of Fas, independently of FasL, as a new mechanism of antitumor ether lipid-induced apoptosis. Int. J. Cancer 2000, 85, 674–682. [Google Scholar] [CrossRef]
- Gajate, C.; Mollinedo, F. The antitumor ether lipid ET-18-OCH(3) induces apoptosis through translocation and capping of Fas/CD95 into membrane rafts in human leukemic cells. Blood 2001, 98, 3860–3863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mollinedo, F.; Gajate, C.; Martin-Santamaria, S.; Gago, F. ET-18-OCH3 (edelfosine): A selective antitumour lipid targeting apoptosis through intracellular activation of Fas/CD95 death receptor. Curr. Med. Chem. 2004, 11, 3163–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuvillier, O.; Mayhew, E.; Janoff, A.S.; Spiegel, S. Liposomal ET-18-OCH(3) induces cytochrome c-mediated apoptosis independently of CD95 (APO-1/Fas) signaling. Blood 1999, 94, 3583–3592. [Google Scholar] [CrossRef] [PubMed]
- van Blitterswijk, W.J.; Klarenbeek, J.B.; van der Luit, A.H.; Alderliesten, M.C.; van Lummel, M.; Verheij, M. Fas/CD95 down-regulation in lymphoma cells through acquired alkyllysophospholipid resistance: Partial role of associated sphingomyelin deficiency. Biochem. J. 2009, 425, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Potier, M.; Chantome, A.; Joulin, V.; Girault, A.; Roger, S.; Besson, P.; Jourdan, M.L.; LeGuennec, J.Y.; Bougnoux, P.; Vandier, C. The SK3/K(Ca)2.3 potassium channel is a new cellular target for edelfosine. Br. J. Pharm. 2011, 162, 464–479. [Google Scholar] [CrossRef] [Green Version]
- Girault, A.; Haelters, J.P.; Potier-Cartereau, M.; Chantome, A.; Pinault, M.; Marionneau-Lambot, S.; Oullier, T.; Simon, G.; Couthon-Gourves, H.; Jaffres, P.A.; et al. New alkyl-lipid blockers of SK3 channels reduce cancer cell migration and occurrence of metastasis. Curr. Cancer Drug Targets 2011, 11, 1111–1125. [Google Scholar] [CrossRef]
- Powis, G.; Seewald, M.J.; Gratas, C.; Melder, D.; Riebow, J.; Modest, E.J. Selective inhibition of phosphatidylinositol phospholipase C by cytotoxic ether lipid analogues. Cancer Res. 1992, 52, 2835–2840. [Google Scholar]
- van der Luit, A.H.; Budde, M.; Ruurs, P.; Verheij, M.; van Blitterswijk, W.J. Alkyl-lysophospholipid accumulates in lipid rafts and induces apoptosis via raft-dependent endocytosis and inhibition of phosphatidylcholine synthesis. J. Biol. Chem. 2002, 277, 39541–39547. [Google Scholar] [CrossRef] [Green Version]
- Wieder, T.; Orfanos, C.E.; Geilen, C.C. Induction of ceramide-mediated apoptosis by the anticancer phospholipid analog, hexadecylphosphocholine. J. Biol. Chem. 1998, 273, 11025–11031. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Guerrico, A.M.; Meshki, J.; Xiao, L.; Benavides, F.; Conti, C.J.; Kazanietz, M.G. Molecular mechanisms of protein kinase C-induced apoptosis in prostate cancer cells. J. Biochem. Mol. Biol. 2005, 38, 639–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musashi, M.; Ota, S.; Shiroshita, N. The role of protein kinase C isoforms in cell proliferation and apoptosis. Int. J. Hematol. 2000, 72, 12–19. [Google Scholar] [PubMed]
- Castro-Galache, M.D.; Menendez-Gutierrez, M.P.; Carrasco Garcia, E.; Garcia-Morales, P.; Martinez-Lacaci, I.; Saceda, M.; Ferragut, J.A. Protein kinase C-alpha antagonizes apoptosis induction by histone deacetylase inhibitors in multidrug resistant leukaemia cells. Int. J. Biochem. Cell Biol. 2007, 39, 1877–1885. [Google Scholar] [CrossRef] [PubMed]
- Hou, Q.; Lin, J.; Huang, W.; Li, M.; Feng, J.; Mao, X. CTRP3 Stimulates Proliferation and Anti-Apoptosis of Prostate Cells through PKC Signaling Pathways. PLoS ONE 2015, 10, e0134006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Z.; Kong, C.; Zhang, Z.; Zhu, Y.; Zhang, Y.; Chen, X. Reduction of protein kinase C alpha (PKC-alpha) promote apoptosis via down-regulation of Dicer in bladder cancer. J. Cell. Mol. Med. 2015, 19, 1085–1093. [Google Scholar] [CrossRef]
- Zheng, J.; Kong, C.; Yang, X.; Cui, X.; Lin, X.; Zhang, Z. Protein kinase C-alpha (PKCalpha) modulates cell apoptosis by stimulating nuclear translocation of NF-kappa-B p65 in urothelial cell carcinoma of the bladder. BMC Cancer 2017, 17, 432. [Google Scholar] [CrossRef]
- Tadokoro, S.; Nakazawa, T.; Kamae, T.; Kiyomizu, K.; Kashiwagi, H.; Honda, S.; Kanakura, Y.; Tomiyama, Y. A potential role for alpha-actinin in inside-out alphaIIbbeta3 signaling. Blood 2011, 117, 250–258. [Google Scholar] [CrossRef] [Green Version]
- Nagano, T.; Ohga, S.; Kishimoto, Y.; Kimura, T.; Yasunaga, K.; Adachi, M.; Ryo, R.; Sato, T. Ultrastructural analysis of platelet-like particles from a human megakaryocytic leukemia cell line (CMK 11-5). Int. J. Hematol. 1992, 56, 67–78. [Google Scholar]
- Shariati, M.; Meric-Bernstam, F. Targeting AKT for cancer therapy. Expert Opin. Investig. Drugs 2019, 28, 977–988. [Google Scholar] [CrossRef]
- Kondapaka, S.B.; Singh, S.S.; Dasmahapatra, G.P.; Sausville, E.A.; Roy, K.K. Perifosine, a novel alkylphospholipid, inhibits protein kinase B activation. Mol. Cancer 2003, 2, 1093–1103. [Google Scholar]
- Pramanik, K.C.; Kudugunti, S.K.; Fofaria, N.M.; Moridani, M.Y.; Srivastava, S.K. Caffeic acid phenethyl ester suppresses melanoma tumor growth by inhibiting PI3K/AKT/XIAP pathway. Carcinogenesis 2013, 34, 2061–2070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gills, J.J.; Dennis, P.A. Perifosine: Update on a novel Akt inhibitor. Curr. Oncol. Rep. 2009, 11, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Hunerdosse, D.M.; Morris, P.J.; Miyamoto, D.K.; Fisher, K.J.; Bateman, L.A.; Ghazaleh, J.R.; Zhong, S.; Nomura, D.K. Chemical Genetics Screening Reveals KIAA1363 as a Cytokine-Lowering Target. ACS Chem. Biol. 2014, 9, 2905–2913. [Google Scholar] [CrossRef] [PubMed]
- Watschinger, K.; Keller, M.A.; Golderer, G.; Hermann, M.; Maglione, M.; Sarg, B.; Lindner, H.H.; Hermetter, A.; Werner-Felmayer, G.; Konrat, R.; et al. Identification of the gene encoding alkylglycerol monooxygenase defines a third class of tetrahydrobiopterin-dependent enzymes. Proc. Natl. Acad. Sci. USA 2010, 107, 13672–13677. [Google Scholar] [CrossRef] [Green Version]
- Watschinger, K.; Keller, M.A.; McNeill, E.; Alam, M.T.; Lai, S.; Sailer, S.; Rauch, V.; Patel, J.; Hermetter, A.; Golderer, G.; et al. Tetrahydrobiopterin and alkylglycerol monooxygenase substantially alter the murine macrophage lipidome. Proc. Natl. Acad. Sci. USA 2015, 112, 2431–2436. [Google Scholar] [CrossRef] [Green Version]
- Dorninger, F.; Forss-Petter, S.; Berger, J. From peroxisomal disorders to common neurodegenerative diseases—The role of ether phospholipids in the nervous system. FEBS Lett. 2017, 591, 2761–2788. [Google Scholar] [CrossRef]
- Dorninger, F.; Gundacker, A.; Zeitler, G.; Pollak, D.D.; Berger, J. Ether Lipid Deficiency in Mice Produces a Complex Behavioral Phenotype Mimicking Aspects of Human Psychiatric Disorders. Int. J. Mol. Sci. 2019, 20, 3929. [Google Scholar] [CrossRef] [Green Version]
- Liegel, R.P.; Ronchetti, A.; Sidjanin, D.J. Alkylglycerone phosphate synthase (AGPS) deficient mice: Models for rhizomelic chondrodysplasia punctate type 3 (RCDP3) malformation syndrome. Mol. Genet. Metab. Rep. 2014, 1, 299–311. [Google Scholar] [CrossRef]
- Teigler, A.; Komljenovic, D.; Draguhn, A.; Gorgas, K.; Just, W.W. Defects in myelination, paranode organization and Purkinje cell innervation in the ether lipid-deficient mouse cerebellum. Hum. Mol. Genet. 2009, 18, 1897–1908. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.; Henry, W.S.; Ricq, E.L.; Graham, E.T.; Phadnis, V.V.; Maretich, P.; Paradkar, S.; Boehnke, N.; Deik, A.A.; Reinhardt, F.; et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020, 585, 603–608. [Google Scholar] [CrossRef]
- Rubio, J.M.; Astudillo, A.M.; Casas, J.; Balboa, M.A.; Balsinde, J. Regulation of Phagocytosis in Macrophages by Membrane Ethanolamine Plasmalogens. Front. Immunol. 2018, 9, 1723. [Google Scholar] [CrossRef] [PubMed]
- Facciotti, F.; Ramanjaneyulu, G.S.; Lepore, M.; Sansano, S.; Cavallari, M.; Kistowska, M.; Forss-Petter, S.; Ni, G.; Colone, A.; Singhal, A.; et al. Peroxisome-derived lipids are self antigens that stimulate invariant natural killer T cells in the thymus. Nat. Immunol. 2012, 13, 474–480. [Google Scholar] [CrossRef] [PubMed]
- Ebenezer, D.L.; Fu, P.; Ramchandran, R.; Ha, A.W.; Putherickal, V.; Sudhadevi, T.; Harijith, A.; Schumacher, F.; Kleuser, B.; Natarajan, V. S1P and plasmalogen derived fatty aldehydes in cellular signaling and functions. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2020, 1865, 158681. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rangholia, N.; Leisner, T.M.; Holly, S.P. Bioactive Ether Lipids: Primordial Modulators of Cellular Signaling. Metabolites 2021, 11, 41. https://doi.org/10.3390/metabo11010041
Rangholia N, Leisner TM, Holly SP. Bioactive Ether Lipids: Primordial Modulators of Cellular Signaling. Metabolites. 2021; 11(1):41. https://doi.org/10.3390/metabo11010041
Chicago/Turabian StyleRangholia, Nikhil, Tina M. Leisner, and Stephen P. Holly. 2021. "Bioactive Ether Lipids: Primordial Modulators of Cellular Signaling" Metabolites 11, no. 1: 41. https://doi.org/10.3390/metabo11010041
APA StyleRangholia, N., Leisner, T. M., & Holly, S. P. (2021). Bioactive Ether Lipids: Primordial Modulators of Cellular Signaling. Metabolites, 11(1), 41. https://doi.org/10.3390/metabo11010041