
Dynamic Evolution and Correlation between Metabolites and Microorganisms during Manufacturing Process and Storage of Fu Brick Tea


Abstract
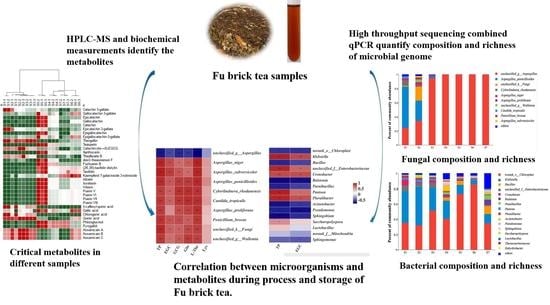
:
1. Introduction
2. Results
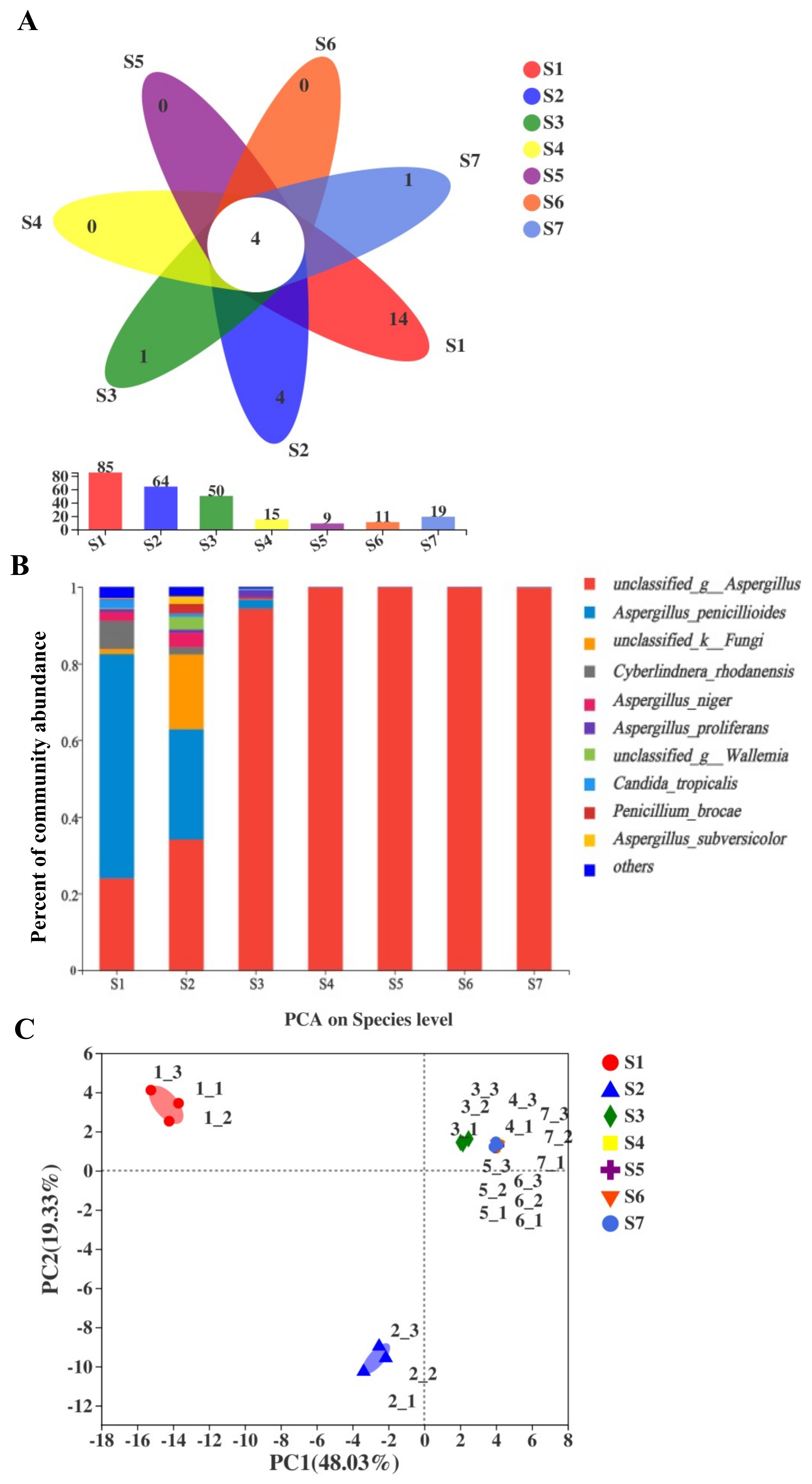
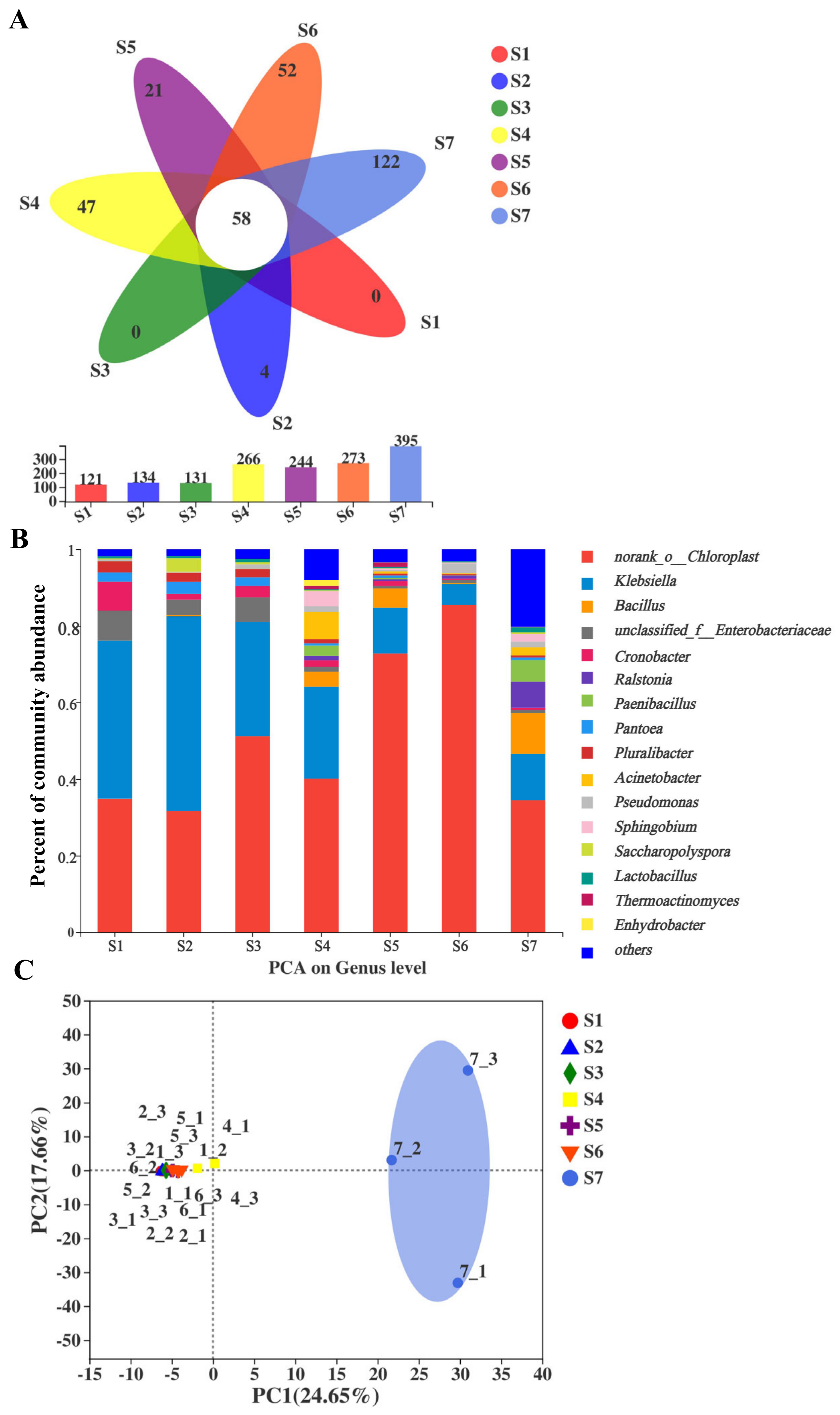
2.1. Sequence Statistics of Fungi and Bacteria of Fu Brick Tea Samples
2.2. Comparisons of Fungal Community and Richness during the Manufacturing Process and Storage
2.3. Comparisons of Bacterial Community and Richness during the Manufacturing Processes and Storage
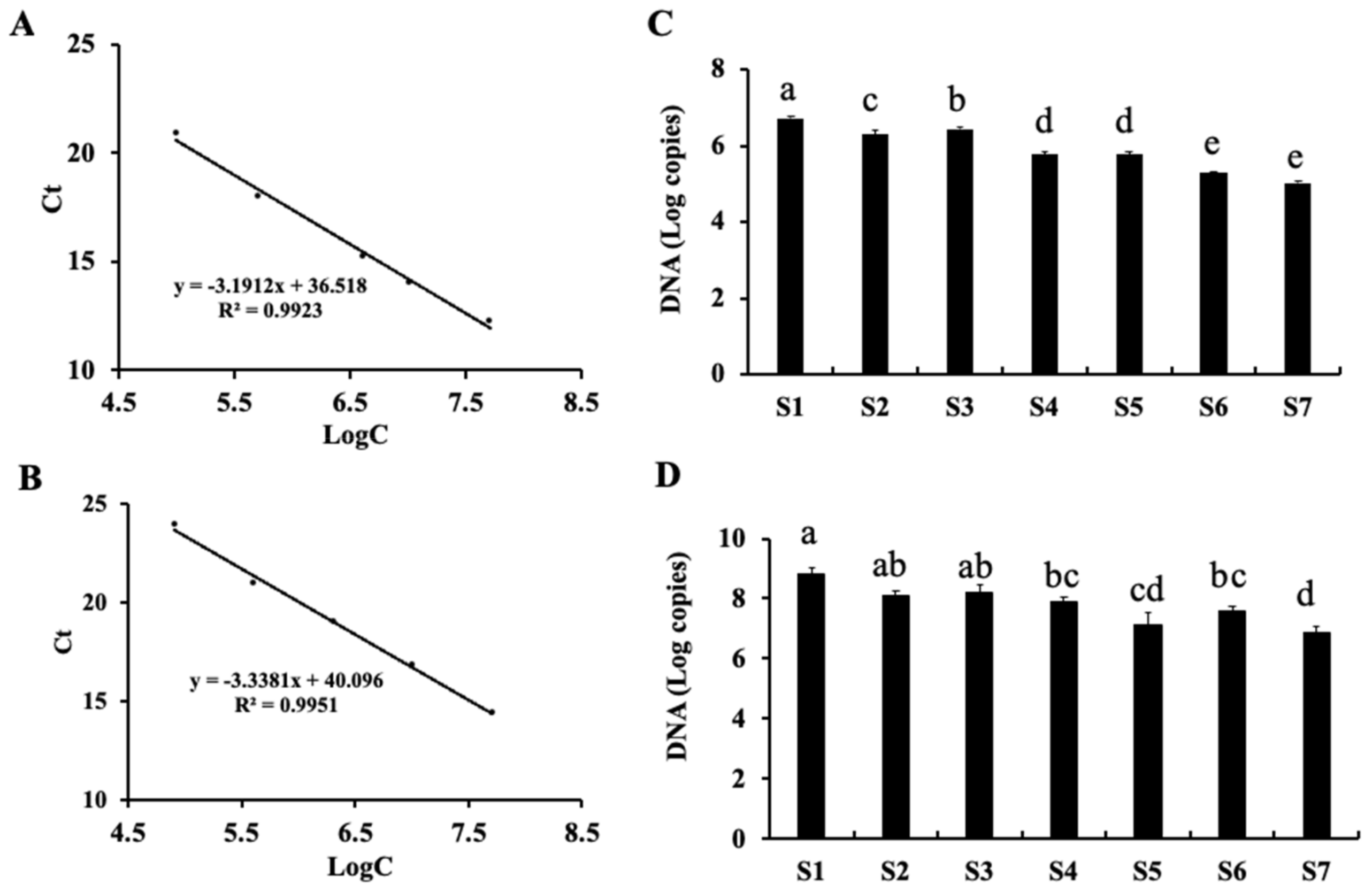
2.4. Quantitative PCR (qPCR) Detection of Fungi and Bacteria during Processing and Storage of Fu Brick Tea
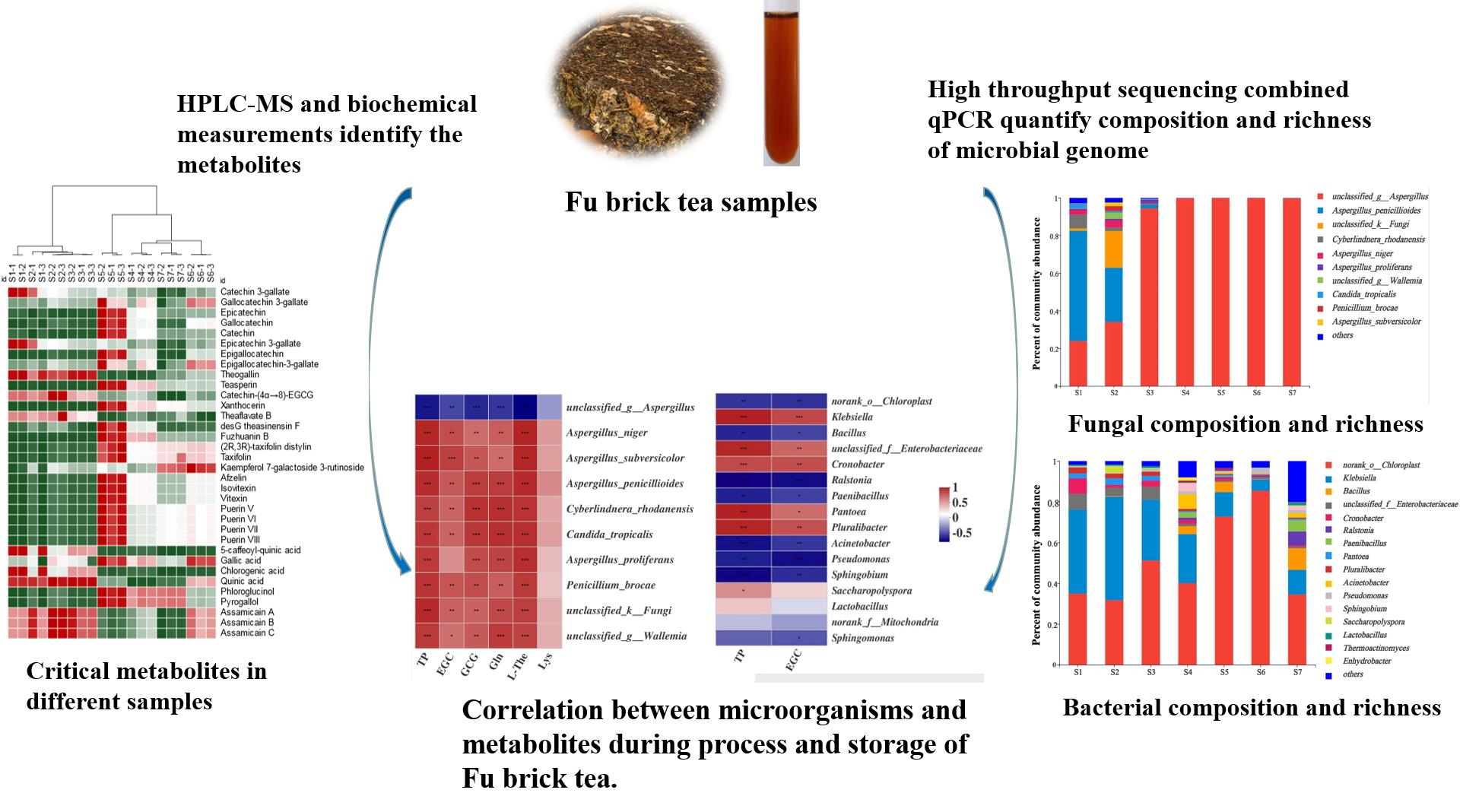
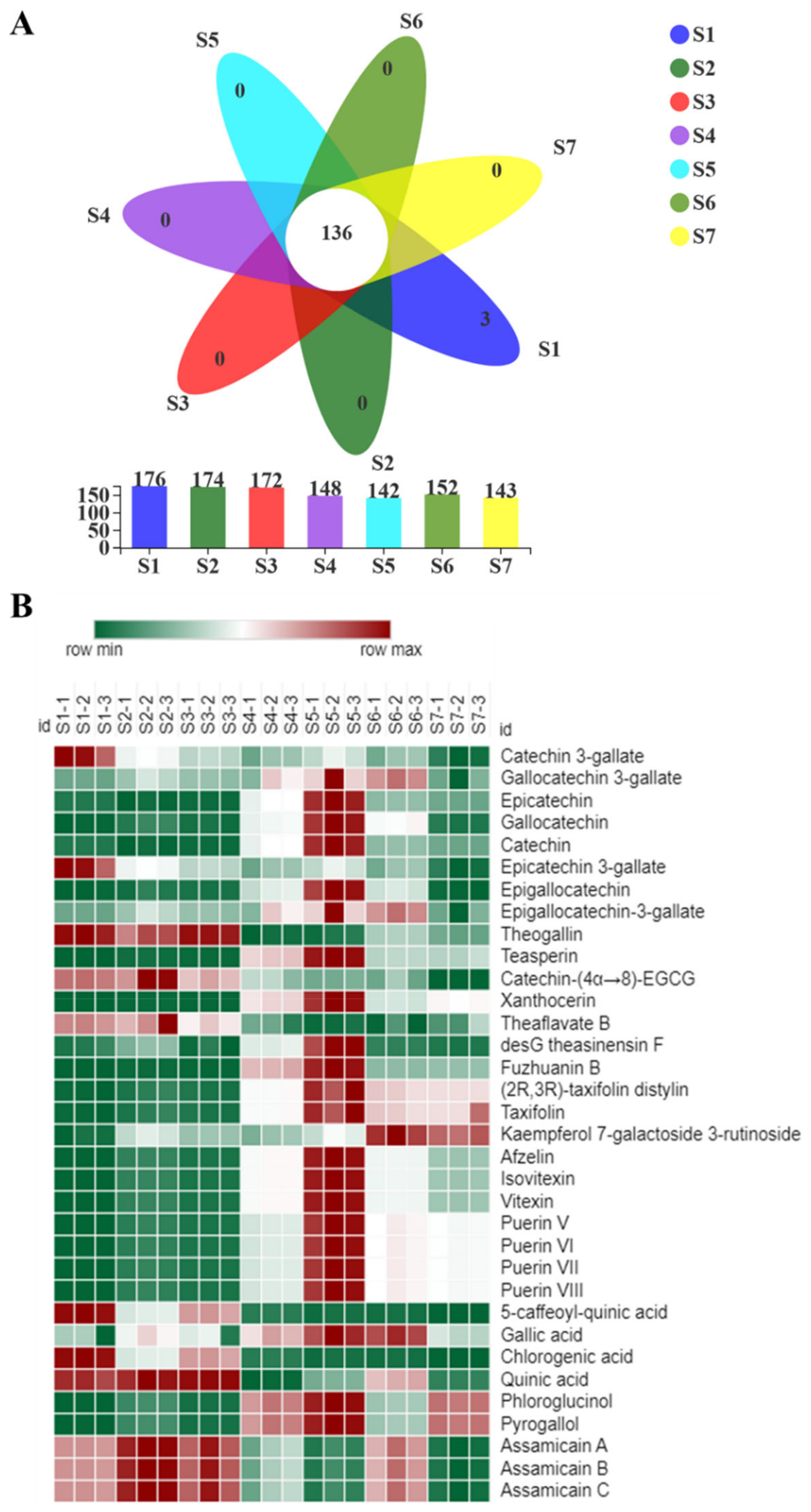
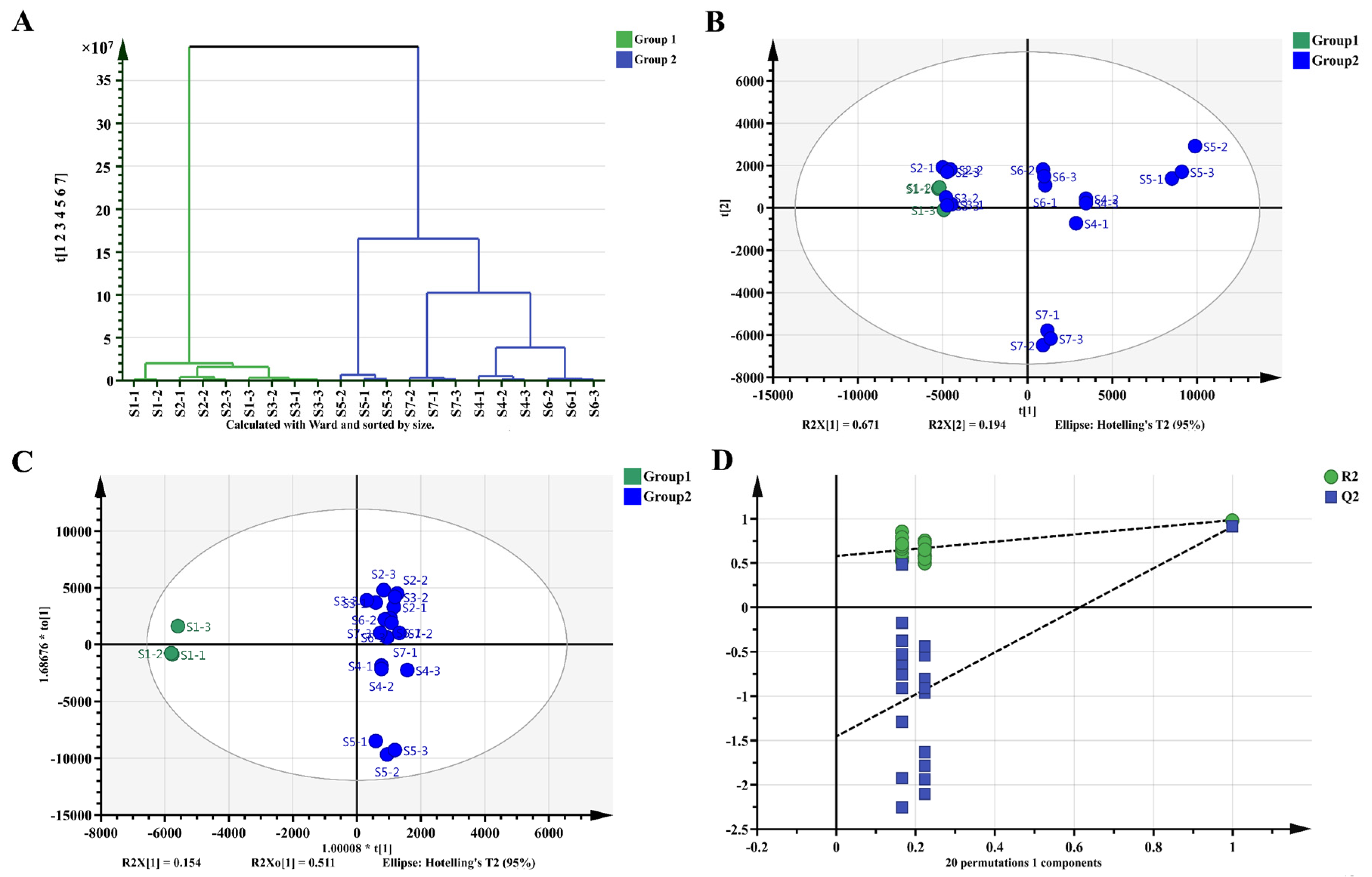
2.5. Metabolomics Analysis of Fu Brick Tea Samples during Processing and Storage
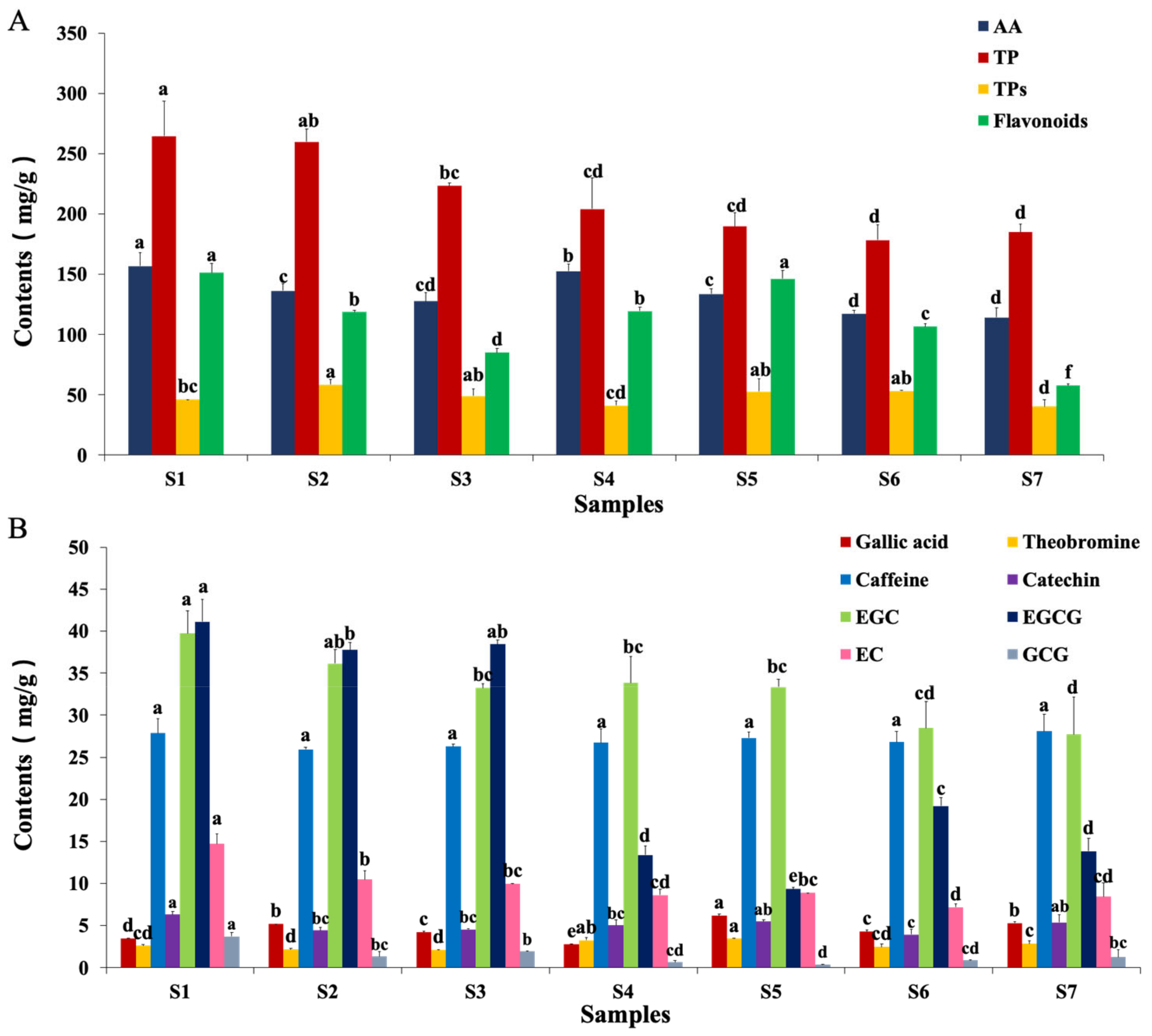
2.6. Measurements of Critical Functional Components of Fu Brick Tea
2.6.1. Amino Acids
2.6.2. Polyphenol
2.6.3. Tea Polysaccharides
2.6.4. Flavonoids
2.6.5. Other Notable Changes
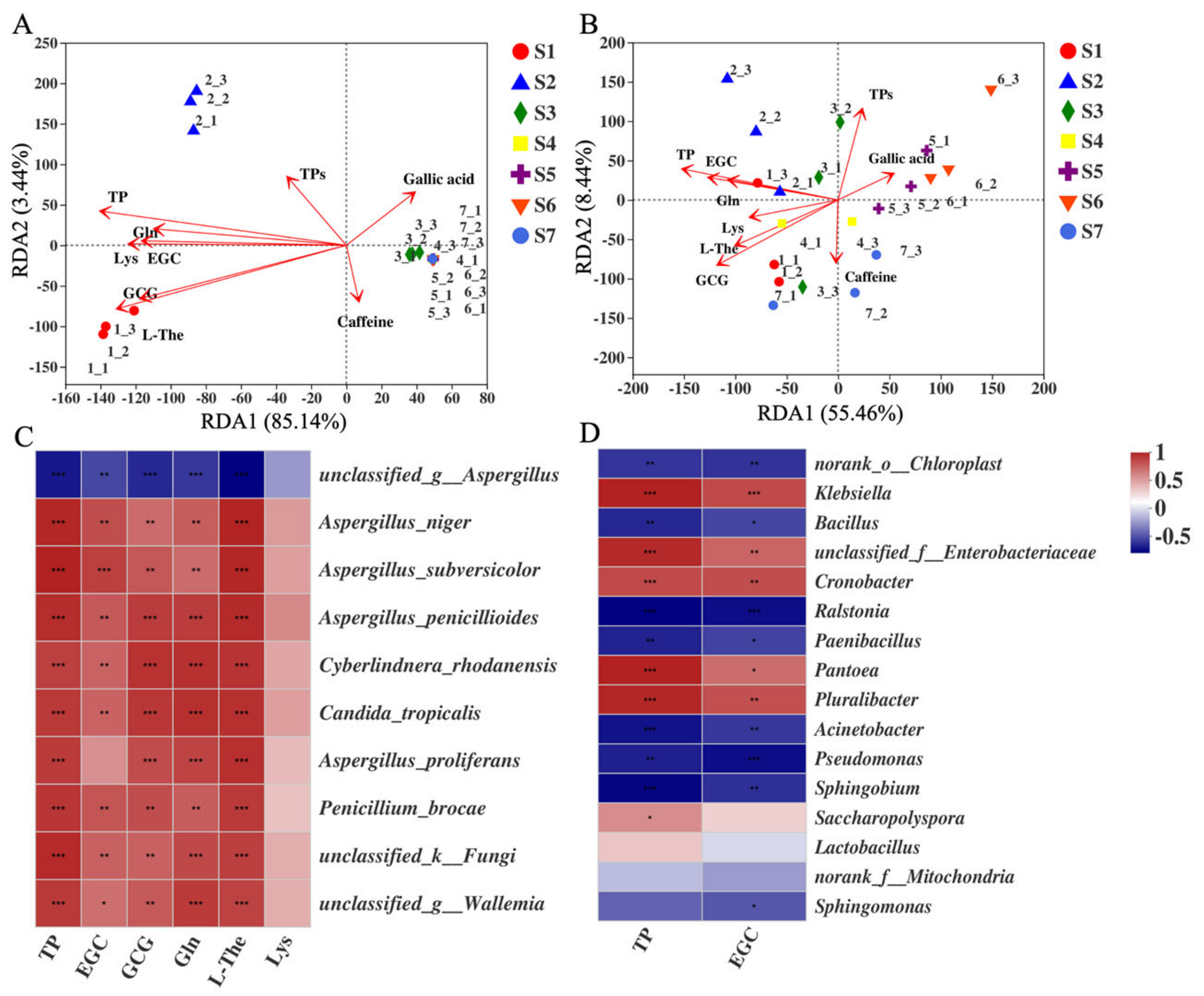
2.7. Correlation between CRITICAL Metabolites and Microorganisms
3. Discussion
4. Experimental Section
4.1. Chemicals and Materials
4.2. DNA Extraction, Polymerase Chain Reaction Amplification, and Sequence
4.3. Sequence Processing and Analysis
4.4. Standard Curve Generation and Quantitative PCR (qPCR) Detection of Fungi and Bacteria during Processing and Storage of Fu Brick Tea
4.5. Sample Preparation
4.5.1. Determination of Main Active Compounds
4.5.2. Determination of Tea Free Amino Acids
4.5.3. Determination of Tea Polysaccharides (TPS)
4.6. HPLC–MS Analysis
4.7. Multivariate Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liang, Z.; Zhang, Z.Z.; Zhou, Y.B.; Ling, T.J.; Wan, X.C. Chinese dark teas: Postfermentation, chemistry and biological activities. Food Res. Int. 2013, 53, 600–607. [Google Scholar]
- Fu, D.; Ryan, E.P.; Huang, J.; Liu, Z.; Weir, T.L.; Snook, R.L.; Ryan, T.P. Fermented Camellia sinensis, Fu Zhuan Tea, regulates hyperlipidemia and transcription factors involved in lipid catabolism. Food Res. Int. 2011, 44, 2999–3005. [Google Scholar] [CrossRef]
- Mo, H.; Yang, Z.; Chen, Z. Microbial fermented tea—a potential source of natural food preservatives. Trends Food Sci. Technol. 2008, 19, 124–130. [Google Scholar] [CrossRef]
- Zhu, M.Z.; Li, N.; Zhou, F.; Ouyang, J.; Wu, J.L. Microbial bioconversion of the chemical components in dark tea. Food Chem. 2020, 312, 126043. [Google Scholar] [CrossRef]
- Wen, Q.; Liu, S. Evolutionary Regulation of Dominant Fungi in Fuzhuan Brick Tea During the Fungus Growing Processes. Available online: https://en.cnki.com.cn/Article_en/CJFDTotal-CYKK1991S1010.htm (accessed on 10 October 2021).
- Li, Q.; Huang, J.; Li, Y.; Zhang, Y.; Luo, Y.; Chen, Y.; Lin, H.; Wang, K.; Liu, Z. Fungal community succession and major components change during manufacturing processes of Fu brick tea. Sci. Rep. 2017, 7, 6947. [Google Scholar] [CrossRef]
- Qi, Z.; Sun, Z. Identification of predominant species in brick tea. Mycosystema 1990, 9, 176–179. [Google Scholar]
- Xu, A.; Wang, Y.; Wen, J.; Liu, P.; Liu, Z.; Li, Z. Fungal community associated with fermentation and storage of Fuzhuan brick-tea. Int. J. Food Microbiol. 2011, 146, 14–22. [Google Scholar] [CrossRef]
- Fu, J.; Lv, H.; Feng, C. Diversity and Variation of Bacterial Community Revealed by MiSeq Sequencing in Chinese Dark Teas. PLoS ONE 2016, 11, e0162719. [Google Scholar] [CrossRef]
- Robin, J.D.; Ludlow, A.T.; Laranger, R.; Wright, W.E.; Shay, J.W. Comparison of DNA Quantification Methods for Next Generation Sequencing. Sci. Rep. 2016, 6, 24067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Postollec, F.; Falentin, H.; Pavan, S.; Combrisson, J.; Sohier, D. Recent advances in quantitative PCR (qPCR) applications in food microbiology. Food Microbiol. 2011, 28, 848–861. [Google Scholar] [CrossRef]
- Want, E.J.; Nordstrom, A.; Morita, H.; Siuzdak, G. From exogenous to endogenous: The inevitable imprint of mass spectrometry in metabolomics. J. Proteome Res. 2007, 6, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Capanoglu, E.; Beekwilder, J.; Boyacioglu, D.; Hall, R.; Vos, R.D. Changes in antioxidant and metabolite profiles during production of tomato paste. J. Agric. Food Chem. 2008, 56, 964–973. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Hu, F.L.; Wang, W.; Wan, X.C.; Bao, G.H. Investigation on biochemical compositional changes during the microbial fermentation processes of Fu brick tea by LC-MS based metabolomics. Food Chem. 2015, 186, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Jin, Y.; Jiang, R.; Xu, Y.; Liu, Z. Dynamic changes in the metabolite profile and taste characteristics of Fu brick tea during the manufacturing processes. Food Chem. 2020, 344, 128576. [Google Scholar] [CrossRef] [PubMed]
- Hda, B.; Hao, L.A.; Zx, A.; Sza, B.; Th, D.; Am, C.; Sxa, B. The mechanism for improving the flesh quality of grass carp (Ctenopharyngodon idella ) following the micro-flowing water treatment using a UPLC-QTOF/MS based metabolomics method. Food Chem. 2020, 327, 126777. [Google Scholar]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Metabolomics: The apogee of the omic triology. Nat. Rev. Mol. Cell Bio. 2013, 13, 263. [Google Scholar] [CrossRef]
- Cheng, L.; Yang, Q.; Chen, Z.; Zhang, J.; Chen, Q.; Wang, Y.; Wei, X. Distinct Changes of Metabolic Profile and Sensory Quality during Qingzhuan Tea Processing Revealed by LC-MS-Based Metabolomics. J. Agric. Food Chem. 2020, 68, 4955–4965. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Ma, Y.; Wei, Z.Z.; Yuan, W.X.; Li, Y.L.; Zhang, C.H.; Xue, X.T.; Zhou, H.J. Determination and Comparison of γ-Aminobutyric Acid (GABA) Content in Pu-erh and Other Types of Chinese Tea. J. Agric. Food Chem. 2011, 59, 3641. [Google Scholar] [CrossRef]
- Chen, Y.S.; Liu, B.L.; Chang, Y.N. Bioactivities and sensory evaluation of Pu-erh teas made from three tea leaves in an improved pile fermentation processes. J. Biosci. Bioeng. 2010, 109, 557–563. [Google Scholar] [CrossRef]
- Ge, Y.; Bian, X.; Sun, B.; Zhao, M.; Ma, Y.; Tang, Y.; Li, N.; Wu, J.-L. Dynamic Profiling of Phenolic Acids during Pu-erh Tea Fermentation Using Derivatization Liquid Chromatography–Mass Spectrometry Approach. J. Agric. Food Chem. 2019, 67, 4568–4577. [Google Scholar] [CrossRef]
- Li, P.L.; Dai, W.D.; Lu, M.; Xie, D.C.; Tan, J.F. Metabolomic analysis reveals the composition differences in 13 Chinese tea cultivars of different manufacturing suitabilities. J. Sci. Food Agric. 2018, 93, 1153–1161. [Google Scholar] [CrossRef]
- Qin, J.H.; Li, N.; Tu, P.F.; Ma, Z.Z.; Zhang, L. Change in Tea Polyphenol and Purine Alkaloid Composition during Solid-State Fungal Fermentation of Postfermented Tea. J. Agric. Food Chem. 2012, 60, 1213–1217. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.P.; Zhong, Q.S.; Lin, Z.; Wang, L.; Tan, J.F.; Guo, L. Aroma characterisation of Pu-erh tea using headspace-solid phase microextraction combined with GC/MS and GC–olfactometry. Food Chem. 2012, 130, 1074–1081. [Google Scholar] [CrossRef]
- Zhang, L.; Li, N.; Ma, Z.Z.; Tu, P.F. Comparison of the chemical constituents of aged pu-erh tea, ripened pu-erh tea, and other teas using HPLC-DAD-ESI-MSn. J. Agric. Food Chem. 2011, 59, 8754. [Google Scholar] [CrossRef]
- Yang, C.; Hu, Z.; Lu, M.; Li, P.; Tan, J.; Chen, M.; Lv, H.; Zhu, Y.; Zhang, Y.; Guo, L. Application of metabolomics profiling in the analysis of metabolites and taste quality in different subtypes of white tea. Food Res. Int. 2018, 106, 909. [Google Scholar] [CrossRef]
- Bourdichon, F.; Casaregola, S.; Farrokh, C.; Frisvad, J.C.; Gerds, M.L.; Hammes, W.P.; Harnett, J.; Huys, G.; Laulund, S.; Ouwehand, A. Food fermentations: Microorganisms with technological beneficial use. Int. J. Food Microbiol. 2012, 154, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Ishikawa, Y.; Morimoto, K.; Hamasaki, T. Flavoglaucin, a metabolite of Eurotium chevalieri, its antioxidation and synergism with tocopherol. J. Am. Oil Chem. Soc. 1984, 61, 1864–1868. [Google Scholar] [CrossRef]
- Slack, G.J.; Puniani, E.; Frisvad, J.C.; Samson, R.A.; Miller, J.D. Secondary metabolites from Eurotium species, Aspergillus calidoustus and A. insuetus common in Canadian homes with a review of their chemistry and biological activities. Mycol. Res. 2009, 113, 480–490. [Google Scholar]
- Ling, T.J.; Wan, X.C.; Ling, W.W.; Zhang, Z.Z.; Xia, T.; Li, D.X.; Hou, R.Y. New Triterpenoids and Other Constituents from a Special Microbial-Fermented Tea—Fuzhuan Brick Tea. J. Agric. Food Chem. 2010, 58, 4945–4950. [Google Scholar] [CrossRef]
- Luo, Z.M.; Ling, T.J.; Li, L.X.; Zhang, Z.Z.; Zhu, H.T.; Zhang, Y.J.; Wan, X.C. A New Norisoprenoid and Other Compounds from Fuzhuan Brick Tea. Molecules 2012, 17, 3539–3546. [Google Scholar] [CrossRef]
- Jiang, H.Y.; Shii, T.; Matsuo, Y.; Tanaka, T.; Jiang, Z.H.; Kouno, I. A new catechin oxidation product and polymeric polyphenols of post-fermented tea. Food Chem. 2011, 129, 830–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, Z.M.; Du, H.X.; Li, L.X.; An, M.Q.; Zhang, Z.Z.; Wan, X.C.; Bao, G.H.; Zhang, L.; Ling, T.J. Fuzhuanins A and B: The B-ring Fission Lactones of Flavan-3-ols from Fuzhuan Brick-Tea. J. Agric. Food Chem. 2013, 61, 6982–6990. [Google Scholar] [CrossRef]
- Zhu, Y.F.; Chen, J.J.; Ji, X.M.; Hu, X.; Ling, T.J.; Zhang, Z.Z.; Bao, G.H.; Wan, X.C. Changes of major tea polyphenols and production of four new B-ring fission metabolites of catechins from post-fermented Jing-Wei Fu brick tea. Food Chem. 2015, 170, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Adams, R.I.; Miletto, M.; Taylor, J.W.; Bruns, T.D. Dispersal in microbes: Fungi in indoor air are dominated by outdoor air and show dispersal limitation at short distances. ISME J. 2013, 7, 1460. [Google Scholar] [CrossRef] [Green Version]
- Xu, N.; Tan, G.C.; Wang, H.Y.; Gai, X.P. Effect of biochar additions to soil on nitrogen leaching, microbial biomass and bacterial community structure. Eur. J. Soil Biol. 2016, 74, 1–8. [Google Scholar] [CrossRef]
- M, B.A.; Marc, L.; Bjoern, U. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinform. NLM 2014, 30, 2144. [Google Scholar]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar]
- Xiaoli, D.; Manuel, K.; E, S.C.; Erin, T.; Carmen, L.; Dan, L.; Marc, S. Fast and Simple Analysis of MiSeq Amplicon Sequencing Data with MetaAmp. Frontiers in Microbiol. 2017, 8, 1461. [Google Scholar]
- Anders, B.A.; Louise, D.M.; Håvard, K. ITS all right mama: Investigating the formation of chimeric sequences in the ITS2 region by DNA metabarcoding analyses of fungal mock communities of different complexities. Mol. Ecol. Resour. 2017, 17. [Google Scholar] [CrossRef]
- Bian, J.; Wang, L.Y.; Wu, J.; Simith, N.; Zhang, L.Z.; Wang, Y.F.; Wu, X.B. MTM1 plays an important role in the regulation of zinc tolerance in Saccharomyces cerevisiae. J. Trace Elem. Med. Biol. 2021, 66, 126759. [Google Scholar]
- Zhang, F.; Bian, J.; Chen, X.; Huang, J.; Smith, N.; Lu, W.; Xu, Y.; Lee, J.; Wu, X. Roles for intracellular cation transporters in respiratory growth of yeast. Metallomics 2019, 11, 1667–1678. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Zhang, L.; Lu, J. A study on chemical estimation of pu-erh tea quality. J. Sci. Food Agric. 2005, 85, 381–390. [Google Scholar] [CrossRef]
- Wang, Q.; Peng, C.; Gong, J. Effects of enzymatic action on the formation of theabrownin during solid state fermentation of Pu-erh tea. J. Sci. Food Agric. 2011, 91, 2412–2418. [Google Scholar] [CrossRef] [PubMed]
- Jabeen, S.; Alam, S.; Saleem, M.; Ahmad, W.; Bibi, R.; Hamid, F.S.; Shah, H.U. Withering Timings Affect the total free amino acids and mineral contents of tea leaves during black tea manufacturing. Arab. J. Chem. 2019, 12, 2411–2417. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amplified Region | Sample | Sequences | Bases (bp) | Average Length |
|---|---|---|---|---|
| ITS1F_ITS2R | 21 | 13,780,204 | 312,864,617 | 226.9 |
| Amplified Region | Sample | Sequences | Bases (bp) | Average Length |
|---|---|---|---|---|
| 338F_806R | 21 | 1,055,795 | 439,213,816 | 416.2 |
| No. | Rt (min) | m/z | Formula | Mass Error (mDa) | VIP | Tentative Identification |
|---|---|---|---|---|---|---|
| 1 | 4.07 | 305.0664 | C15H14O7 | −0.3 | 3.63871 | Gallocatechin |
| 2 | 4.7 | 289.0712 | C15H14O6 | −0.5 | 3.33493 | Epicatechin |
| 3 | 4.7 | 289.0712 | C15H14O6 | −0.5 | 3.33493 | Catechin |
| 4 | 5.85 | 441.0825 | C22H18O10 | −0.2 | 2.87242 | Catechin 3-gallate |
| 5 | 5.85 | 441.0825 | C22H18O10 | −0.2 | 2.87242 | Epicatechin 3-gallate |
| 6 | 4.74 | 457.0775 | C22H18O11 | −0.2 | 2.16752 | Gallocatechin 3-gallate |
| 7 | 4.74 | 457.0775 | C22H18O11 | −0.2 | 2.16752 | Epigallocatechin-3-gallate |
| 8 | 4.39 | 311.0768 | C13H14O6 | −0.4 | 2.06244 | Fuzhuanin B |
| 9 | 4.75 | 915.1625 | C44H36O22 | 0 | 1.94329 | Assamicain A |
| 10 | 4.75 | 915.1625 | C44H36O22 | 0 | 1.94329 | Assamicain B |
| 11 | 4.75 | 915.1625 | C44H36O22 | 0 | 1.94329 | Assamicain C |
| 12 | 3.78 | 305.0666 | C15H14O7 | −0.1 | 1.91098 | Epigallocatechin |
| 13 | 4.49 | 349.058 | C15H12O7 | 1.5 | 1.82695 | Taxifolin |
| 14 | 4.49 | 349.058 | C15H12O7 | 1.5 | 1.81141 | (2R,3R)-taxifolin distylin |
| 15 | 6.08 | 755.2041 | C33H40O20 | 0.1 | 1.80491 | Kaempferol 7-galactoside 3-rutinoside |
| 16 | 2.65 | 125.0241 | C6H6O3 | −0.3 | 1.74387 | Phloroglucinol |
| 17 | 2.65 | 125.0241 | C6H6O3 | −0.3 | 1.74387 | Pyrogallol |
| 18 | 3.46 | 343.0668 | C14H16O10 | −0.3 | 1.67692 | Theogallin |
| 19 | 4.3 | 353.0874 | C16H18O9 | −0.4 | 1.57482 | 5-caffeoyl-quinic acid |
| 20 | 4.3 | 353.0874 | C16H18O9 | −0.4 | 1.57482 | Chlorogenic acid |
| 21 | 0.8 | 191.0556 | C7H12O6 | −0.5 | 1.52054 | Quinic acid |
| 22 | 4.75 | 169.0137 | C7H6O5 | −0.6 | 1.50629 | Gallic acid |
| 23 | 6.31 | 231.0656 | C13H12O4 | −0.7 | 1.49003 | Teasperin |
| 24 | 5.92 | 431.0983 | C21H20O10 | −0.1 | 1.35541 | Afzelin |
| 25 | 5.92 | 431.0983 | C21H20O10 | −0.1 | 1.35541 | Isovitexin |
| 26 | 5.92 | 431.0983 | C21H20O10 | −0.1 | 1.35541 | Vitexin |
| 27 | 5.94 | 293.0663 | C13H12O5 | −0.3 | 1.2387 | Xanthocerin |
| 28 | 4.49 | 745.1406 | C37H30O17 | −0.4 | 1.06786 | Catechin-(4α→8)-EGCG |
| 29 | 4.67 | 416.1347 | C21H23NO8 | −0.4 | 1.03255 | Puerin V |
| 30 | 4.67 | 416.1347 | C21H23NO8 | −0.4 | 1.03255 | Puerin VI |
| 31 | 4.67 | 416.1347 | C21H23NO8 | −0.4 | 1.03255 | Puerin VII |
| 32 | 4.67 | 416.1347 | C21H23NO8 | −0.4 | 1.03255 | Puerin VIII |
| 33 | 4.05 | 593.1302 | C30H26O13 | 0.2 | 1.03186 | desG theasinensin F |
| 34 | 4.49 | 745.1406 | C36H28O15 | −0.5 | 1.0057 | Theaflavate B |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, J.; Xu, R.; Zong, L.; Brake, J.; Cheng, L.; Wu, J.; Wu, X. Dynamic Evolution and Correlation between Metabolites and Microorganisms during Manufacturing Process and Storage of Fu Brick Tea. Metabolites 2021, 11, 703. https://doi.org/10.3390/metabo11100703
Li J, Xu R, Zong L, Brake J, Cheng L, Wu J, Wu X. Dynamic Evolution and Correlation between Metabolites and Microorganisms during Manufacturing Process and Storage of Fu Brick Tea. Metabolites. 2021; 11(10):703. https://doi.org/10.3390/metabo11100703
Chicago/Turabian StyleLi, Jing, Ran Xu, Lixuan Zong, Joseph Brake, Lizeng Cheng, Jie Wu, and Xiaobin Wu. 2021. "Dynamic Evolution and Correlation between Metabolites and Microorganisms during Manufacturing Process and Storage of Fu Brick Tea" Metabolites 11, no. 10: 703. https://doi.org/10.3390/metabo11100703
APA StyleLi, J., Xu, R., Zong, L., Brake, J., Cheng, L., Wu, J., & Wu, X. (2021). Dynamic Evolution and Correlation between Metabolites and Microorganisms during Manufacturing Process and Storage of Fu Brick Tea. Metabolites, 11(10), 703. https://doi.org/10.3390/metabo11100703