
AMPK-Mediated Metabolic Switching Is High Effective for Phytochemical Levo-Tetrahydropalmatine (l-THP) to Reduce Hepatocellular Carcinoma Tumor Growth

 ,
, 
Abstract
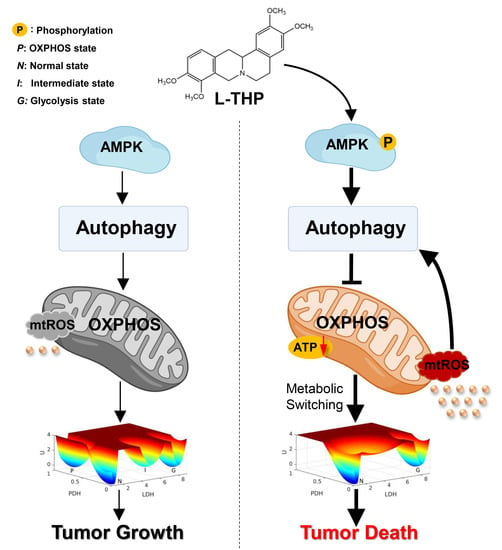
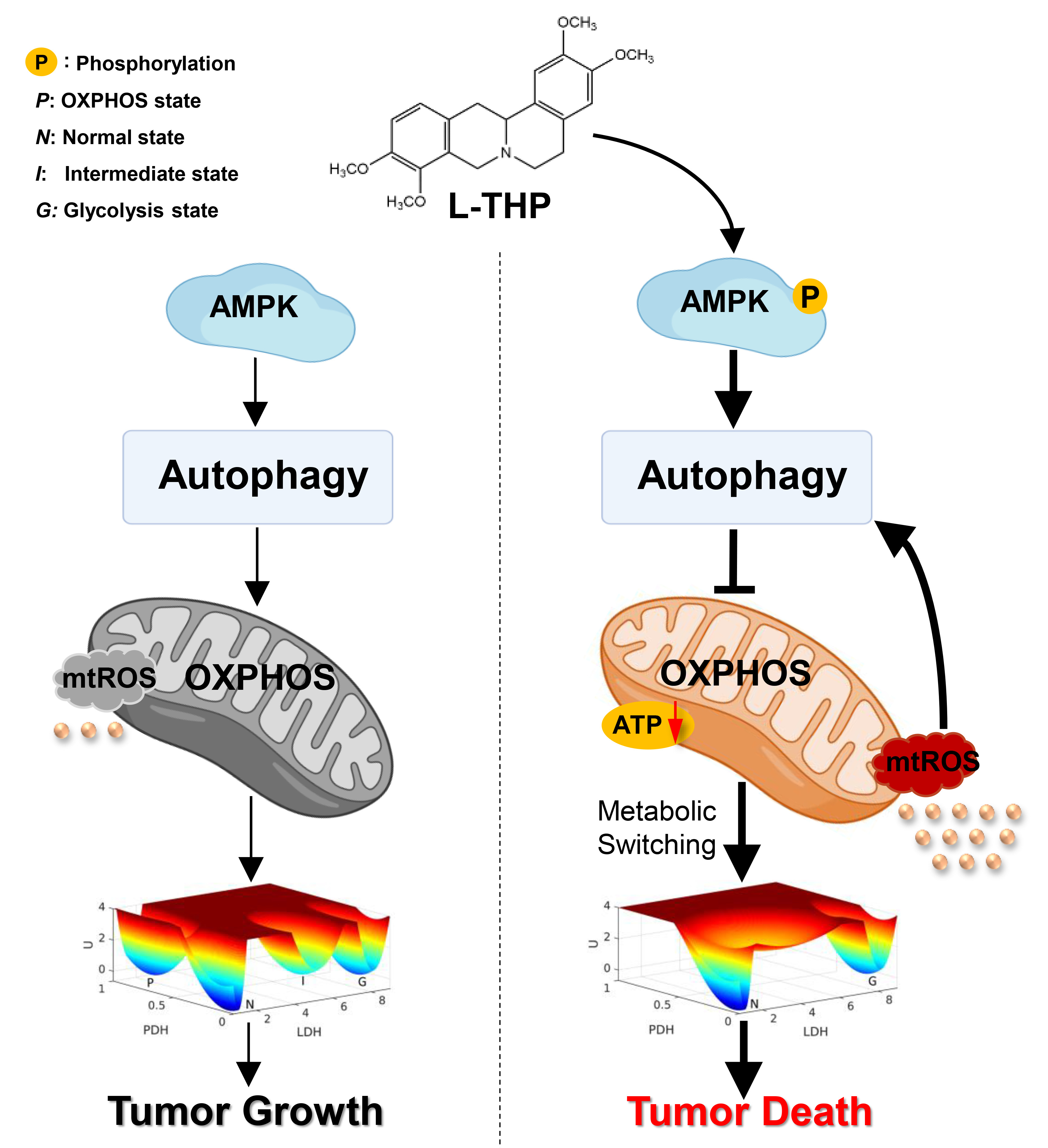
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. AMPK Is a Key Metabolic Hub in the Cancer Gene-Metabolism Integrative Network
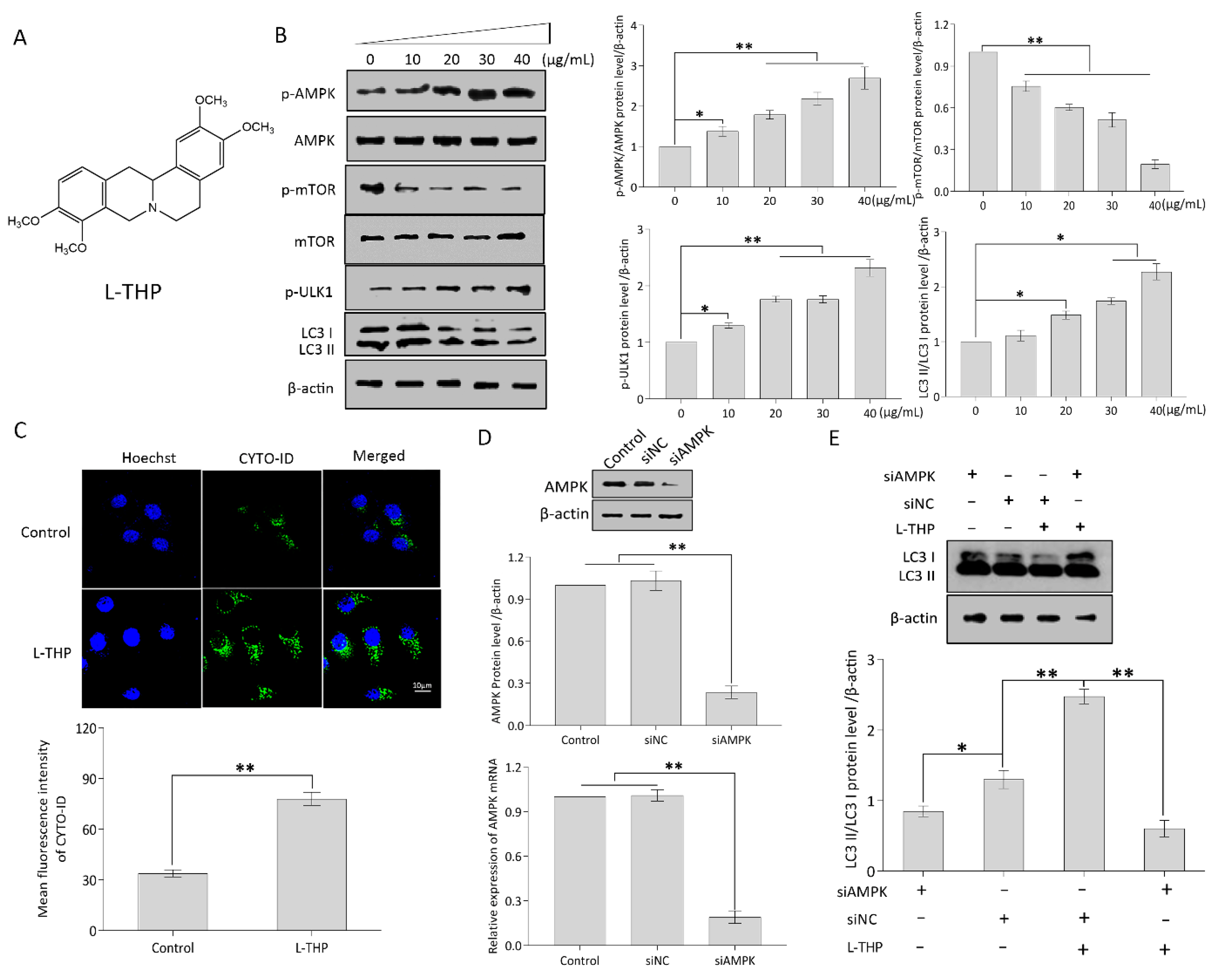
2.2. l-THP Enhances Autophagy by Activating the AMPK-mTOR-ULK1 Axis
2.2.1. l-THP Activates the AMPK-mTOR-ULK1 Signaling Pathway
2.2.2. l-THP Enhances the Autophagic Flux in HepG2 Cells
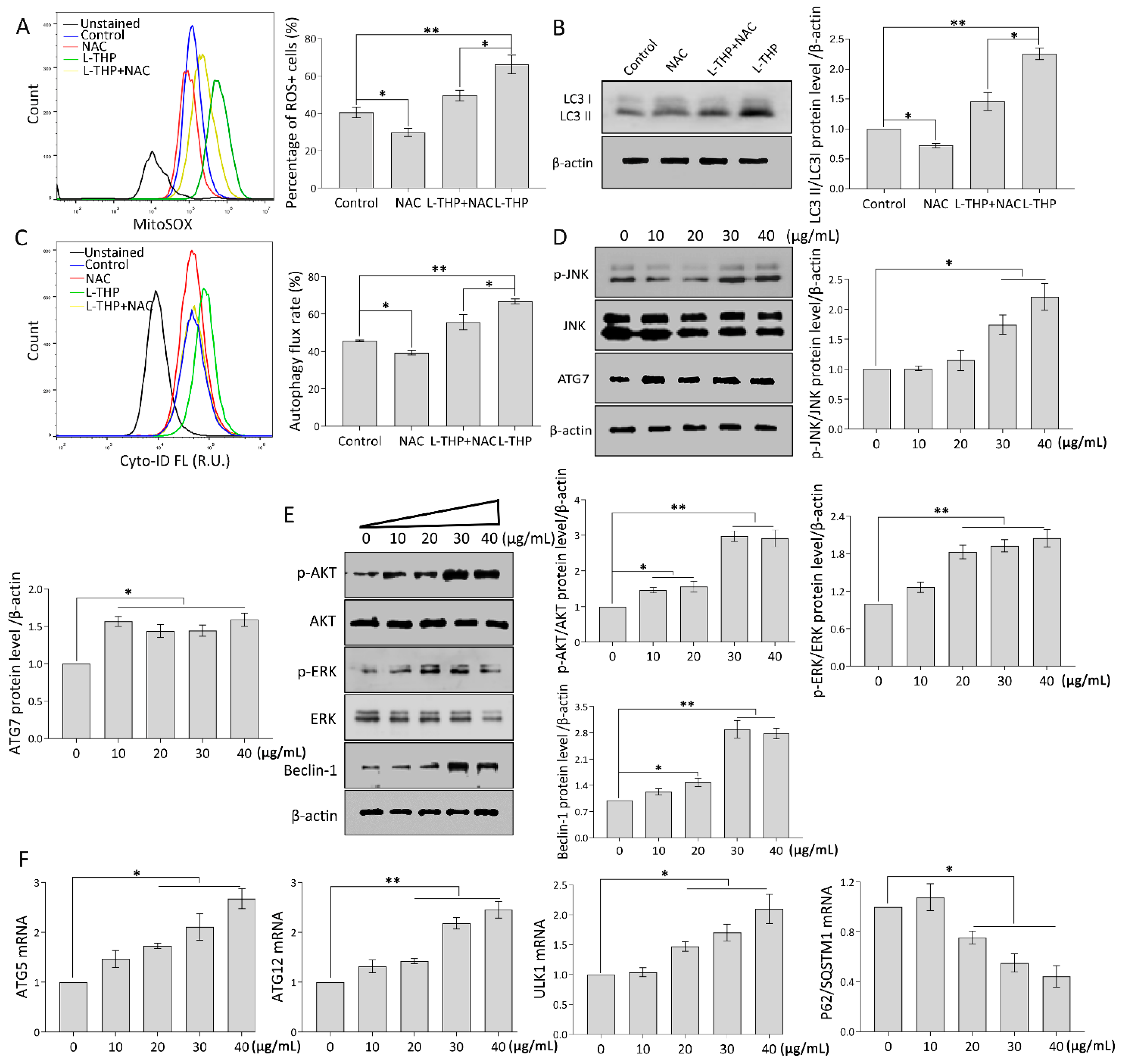
2.3. l-THP Increases the Autophagic Flux via the ROS-JNK-ATG Cascade
2.3.1. mtROS Molecular Pathway Is Essential to Autophagy Induction
2.3.2. Specific Genes Trigger Autophagy Occurrence
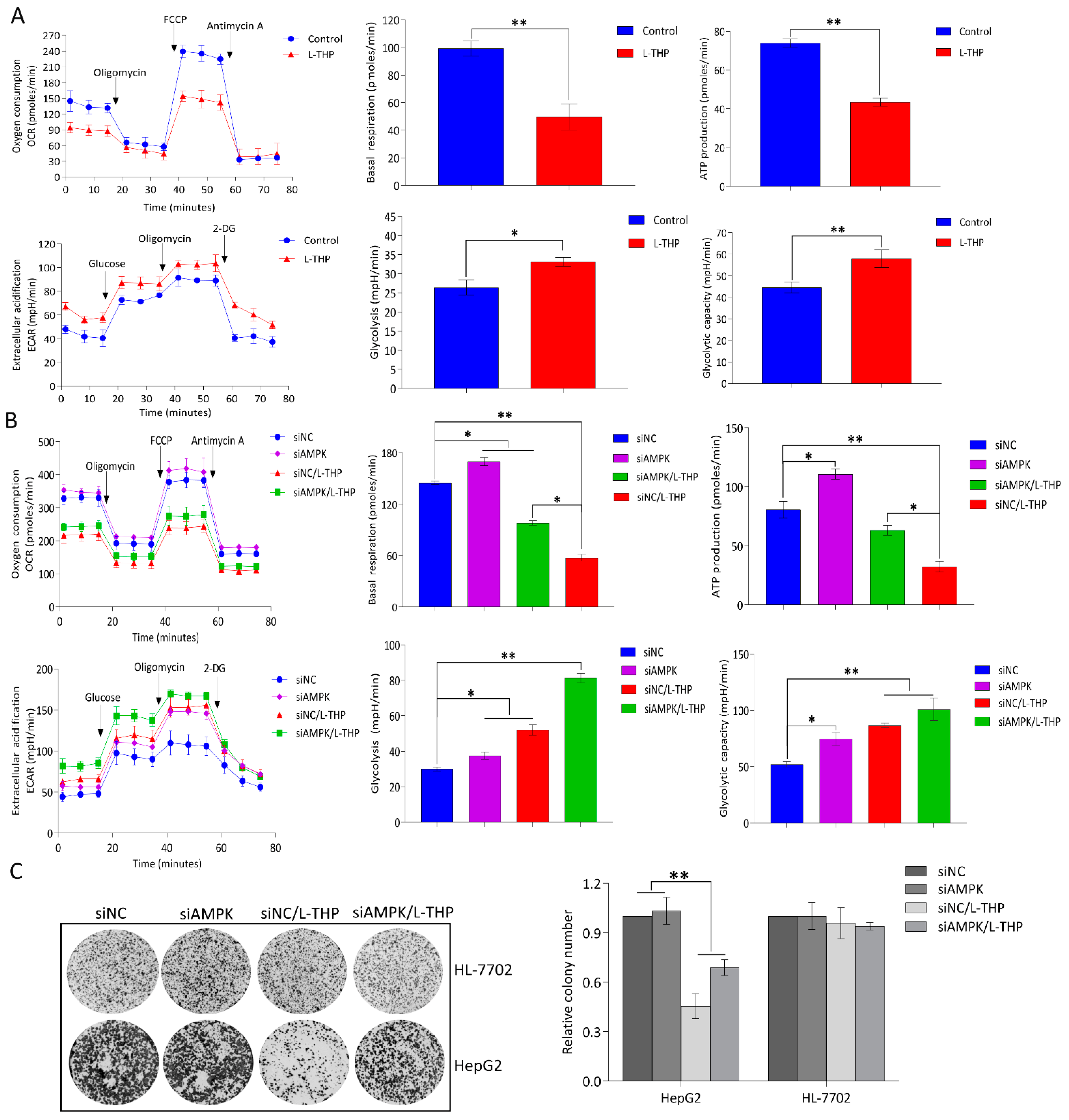
2.4. AMPK Activation by l-THP Decreased OXPHOS and Increased Glycolysis
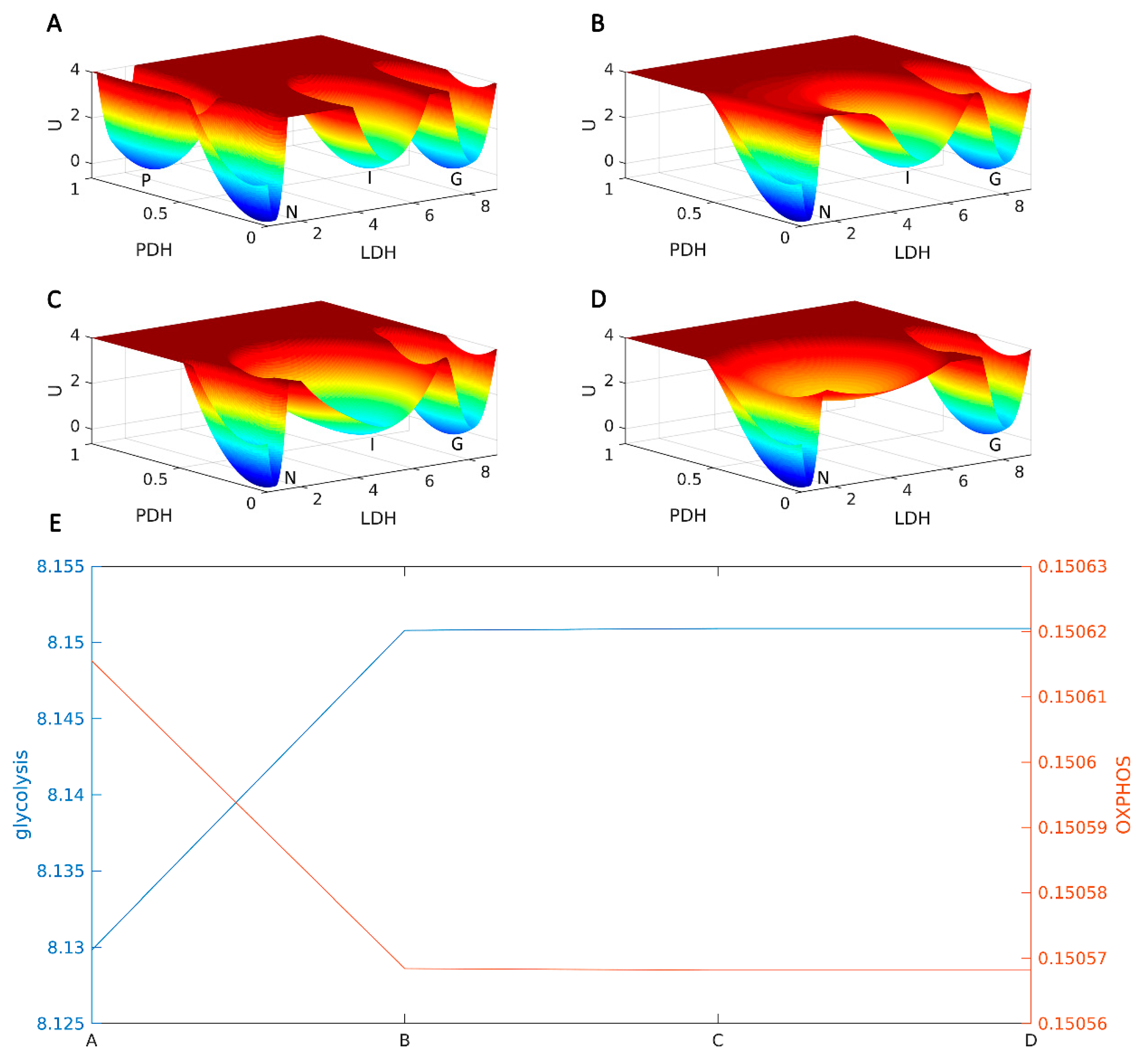
2.4.1. l-THP Changes the Metabolic Homeostasis between OXPHOS and Glycolysis
2.4.2. AMPK Is a Hub for Metabolic Switching and Proliferation in HepG2 Cells
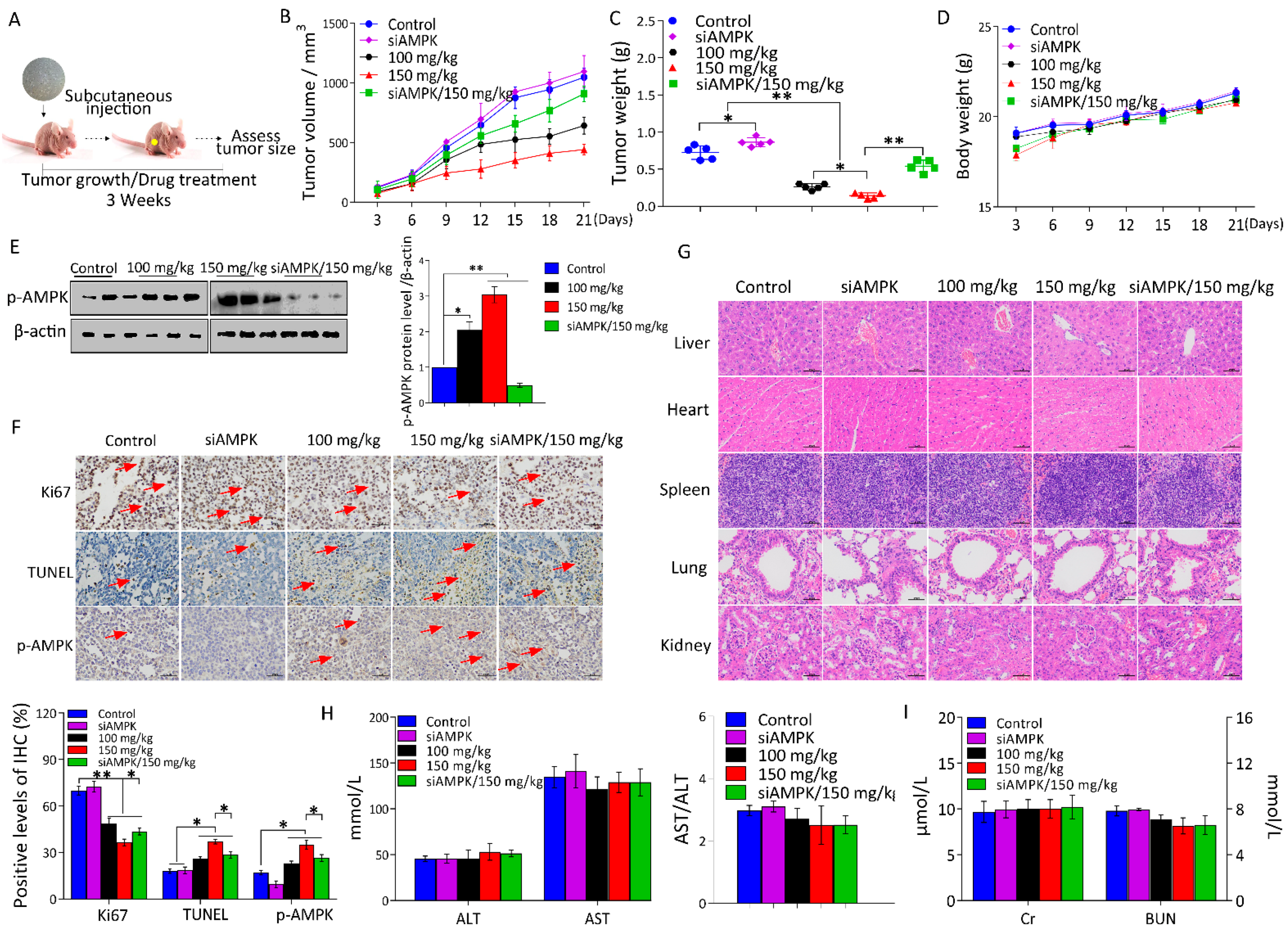
2.5. l-THP Inhibits Tumor Growth in the Nude Mice Model
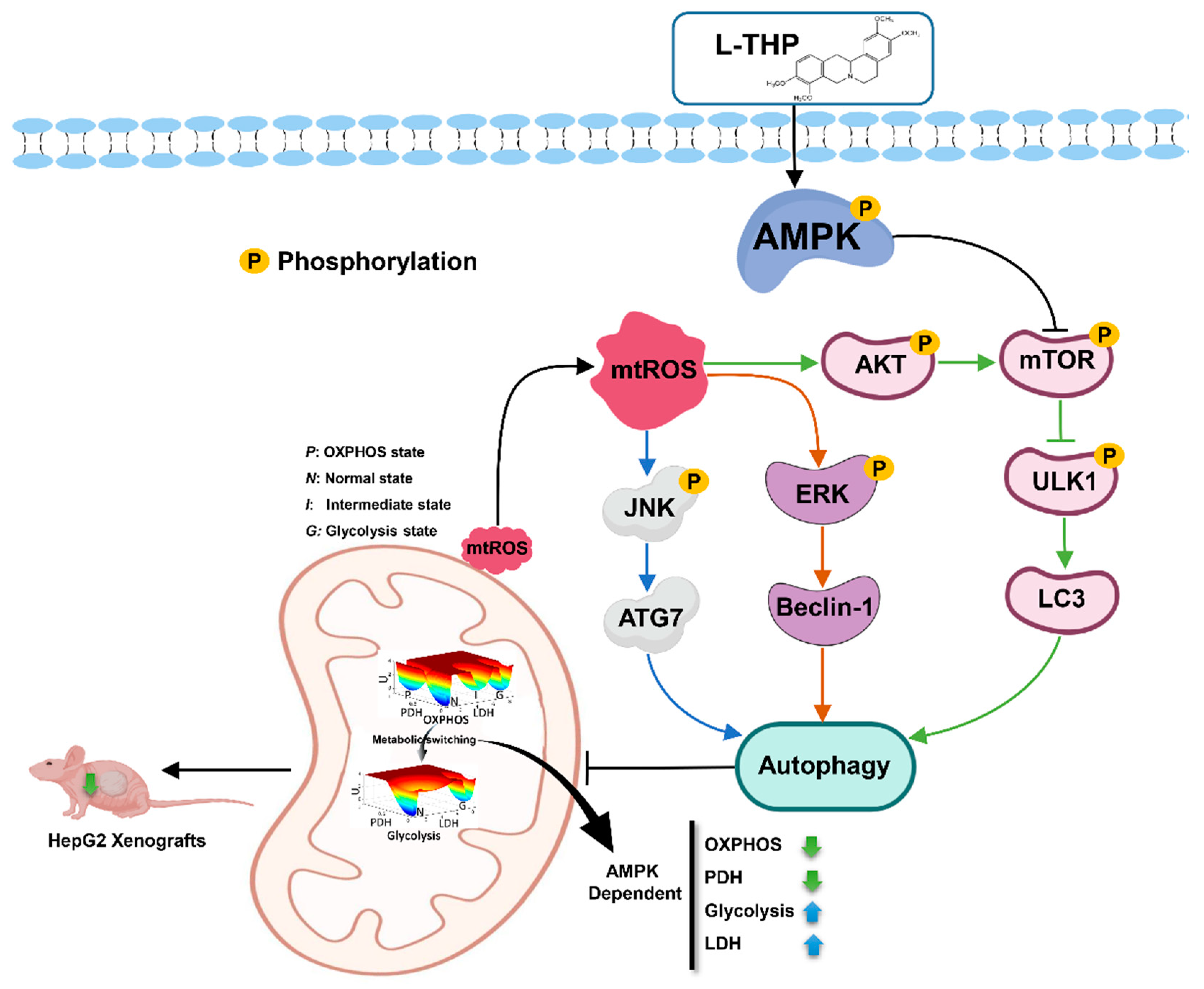
3. Conclusions
4. Materials and Methods
4.1. Materials and Reagents
4.2. Cell Culture
4.3. Self-Consistent Mean Field Approximation Approach
4.4. Detection of Autophagy Using Confocal Microscopy
4.5. Detection of Mitochondrial ROS Production by MitoSOX
4.6. Autophagic Flux Analysis
4.7. Western Blotting Analysis
4.8. siRNA Transfection
4.9. RNA Extraction and Quantitative Reverse Transcription-Polymerase Chain Reaction (qRT-PCR) Analysis
4.10. Colony Formation Assay
4.11. Cell Metabolism Measurements
4.12. Xenograft Tumor Model BALB/c Nude Mice and Treatment Strategies
4.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca-Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allemani, C.; Matsuda, T.; Di Carlo, V.; Harewood, R.; Matz, M.; Niksic, M.; Bonaventure, A.; Valkov, M.; Johnson, C.J.; Esteve, J.; et al. Global surveillance of trends in cancer survival 2000-14 (CONCORD-3): Analysis of individual records for 37 513 025 patients diagnosed with one of 18 cancers from 322 population-based registries in 71 countries. Lancet 2018, 391, 1023–1075. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; He, S.; Ma, B. Autophagy and autophagy-related proteins in cancer. Mol. Cancer 2020, 19, 12. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.M.; Macleod, K.F. Autophagy and cancer cell metabolism. Int. Rev. Cell Mol. Biol. 2019, 347, 145–190. [Google Scholar] [CrossRef] [PubMed]
- Russo, M.; Russo, G.L. Autophagy inducers in cancer. Biochem. Pharmacol. 2018, 153, 51–61. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, X.; Huang, X.; Hong, T.; Zhang, K.; Qi, W.; Guo, M.; Nie, S. Lysosome-mediated cytotoxic autophagy contributes to tea polysaccharide-induced colon cancer cell death via mTOR-TFEB signaling. J. Agric. Food Chem. 2021, 69, 686–697. [Google Scholar] [CrossRef]
- Sabharwal, S.S.; Schumacker, P.T. Mitochondrial ROS in cancer: Initiators, amplifiers or an Achilles’ heel? Nat. Rev. Cancer 2014, 14, 709–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Polson, E.S.; Kuchler, V.B.; Abbosh, C.; Ross, E.M.; Mathew, R.K.; Beard, H.A.; da Silva, B.; Holding, A.N.; Ballereau, S.; Chuntharpursat-Bon, E.; et al. KHS101 disrupts energy metabolism in human glioblastoma cells and reduces tumor growth in mice. Sci. Transl. Med. 2018, 10, 2718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patra, K.C.; Weerasekara, V.K.; Bardeesy, N. AMPK-mediated lysosome biogenesis in lung cancer growth. Cell Metab. 2019, 29, 238–240. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Wang, J. Uncovering the underlying mechanisms of cancer metabolism through the landscapes and probability flux quantifications. iScience 2020, 23, 101002. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.B.; Zhou, G.; Li, C. AMPK: An emerging drug target for diabetes and the metabolic syndrome. Cell Metab. 2009, 9, 407–416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia, D.; Shaw, R.J. AMPK: Mechanisms of cellular energy sensing and restoration of metabolic balance. Mol. Cell 2017, 66, 789–800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, R.; Zhang, W.; Tang, K.; Zhang, H.; Zhang, Y.; Li, D.; Li, Y.; Xu, P.; Luo, S.; Cai, W.; et al. Switch of glycolysis to gluconeogenesis by dexamethasone for treatment of hepatocarcinoma. Nat. Commun. 2013, 4, 2508. [Google Scholar] [CrossRef] [Green Version]
- Wu, H.; Waldbauer, K.; Tang, L.; Xie, L.; McKinnon, R.; Zehl, M.; Yang, H.; Xu, H.; Kopp, B. Influence of vinegar and wine processing on the alkaloid content and composition of the traditional Chinese medicine Corydalis Rhizoma (Yanhusuo). Molecules 2014, 19, 11487–11504. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Gao, J.L.; Ji, J.W.; Gao, M.; Yin, Q.S.; Qiu, Q.L.; Wang, C.; Chen, S.Z.; Xu, J.; Liang, R.S.; et al. Cytotoxicity enhancement in MDA-MB-231 cells by the combination treatment of tetrahydropalmatine and berberine derived from Corydalis yanhusuo W. T. Wang. J. Intercult. Ethnopharmacol. 2014, 3, 68–72. [Google Scholar] [CrossRef]
- Chu, H.; Jin, G.; Friedman, E.; Zhen, X. Recent development in studies of tetrahydroprotoberberines: Mechanism in antinociception and drug addiction. Cell Mol. Neurobiol. 2008, 28, 491–499. [Google Scholar] [CrossRef]
- Rabinovitch, R.C.; Samborska, B.; Faubert, B.; Ma, E.H.; Gravel, S.P.; Andrzejewski, S.; Raissi, T.C.; Pause, A.; St-Pierre, J.; Jones, R.G. AMPK maintains cellular metabolic homeostasis through regulation of mitochondrial reactive oxygen species. Cell Rep. 2017, 21, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Sarkissyan, M.; Mcghee, E.; Lee, S.; Vadgama, J.V. Combined inhibition of glycolysis and AMPK induces synergistic breast cancer cell killing. Breast Cancer Res. Treat. 2015, 151, 529–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, Y.; Shao, D.; Zhao, Y.; Zhang, F.; Zheng, X.; Tan, Y.; He, K.; Li, J.; Chen, L. Berberine reverses hypoxia-induced chemoresistance in breast cancer through the inhibition of AMPK-HIF-1α. Int. J. Biol. Sci. 2017, 13, 794–803. [Google Scholar] [CrossRef] [PubMed]
- Barth, S.; Glick, D.; Macleod, K.F. Autophagy: Assays and artifacts. J. Pathol. 2010, 221, 117–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunlop, E.A.; Hunt, D.K.; Acosta-Jaquez, H.A.; Fingar, D.C.; Tee, A.R. ULK1 inhibits mTORC1 signaling, promotes multisite Raptor phosphorylation and hinders substrate binding. Autophagy 2011, 7, 737–747. [Google Scholar] [CrossRef]
- Kim, Y.C.; Guan, K.L. mTOR: A pharmacologic target for autophagy regulation. J. Clin. Investig. 2015, 125, 25–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurley, J.H.; Young, L.N. Mechanisms of autophagy initiation. Annu. Rev. Biochem. 2017, 86, 225–244. [Google Scholar] [CrossRef]
- Jaber, N.; Dou, Z.; Chen, J.S.; Catanzaro, J.; Jiang, Y.P.; Ballou, L.M.; Selinger, E.; Ouyang, X.; Lin, R.Z.; Zhang, J. Class III PI3K Vps34 plays an essential role in autophagy and in heart and liver function. Proc. Natl. Acad. Sci. USA 2012, 109, 2003–2008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellot, G.L.; Liu, D.; Pervaiz, S. ROS, autophagy, mitochondria and cancer: Ras, the hidden master? Mitochondrion 2013, 13, 155–162. [Google Scholar] [CrossRef]
- Lim, H.; Lim, Y.M.; Kim, K.H.; Jeon, Y.E.; Lee, M.S. A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat. Commun. 2018, 9, 1438. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Levine, B. Autophagic cell death: The story of a misnomer. Nat. Rev. Mol. Cell Bio. 2008, 9, 1004–1010. [Google Scholar] [CrossRef] [PubMed]
- Gibson, S.B.; Azad, M.B.; Chen, Y. Regulation of autophagy by Reactive Oxygen Species (ROS): Implications for cancer progression and treatment. Antioxid. Redox Signal. 2009, 11, 777–790. [Google Scholar] [CrossRef]
- Spiegel, J.; Cromm, P.M.; Zimmermann, G.; Grossmann, T.N.; Waldmann, H. Small-molecule modulation of Ras signaling. Nat. Chem. Biol. 2014, 10, 613–622. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, S.; Mano, S.; Oikawa, K.; Hikino, K.; Teshima, K.M.; Kimori, Y.; Nishimura, M.; Shimazaki, K.I.; Takemiya, A. Autophagy controls reactive oxygen species homeostasis in guard cells that is essential for stomatal opening. Proc. Natl. Acad. Sci. USA 2019, 116, 19187–19192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Byun, J.Y.; Yoon, C.-H.; An, S.; Park, I.C.; Kang, C.M.; Kim, M.J.; Lee, S.J. The Rac1/MKK7/JNK pathway signals upregulation of Atg5 and subsequent autophagic cell death in response to oncogenic Ras. Carcinogenesis 2009, 30, 1880–1888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poillet-Perez, L.; Despouy, G.; Delage-Mourroux, R.; Boyer-Guittaut, M. Interplay between ROS and autophagy in cancer cells, from tumor initiation to cancer therapy. Redox Biol. 2015, 4, 184–192. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Dou, Z.C.; Ren, W.H.; Li, S.M.; Liang, X.; Zhi, K.A.O. CircCDR1as upregulates autophagy under hypoxia to promote tumor cell survival via AKT/ERK(½)/mTOR signaling pathways in oral squamous cell carcinomas. Cell Death Dis. 2019, 10, 745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Fan, D.; Ru, B.; Cheng, K.W.; Hu, S.; Zhang, J.; Li, E.T.; Wang, M. 6-C-(E-phenylethenyl)naringenin induces cell growth inhibition and cytoprotective autophagy in colon cancer cells. Eur. J. Cancer 2016, 68, 38–50. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Chang, J.T.; Guo, B.; Hansen, M.; Jia, K.L.; Kovacs, A.L.; Kumsta, C.; Lapierre, L.R.; Legouis, R.; Lin, L.; et al. Guidelines for monitoring autophagy in Caenorhabditis elegans. Autophagy 2015, 11, 9–27. [Google Scholar] [CrossRef] [PubMed]
- Kimmelman, A.C.; White, E. Autophagy and tumor metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [Green Version]
- Bonora, M.; Wieckowsk, M.R.; Chinopoulos, C.; Kepp, O.; Kroemer, G.; Galluzzi, L.; Pinton, P. Molecular mechanisms of cell death: Central implication of ATP synthase in mitochondrial permeability transition. Oncogene 2015, 34, 1608. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Hao, B.; Zhang, M.; Reiter, R.J.; Lin, S.; Zheng, T.; Chen, X.; Ren, Y.; Yue, L.; Abay, B.; et al. Melatonin enhances radiofrequency-induced NK antitumor immunity, causing cancer metabolism reprogramming and inhibition of multiple pulmonary tumor development. Signal Transduct. Target. Ther. 2021, 6, 330. [Google Scholar] [CrossRef]
- Dai, W.; Xu, Y.; Mo, S.; Li, Q.; Yu, J.; Wang, R.; Ma, Y.; Ni, Y.; Xiang, W.; Han, L.; et al. GLUT3 induced by AMPK/CREB1 axis is key for withstanding energy stress and augments the efficacy of current colorectal cancer therapies. Signal Transduct. Target. Ther. 2020, 5, 177. [Google Scholar] [CrossRef]
- Seyfried, T.N.; Chinopoulos, C. Can the mitochondrial metabolic theory explain better the origin and management of cancer than can the somatic mutation theory? Metabolites 2021, 11, 572. [Google Scholar] [CrossRef] [PubMed]
- Liang, J.; Cao, R.; Zhang, Y.; Xia, Y.; Zheng, Y.; Li, X.; Wang, L.; Yang, W.; Lu, Z. PKM2 dephosphorylation by Cdc25A promotes the Warburg effect and tumorigenesis. Nat. Commun. 2016, 7, 12431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elgendy, M.; Ciro, M.; Hosseini, A.; Weiszmann, J.; Mazzarella, L.; Ferrari, E.; Cazzoli, R.; Curigliano, G.; DeCensi, A.; Bonanni, B.; et al. Combination of hypoglycemia and metformin impairs tumor metabolic plasticity and growth by modulating the PP2A-GSK3 beta-MCL-1 axis. Cancer Cell 2019, 35, 798–815. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, D.B.; Shaw, R.J. The LKB1-AMPK pathway: Metabolism and growth control in tumour suppression. Nat. Rev. Cancer 2009, 9, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Eichner, L.J.; Brun, S.N.; Herzig, S.; Young, N.P.; Curtis, S.D.; Shackelford, D.B.; Shokhirev, M.N.; Leblanc, M.; Vera, L.I.; Hutchins, A.; et al. Genetic analysis reveals AMPK is required to support tumor growth in murine Kras-dependent lung cancer models. Cell Metab. 2019, 29, 285–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yin, X.; Li, W.; Zhang, J.; Zhao, W.; Cai, H.; Zhang, C.; Liu, Z.; Guo, Y.; Wang, J. AMPK-Mediated Metabolic Switching Is High Effective for Phytochemical Levo-Tetrahydropalmatine (l-THP) to Reduce Hepatocellular Carcinoma Tumor Growth. Metabolites 2021, 11, 811. https://doi.org/10.3390/metabo11120811
Yin X, Li W, Zhang J, Zhao W, Cai H, Zhang C, Liu Z, Guo Y, Wang J. AMPK-Mediated Metabolic Switching Is High Effective for Phytochemical Levo-Tetrahydropalmatine (l-THP) to Reduce Hepatocellular Carcinoma Tumor Growth. Metabolites. 2021; 11(12):811. https://doi.org/10.3390/metabo11120811
Chicago/Turabian StyleYin, Xunzhe, Wenbo Li, Jiaxin Zhang, Wenjing Zhao, Huaxing Cai, Chi Zhang, Zuojia Liu, Yan Guo, and Jin Wang. 2021. "AMPK-Mediated Metabolic Switching Is High Effective for Phytochemical Levo-Tetrahydropalmatine (l-THP) to Reduce Hepatocellular Carcinoma Tumor Growth" Metabolites 11, no. 12: 811. https://doi.org/10.3390/metabo11120811
APA StyleYin, X., Li, W., Zhang, J., Zhao, W., Cai, H., Zhang, C., Liu, Z., Guo, Y., & Wang, J. (2021). AMPK-Mediated Metabolic Switching Is High Effective for Phytochemical Levo-Tetrahydropalmatine (l-THP) to Reduce Hepatocellular Carcinoma Tumor Growth. Metabolites, 11(12), 811. https://doi.org/10.3390/metabo11120811