
Microbial Metabolites in Colorectal Cancer: Basic and Clinical Implications


Abstract
:1. Introduction
2. Measurement of Microbial Metabolites by Metabolomics
3. Function of Microbial Metabolites in CRC
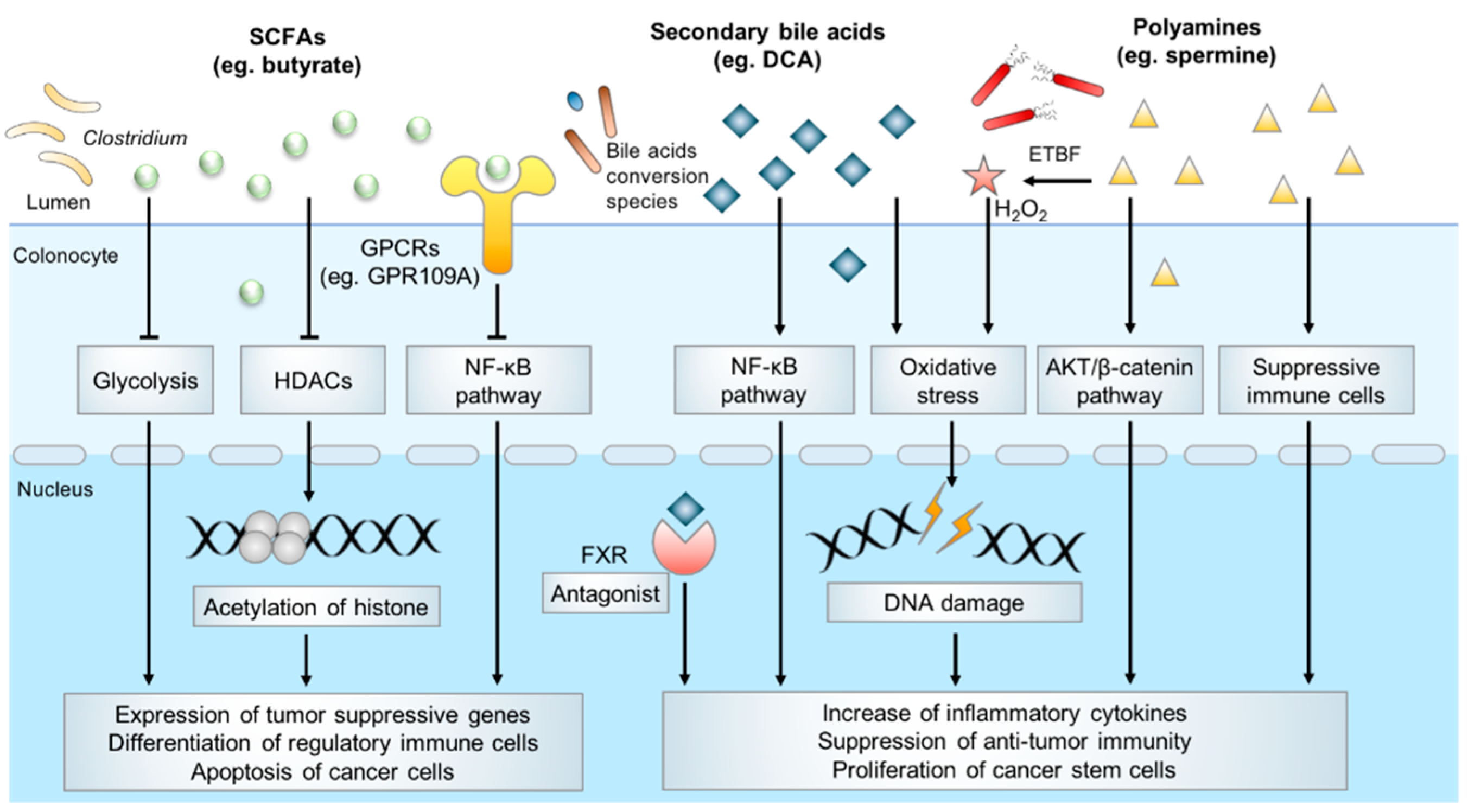
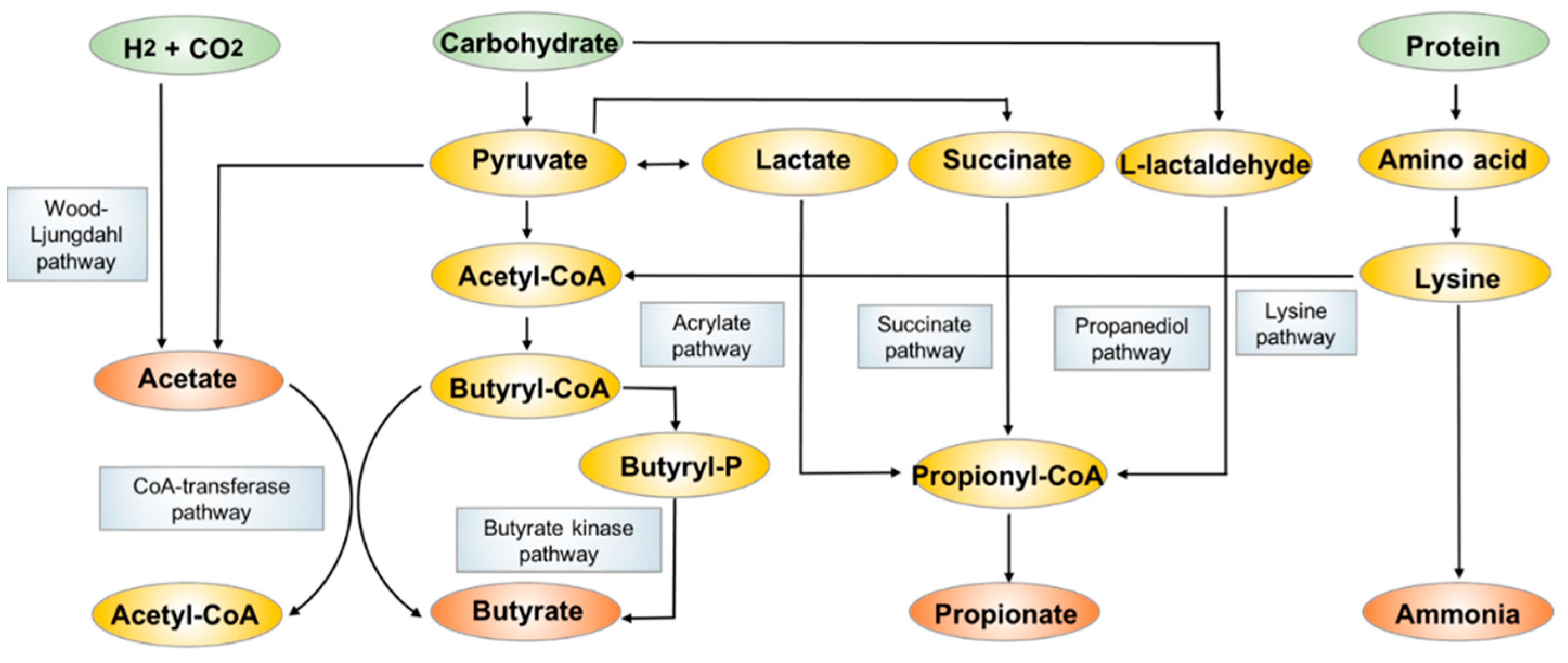
3.1. Short-Chain Fatty Acids (SCFAs)
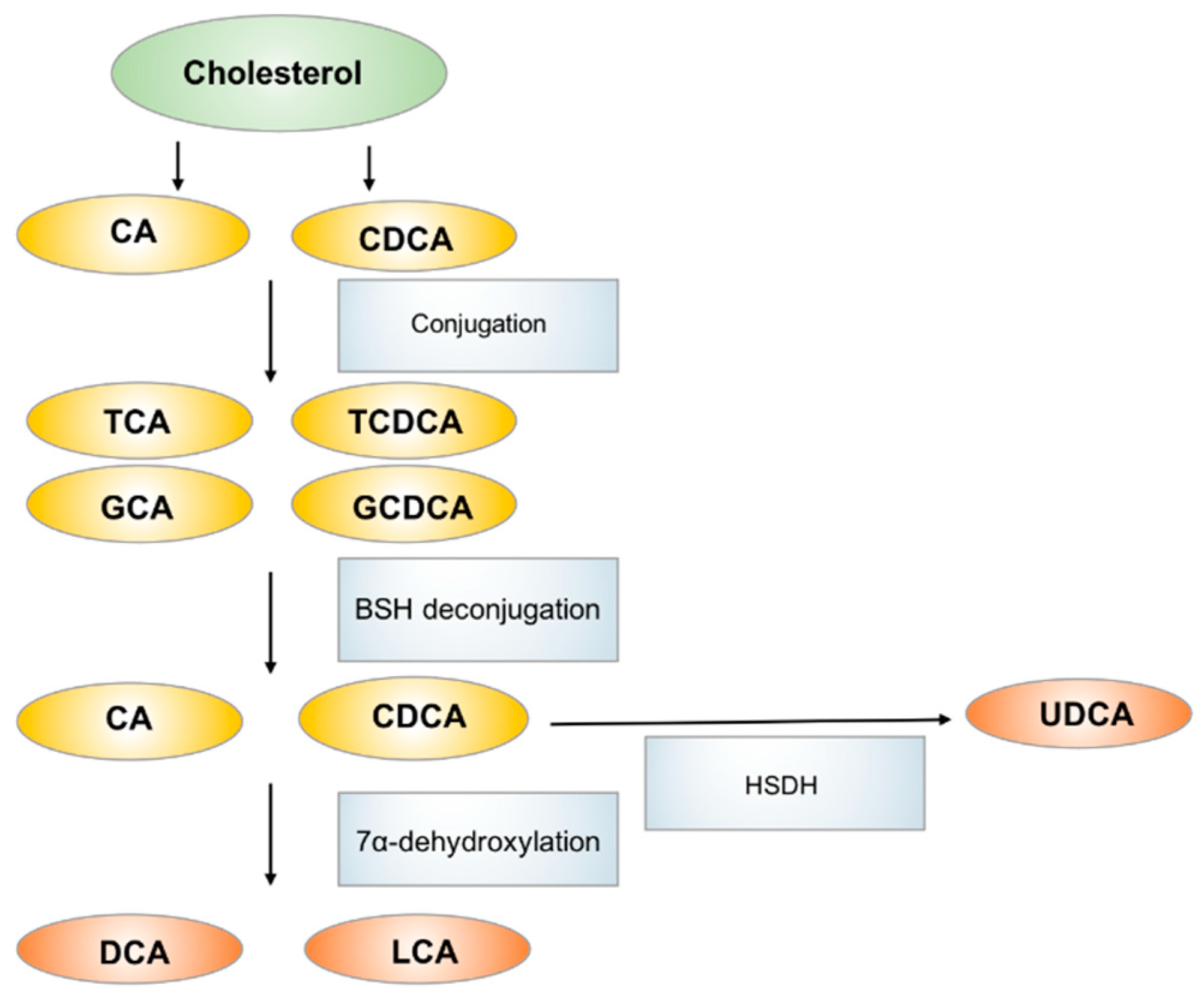
3.2. Bile Acids
3.3. Polyamines
3.4. Other Microbial Metabolites in CRC
4. Implication for Clinical Applications of Microbial Metabolites in CRC
4.1. Use of Microbial Metabolites as Biomarkers
4.2. Modulation of Microbial Metabolites for CRC Prevention and Treatment
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Mph, K.D.M.; Sauer, A.G.; Fedewa, S.A.; Butterly, L.F.; Anderson, J.C.; Cercek, A.; Smith, R.A.; Jemal, A. Colorectal cancer statistics, 2020. CA A Cancer J. Clin. 2020, 70, 145–164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keum, N.; Giovannucci, E. Global burden of colorectal cancer: Emerging trends, risk factors and prevention strategies. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 713–732. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of in-cidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dekker, E.; Tanis, P.J.; Vleugels, J.L.A.; Kasi, P.M.; Wallace, M.B. Colorectal cancer. Lancet 2019, 394, 1467–1480. [Google Scholar] [CrossRef]
- Mousavi, S.M.; Fallah, M.; Sundquist, K.; Hemminki, K. Age- and time-dependent changes in cancer incidence among immigrants to Sweden: Colorectal, lung, breast and prostate cancers. Int. J. Cancer 2012, 131, E122–E128. [Google Scholar] [CrossRef] [PubMed]
- Kune, G.A. The Melbourne Colorectal Cancer Study: Reflections on a 30-year experience. Med. J. Aust. 2010, 193, 648–652. [Google Scholar] [CrossRef]
- Song, M.; Chan, A.T.; Sun, J. Influence of the Gut Microbiome, Diet, and Environment on Risk of Colorectal Cancer. Gastroenterology 2020, 158, 322–340. [Google Scholar] [CrossRef]
- Claus, S.P.; Guillou, H.; Ellero-Simatos, S. The gut microbiota: A major player in the toxicity of environmental pollutants? NPJ Biofilms Microbiomes 2016, 2, 16003. [Google Scholar] [CrossRef]
- Drewes, J.L.; Housseau, F.; Sears, C.L. Sporadic colorectal cancer: Microbial contributors to disease prevention, development and therapy. Br. J. Cancer 2016, 115, 273–280. [Google Scholar] [CrossRef]
- Frank, D.N.; Zhu, W.; Sartor, R.B.; Li, E. Investigating the biological and clinical significance of human dysbioses. Trends Microbiol. 2011, 19, 427–434. [Google Scholar] [CrossRef] [Green Version]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current Understanding of Dysbiosis in Disease in Human and Animal Models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.H.; Yu, J. Gut microbiota in colorectal cancer: Mechanisms of action and clinical applications. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 690–704. [Google Scholar] [CrossRef]
- Castellarin, M.; Warren, R.L.; Freeman, J.D.; Dreolini, L.; Krzywinski, M.; Strauss, J.; Barnes, R.; Watson, P.; Allen-Vercoe, E.; Moore, R.A.; et al. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 2012, 22, 299–306. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum Promotes Colorectal Carcinogenesis by Modulating E-Cadherin/β-Catenin Signaling via its FadA Adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum Potentiates Intestinal Tumorigenesis and Modulates the Tumor-Immune Microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Gur, C.; Ibrahim, Y.; Isaacson, B.; Yamin, R.; Abed, J.; Gamliel, M.; Enk, J.; Bar-On, Y.; Stanietsky-Kaynan, N.; Coppenhagen-Glazer, S.; et al. Binding of the Fap2 Protein of Fusobacterium nucleatum to Human Inhibitory Receptor TIGIT Protects Tumors from Immune Cell Attack. Immunity 2015, 42, 344–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zeng, H.; Umar, S.; Rust, B.; Lazarova, D.; Bordonaro, M. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. Int. J. Mol. Sci. 2019, 20, 1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, A.J.; Moore, S.C.; Boca, S.; Huang, W.-Y.; Xiong, X.; Stolzenberg-Solomon, R.; Sinha, R.; Sampson, J.N. A prospective study of serum metabolites and colorectal cancer risk. Cancer 2014, 120, 3049–3057. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Yu, Y.; Wang, Y.; Wang, J.; Guan, R.; Sun, Y.; Shi, F.; Gao, J.; Fu, X. Role of SCFAs in gut microbiome and glycolysis for colorectal cancer therapy. J. Cell. Physiol. 2019, 234, 17023–17049. [Google Scholar] [CrossRef] [PubMed]
- Xie, G.; Zhang, S.; Zheng, X.; Jia, W. Metabolomics approaches for characterizing metabolic interactions between host and its commensal microbes. Electrophor. 2013, 34, 2787–2798. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.X.; Wang, S.-Y.; Kuo, C.-H.; Tsai, I.-L. Metabolome analysis for investigating host-gut microbiota interactions. J. Formos. Med Assoc. 2019, 118, S10–S22. [Google Scholar] [CrossRef]
- Nordström, A.; Lewensohn, R. Metabolomics: Moving to the Clinic. J. Neuroimmune Pharmacol. 2010, 5, 4–17. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S. Emerging applications of metabolomics in drug discovery and precision medicine. Nat. Rev. Drug Discov. 2016, 15, 473–484. [Google Scholar] [CrossRef] [PubMed]
- Krautkramer, K.A.; Fan, J.; Bäckhed, F. Gut microbial metabolites as multi-kingdom intermediates. Nat. Rev. Microbiol. 2020, 19, 77–94. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, H.; Zhu, M.-J. A sensitive GC/MS detection method for analyzing microbial metabolites short chain fatty acids in fecal and serum samples. Talanta 2019, 196, 249–254. [Google Scholar] [CrossRef]
- Ibáñez, C.; Simó, C.; Palazoglu, M.; Cifuentes, A. GC-MS based metabolomics of colon cancer cells using different extraction solvents. Anal. Chim. Acta 2017, 986, 48–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chetwynd, A.J.; Ogilvie, L.A.; Nzakizwanayo, J.; Pazdirek, F.; Hoch, J.; Dedi, C. The potential of nanoflow liquid chromatog-raphy-nano electrospray ionisation-mass spectrometry for global profiling the faecal metabolome. J. Chromatogr. A 2019, 1600, 127–136. [Google Scholar] [CrossRef]
- Röth, D.; Chiang, A.J.; Hu, W.; Gugiu, G.B.; Morra, C.N.; Versalovic, J.; Kalkum, M. Two-carbon folate cycle of commensal Lactobacillus reuteri 6475 gives rise to immunomodulatory ethionine, a source for histone ethylation. FASEB J. 2019, 33, 3536–3548. [Google Scholar] [CrossRef] [PubMed]
- Lamichhane, S.; Yde, C.C.; Schmedes, M.S.; Jensen, H.M.; Meier, S.; Bertram, H.C. Strategy for Nuclear-Magnetic-Resonance-Based Metabolomics of Human Feces. Anal. Chem. 2015, 87, 5930–5937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schrimpe-Rutledge, A.C.; Codreanu, S.G.; Sherrod, S.D.; McLean, J.A. Untargeted Metabolomics Strategies—Challenges and Emerging Directions. J. Am. Soc. Mass Spectrom. 2016, 27, 1897–1905. [Google Scholar] [CrossRef] [Green Version]
- Rath, C.M.; Alexandrov, T.; Higginbottom, S.K.; Song, J.; Milla, M.E.; Fischbach, M.A.; Sonnenburg, J.L.; Dorrestein, P.C. Molecular Analysis of Model Gut Microbiotas by Imaging Mass Spectrometry and Nanodesorption Electrospray Ionization Reveals Dietary Metabolite Transformations. Anal. Chem. 2012, 84, 9259–9267. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, S.; Wong, A.C.-Y.; Yan, X.; Wu, B.; Zhao, H.; Tibshirani, R.J.; Zare, R.N.; Brooks, J.D. Early detection of unilateral ureteral obstruction by desorption electrospray ionization mass spectrometry. Sci. Rep. 2019, 9, 11007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Xie, H.; Ren, J.; Chen, Y.; Li, X.; Chen, X.; Chan, T.-W.D. Metabolomic approach for rapid differentiation of Fritillaria bulbs by matrix-assisted laser desorption/ionization mass spectrometry and multivariate statistical analysis. J. Pharm. Biomed. Anal. 2020, 185, 113177. [Google Scholar] [CrossRef]
- Mayali, X. NanoSIMS: Microscale Quantification of Biogeochemical Activity with Large-Scale Impacts. Ann. Rev. Mar. Sci. 2020, 12, 449–467. [Google Scholar] [CrossRef] [PubMed]
- Liebal, U.W.; Phan, A.N.T.; Sudhakar, M.; Raman, K.; Blank, L.M. Machine Learning Applications for Mass Spectrometry-Based Metabolomics. Metabolites 2020, 10, 243. [Google Scholar] [CrossRef]
- Sen, P.; Lamichhane, S.; Mathema, V.B.; McGlinchey, A.; Dickens, A.M.; Khoomrung, S.; Orešič, M. Deep learning meets metabolomics: A methodological perspective. Briefings Bioinform. 2020. [Google Scholar] [CrossRef]
- Fan, Y.; Pedersen, O. Gut microbiota in human metabolic health and disease. Nat. Rev. Microbiol. 2021, 19, 55–71. [Google Scholar] [CrossRef]
- Martin, F.P.; Dumas, M.E.; Wang, Y.; Legido-Quigley, C.; Yap, I.K.; Tang, H. A top-down systems biology view of microbiome-mammalian metabolic interactions in a mouse model. Mol. Syst. Biol. 2007, 3, 112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sender, R.; Fuchs, S.; Milo, R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016, 14, e1002533. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, S.; Huang, J.; Chen, Z.; Jiang, Z.; Li, X.; Chen, Z. Metabolomics in gut microbiota: Applications and challenges. Sci. Bull. 2016, 61, 1151–1153. [Google Scholar] [CrossRef]
- Louis, P.; Hold, G.L.; Flint, H.J. The gut microbiota, bacterial metabolites and colorectal cancer. Nat. Rev. Genet. 2014, 12, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Koh, A.; De Vadder, F.; Kovatcheva-Datchary, P.; Bäckhed, F. From Dietary Fiber to Host Physiology: Short-Chain Fatty Acids as Key Bacterial Metabolites. Cell 2016, 165, 1332–1345. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Cooper, D.E.; Cluntun, A.A.; Warmoes, M.O.; Zhao, S.; Reid, M.A. Acetate production from glucose and coupling to mi-tochondrial metabolism in mammals. Cell 2018, 175, 502–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esposito, A.; Tamburini, S.; Triboli, L.; Ambrosino, L.; Chiusano, M.L.; Jousson, O. Insights into the genome structure of four acetogenic bacteria with specific reference to the Wood–Ljungdahl pathway. Microbiology 2019, 8, e938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichardt, N.; Duncan, S.H.; Young, P.; Belenguer, A.; Leitch, C.M.; Scott, K.P.; Flint, H.J.; Louis, P. Phylogenetic distribution of three pathways for propionate production within the human gut microbiota. ISME J. 2014, 8, 1323–1335. [Google Scholar] [CrossRef] [Green Version]
- Koliarakis, I.; Psaroulaki, A.; Nikolouzakis, T.K.; Kokkinakis, M.; Sgantzos, M.; Goulielmos, G.; Androutsopoulos, V.P.; Tsatsakis, A.; Tsiaoussis, J. Intestinal microbiota and colorectal cancer: A new aspect of research. J. B.U.ON. Off. J. Balk. Union Oncol. 2018, 23, 1216–1234. [Google Scholar]
- Louis, P.; Young, P.; Holtrop, G.; Flint, H.J. Diversity of human colonic butyrate-producing bacteria revealed by analysis of the butyryl-CoA:acetate CoA-transferase gene. Environ. Microbiol. 2010, 12, 304–314. [Google Scholar] [CrossRef]
- Louis, P.; Duncan, S.H.; McCrae, S.I.; Millar, J.; Jackson, M.S.; Flint, H.J. Restricted Distribution of the Butyrate Kinase Pathway among Butyrate-Producing Bacteria from the Human Colon. J. Bacteriol. 2004, 186, 2099–2106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bui, T.P.N.; Ritari, J.; Boeren, S.; De Waard, P.; Plugge, C.M.; De Vos, W.M. Production of butyrate from lysine and the Amadori product fructoselysine by a human gut commensal. Nat. Commun. 2015, 6, 10062. [Google Scholar] [CrossRef] [Green Version]
- Cummings, J.H.; Pomare, E.W.; Branch, W.J.; Naylor, C.P.; Macfarlane, G.T. Short chain fatty acids in human large intestine, portal, hepatic and venous blood. Gut 1987, 28, 1221–1227. [Google Scholar] [CrossRef] [Green Version]
- Den Besten, G.; van Eunen, K.; Groen, A.K.; Venema, K.; Reijngoud, D.J.; Bakker, B.M. The role of short-chain fatty acids in the interplay between diet, gut microbiota, and host energy metabolism. J. Lipid Res. 2013, 54, 2325–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Beek, C.M.; Dejong, C.H.C.; Troost, F.J.; Masclee, A.A.M.; Lenaerts, K. Role of short-chain fatty acids in colonic inflammation, carcinogenesis, and mucosal protection and healing. Nutr. Rev. 2017, 75, 286–305. [Google Scholar] [CrossRef]
- McNabney, S.M.; Henagan, T.M. Short Chain Fatty Acids in the Colon and Peripheral Tissues: A Focus on Butyrate, Colon Cancer, Obesity and Insulin Resistance. Nutrient 2017, 9, 1348. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.-M.; Yu, Y.-N.; Wang, J.-L.; Lin, Y.-W.; Kong, X.; Yang, C.-Q.; Yang, L.; Liu, Z.-J.; Yuan, Y.-Z.; Liu, F.; et al. Decreased dietary fiber intake and structural alteration of gut microbiota in patients with advanced colorectal adenoma. Am. J. Clin. Nutr. 2013, 97, 1044–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, J.; Carbonero, F.; Zoetendal, E.G.; Delany, J.P.; Wang, M.; Newton, K.; Gaskins, H.R.; O’Keefe, S.J.D. Diet, microbiota, and microbial metabolites in colon cancer risk in rural Africans and African Americans. Am. J. Clin. Nutr. 2013, 98, 111–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Keefe, S.J.D. Diet, microorganisms and their metabolites, and colon cancer. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 691–706. [Google Scholar] [CrossRef]
- Mariadason, J.M. HDACs and HDAC inhibitors in colon cancer. Epigenetics 2008, 3, 28–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donohoe, D.R.; Holley, D.; Collins, L.B.; Montgomery, S.A.; Whitmore, A.C.; Hillhouse, A.; Curry, K.P.; Renner, S.W.; Greenwalt, A.; Ryan, E.P.; et al. A Gnotobiotic Mouse Model Demonstrates That Dietary Fiber Protects against Colorectal Tumorigenesis in a Microbiota- and Butyrate-Dependent Manner. Cancer Discov. 2014, 4, 1387–1397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.J.; Chueh, A.C.; Togel, L.; Corner, G.A.; Ahmed, N.; Goel, S. Apoptotic sensitivity of colon cancer cells to histone deacetylase inhibitors is mediated by an Sp1/Sp3-activated transcriptional program involving immediate-early gene induc-tion. Cancer Res. 2010, 70, 609–620. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.; Kim, B.G.; Kim, J.H.; Chun, J.; Im, J.P.; Kim, J.S. Sodium butyrate inhibits the NF-kappa B signaling pathway and histone deacetylation, and attenuates experimental colitis in an IL-10 independent manner. Int. Immunopharmacol. 2017, 51, 47–56. [Google Scholar] [CrossRef]
- Furusawa, Y.; Obata, Y.; Fukuda, S.; Endo, T.A.; Nakato, G.; Takahashi, D.; Nakanishi, Y.; Uetake, C.; Kato, K.; Kato, T.; et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013, 504, 446–450. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.V.; Hao, L.; Offermanns, S.; Medzhitov, R. The microbial metabolite butyrate regulates intestinal macrophage function via histone deacetylase inhibition. Proc. Natl. Acad. Sci. USA 2014, 111, 2247–2252. [Google Scholar] [CrossRef] [Green Version]
- Hamer, H.; Jonkers, D.; Venema, K.; Vanhoutvin, S.; Troost, F.J.; Brummer, R. Review article: The role of butyrate on colonic function. Aliment. Pharmacol. Ther. 2008, 27, 104–119. [Google Scholar] [CrossRef]
- Li, Q.; Cao, L.; Tian, Y.; Zhang, P.; Ding, C.; Lu, W.; Jia, C.; Shao, C.; Liu, W.; Wang, D. Butyrate Suppresses the Proliferation of Colorectal Cancer Cells via Targeting Pyruvate Kinase M2 and Metabolic Reprogramming. Mol. Cell. Proteom. 2018, 17, 1531–1545. [Google Scholar] [CrossRef] [Green Version]
- Thangaraju, M.; Cresci, G.A.; Liu, K.; Ananth, S.; Gnanaprakasam, J.P.; Browning, D.D.; Mellinger, J.D.; Smith, S.B.; Digby, G.J.; Lambert, N.A.; et al. GPR109A Is a G-protein–Coupled Receptor for the Bacterial Fermentation Product Butyrate and Functions as a Tumor Suppressor in Colon. Cancer Res. 2009, 69, 2826–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, N.; Gurav, A.; Sivaprakasam, S.; Brady, E.; Padia, R.; Shi, H.; Thangaraju, M.; Prasad, P.D.; Manicassamy, S.; Munn, D.H.; et al. Activation of Gpr109a, receptor for niacin and the commensal metabolite butyrate, suppresses colonic inflammation and carcinogenesis. Immunity 2014, 40, 128–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, D.; Jin, D.; Huang, S.; Wu, J.; Xu, M.; Liu, T.; Dong, W.; Liu, X.; Wang, S.; Zhong, W.; et al. Clostridium butyricum, a butyrate-producing probiotic, inhibits intestinal tumor development through modulating Wnt signaling and gut microbiota. Cancer Lett. 2020, 469, 456–467. [Google Scholar] [CrossRef]
- Hosseini, E.; Grootaert, C.; Verstraete, W.; Van De Wiele, T. Propionate as a health-promoting microbial metabolite in the human gut. Nutr. Rev. 2011, 69, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Tong, L.-C.; Wang, Y.; Wang, Z.-B.; Liu, W.-Y.; Sun, S.; Li, L.; Su, D.-F.; Zhang, L.-C. Propionate Ameliorates Dextran Sodium Sulfate-Induced Colitis by Improving Intestinal Barrier Function and Reducing Inflammation and Oxidative Stress. Front. Pharmacol. 2016, 7, 253. [Google Scholar] [CrossRef]
- Bajic, D.; Niemann, A.; Hillmer, A.-K.; Mejias-Luque, R.; Bluemel, S.; Docampo, M.; Funk, M.C.; Tonin, E.; Boutros, M.; Schnabl, B.; et al. Gut Microbiota-Derived Propionate Regulates the Expression of Reg3 Mucosal Lectins and Ameliorates Experimental Colitis in Mice. J. Crohn’s Coliti 2020, 14, 1462–1472. [Google Scholar] [CrossRef]
- Arpaia, N.; Campbell, C.; Fan, X.; Dikiy, S.; Van Der Veeken, J.; DeRoos, P.; Liu, H.; Cross, J.R.; Pfeffer, K.; Coffer, P.J.; et al. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature 2013, 504, 451–455. [Google Scholar] [CrossRef]
- Bose, S.; Ramesh, V.; Locasale, J.W. Acetate Metabolism in Physiology, Cancer, and Beyond. Trends Cell Biol. 2019, 29, 695–703. [Google Scholar] [CrossRef] [PubMed]
- Gomes, S.D.; Oliveira, C.S.; Azevedo-Silva, J.; Casanova, M.R.; Barreto, J.; Pereira, H.; Chaves, S.R.; Rodrigues, L.R.; Casal, M.; Côrte-Real, M.; et al. The Role of Diet Related Short-Chain Fatty Acids in Colorectal Cancer Metabolism and Survival: Prevention and Therapeutic Implications. Curr. Med. Chem. 2020, 27, 4087–4108. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Xie, G.; Jia, W. Bile acid-microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ticho, A.L.; Malhotra, P.; Dudeja, P.K.; Gill, R.K.; Alrefai, W.A. Intestinal Absorption of Bile Acids in Health and Disease. Compr. Physiol. 2019, 10, 21–56. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocvirk, S.; Wilson, A.S.; Posma, J.M.; Li, J.V.; Koller, K.R.; Day, G.M.; Flanagan, A.C.; Otto, J.E.; Sacco, E.P.; Sacco, F.D.; et al. A prospective cohort analysis of gut microbial co-metabolism in Alaska Native and rural African people at high and low risk of colorectal cancer. Am. J. Clin. Nutr. 2020, 111, 406–419. [Google Scholar] [CrossRef]
- Yachida, S.; Mizutani, S.; Shiroma, H.; Shiba, S.; Nakajima, T.; Sakamoto, T.; Watanabe, H.; Masuda, K.; Nishimoto, Y.; Kubo, M.; et al. Metagenomic and metabolomic analyses reveal distinct stage-specific phenotypes of the gut microbiota in colorectal cancer. Nat. Med. 2019, 25, 968–976. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.W.; Kennaway, E.L.; Kennaway, N.M. Production of Tumours in Mice by Deoxycholic Acid. Nat. Cell Biol. 1940, 145, 627. [Google Scholar] [CrossRef]
- Bernstein, C.; Holubec, H.; Bhattacharyya, A.K.; Nguyen, H.; Payne, C.M.; Zaitlin, B.; Bernstein, H. Carcinogenicity of deoxycholate, a secondary bile acid. Arch. Toxicol. 2011, 85, 863–871. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Dong, W.; Wang, S.; Zhang, Y.; Liu, T.; Xie, R.; Wang, B.; Cao, H. Deoxycholic acid disrupts the intestinal mucosal barrier and promotes intestinal tumorigenesis. Food Funct. 2018, 9, 5588–5597. [Google Scholar] [CrossRef] [Green Version]
- Powolny, A.; Xu, J.; Loo, G. Deoxycholate induces DNA damage and apoptosis in human colon epithelial cells expressing either mutant or wild-type p53. Int. J. Biochem. Cell Biol. 2001, 33, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Payne, C.M.; Weber, C.; Crowley-Skillicorn, C.; Dvorak, K.; Bernstein, H.; Bernstein, C.; Holubec, H.; Dvorakova, B.; Garewal, H. Deoxycholate induces mitochondrial oxidative stress and activates NF-kappaB through multiple mechanisms in HCT-116 colon epithelial cells. Carcinogenesis 2007, 28, 215–222. [Google Scholar] [CrossRef]
- Fu, T.; Coulter, S.; Yoshihara, E.; Oh, T.G.; Fang, S.; Cayabyab, F.; Zhu, Q.; Zhang, T.; Leblanc, M.; Liu, S.; et al. FXR Regulates Intestinal Cancer Stem Cell Proliferation. Cell 2019, 176, 1098–1112. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.K.; Cho, J.H.; Kim, E.; Kim, Y.J. Ursodeoxycholic acid inhibits the proliferation of colon cancer cells by regulating oxidative stress and cancer stem-like cell growth. PLoS ONE 2017, 12, e0181183. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.H.; Kim, J.H.; Kim, B.G.; Lee, K.L.; Kim, J.W.; Koh, S.-J. Tauroursodeoxycholic acid attenuates colitis-associated colon cancer by inhibiting nuclear factor kappaB signaling. J. Gastroenterol. Hepatol. 2019, 34, 544–551. [Google Scholar] [CrossRef]
- Centuori, S.M.; Martinez, J.D. Differential Regulation of EGFR–MAPK Signaling by Deoxycholic Acid (DCA) and Ursodeoxycholic Acid (UDCA) in Colon Cancer. Dig. Dis. Sci. 2014, 59, 2367–2380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casero, R.A., Jr.; Stewart, T.M.; Pegg, A.E. Polyamine metabolism and cancer: Treatments, challenges and opportunities. Nat. Rev. Cancer 2018, 18, 681–695. [Google Scholar] [CrossRef]
- Sánchez-Jiménez, F.; Medina, M.Á.; Villalobos-Rueda, L.; Urdiales, J.L. Polyamines in mammalian pathophysiology. Cell. Mol. Life Sci. 2019, 76, 3987–4008. [Google Scholar] [CrossRef] [PubMed]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Pegg, A.E. Functions of Polyamines in Mammals. J. Biol. Chem. 2016, 291, 14904–14912. [Google Scholar] [CrossRef] [Green Version]
- Shah, P.; Swiatlo, E. A multifaceted role for polyamines in bacterial pathogens. Mol. Microbiol. 2008, 68, 4–16. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, T.; Katsumata, K.; Kuwabara, H.; Soya, R.; Enomoto, M.; Ishizaki, T.; Tsuchida, A.; Mori, M.; Hiwatari, K.; Soga, T.; et al. Urinary Polyamine Biomarker Panels with Machine-Learning Differentiated Colorectal Cancers, Benign Disease, and Healthy Controls. Int. J. Mol. Sci. 2018, 19, 756. [Google Scholar] [CrossRef]
- Johnson, C.H.; Dejea, C.M.; Edler, D.; Hoang, L.T.; Santidrian, A.F.; Felding, B.H.; Ivanisevic, J.; Cho, K.; Wick, E.C.; Hechenbleikner, E.M.; et al. Metabolism Links Bacterial Biofilms and Colon Carcinogenesis. Cell Metab. 2015, 21, 891–897. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, A.C.; Shields, C.E.D.; Wu, S.; Huso, D.L.; Wu, X.; Murray-Stewart, T.R.; Hacker-Prietz, A.; Rabizadeh, S.; Woster, P.M.; Sears, C.L.; et al. Polyamine catabolism contributes to enterotoxigenic Bacteroides fragilis-induced colon tumorigenesis. Proc. Natl. Acad. Sci. USA 2011, 108, 15354–15359. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Ruan, P.; Zhao, Y.; Li, X.; Wang, J.; Wu, X. Spermidine/spermine N1-acetyltransferase regulates cell growth and metastasis via AKT/beta-catenin signaling pathways in hepatocellular and colorectal carcinoma cells. Oncotarget 2017, 8, 1092–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, C.S.; Shicora, A.C.; Keough, M.P.; Snook, A.E.; Burns, M.R.; Gilmour, S.K. Polyamine-blocking therapy reverses immunosup-pression in the tumor microenvironment. Cancer Immunol. Res. 2014, 2, 274–285. [Google Scholar] [CrossRef] [Green Version]
- Agus, A.; Planchais, J.; Sokol, H. Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease. Cell Host Microbe 2018, 23, 716–724. [Google Scholar] [CrossRef] [Green Version]
- Aoki, R.; Aoki-Yoshida, A.; Suzuki, C.; Takayama, Y. Indole-3-Pyruvic Acid, an Aryl Hydrocarbon Receptor Activator, Suppresses Experimental Colitis in Mice. J. Immunol. 2018, 201, 3683–3693. [Google Scholar] [CrossRef] [Green Version]
- Guertin, K.A.; Li, X.S.; Graubard, B.I.; Albanes, D.; Weinstein, S.J.; Goedert, J.J.; Wang, Z.; Hazen, S.L.; Sinha, R. Serum Trimethylamine N-oxide, Carnitine, Choline, and Betaine in Relation to Colorectal Cancer Risk in the Alpha Tocopherol, Beta Carotene Cancer Prevention Study. Cancer Epidemiol. Biomarkers Prev. 2017, 26, 945–952. [Google Scholar] [CrossRef] [Green Version]
- Thomas, A.M.; Manghi, P.; Asnicar, F.; Pasolli, E.; Armanini, F.; Zolfo, M.; Beghini, F.; Manara, S.; Karcher, N.; Pozzi, C.; et al. Metagenomic analysis of colorectal cancer datasets identifies cross-cohort microbial diagnostic signatures and a link with choline degradation. Nat. Med. 2019, 25, 667–678. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.W.H.; Law, B.M.H.; Waye, M.M.Y.; Chan, J.Y.W.; So, W.K.W.; Chow, K.M. Trimethylamine-N-oxide as One Hypothetical Link for the Relationship between Intestinal Microbiota and Cancer—Where We Are and Where Shall We Go? J. Cancer 2019, 10, 5874–5882. [Google Scholar] [CrossRef] [PubMed]
- Ni, Y.; Xie, G.; Jia, W. Metabonomics of Human Colorectal Cancer: New Approaches for Early Diagnosis and Biomarker Discovery. J. Proteome Res. 2014, 13, 3857–3870. [Google Scholar] [CrossRef]
- Provenzale, D.; Gupta, S.; Ahnen, D.J.; Markowitz, A.J.; Chung, D.C.; Mayer, R.J.; Regenbogen, S.E.; Blanco, A.M.; Bray, T.; Cooper, G.; et al. NCCN Guidelines Insights: Colorectal Cancer Screening, Version 1.2018. J. Natl. Compr. Cancer Netw. 2018, 16, 939–949. [Google Scholar] [CrossRef] [PubMed]
- Kelley, R.K.; Van Bebber, S.L.; Phillips, K.A.; Venook, A.P. Personalized medicine and oncology practice guidelines: A case study of contemporary biomarkers in colorectal cancer. J. Natl. Compr. Cancer Netw. 2011, 9, 13–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weir, T.L.; Manter, D.K.; Sheflin, A.M.; Barnett, B.A.; Heuberger, A.L.; Ryan, E.P. Stool Microbiome and Metabolome Differences between Colorectal Cancer Patients and Healthy Adults. PLoS ONE 2013, 8, e70803. [Google Scholar] [CrossRef] [Green Version]
- Brown, D.G.; Rao, S.; Weir, T.L.; O’Malia, J.; Bazan, M.; Brown, R.J.; Ryan, E.P. Metabolomics and metabolic pathway networks from human colorectal cancers, adjacent mucosa, and stool. Cancer Metab. 2016, 4, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Ma, C.; Bezabeh, T.; Wang, Z.; Liang, J.; Huang, Y.; Zhao, J.; Liu, X.; Ye, W.; Tang, W.; et al. 1H NMR-based metabolomics reveal overlapping discriminatory metabolites and metabolic pathway disturbances between colorectal tumor tissues and fecal samples. Int. J. Cancer 2019, 145, 1679–1689. [Google Scholar] [CrossRef]
- Manna, S.K.; Tanaka, N.; Krausz, K.W.; Haznadar, M.; Xue, X.; Matsubara, T.; Bowman, E.D.; Fearon, E.R.; Harris, C.C.; Shah, Y.M.; et al. Biomarkers of Coordinate Metabolic Reprogramming in Colorectal Tumors in Mice and Humans. Gastroenterol. 2014, 146, 1313–1324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Misra, B.B.; Liang, L.; Bi, D.; Weng, W.; Wu, W.; Cai, S.; Qin, H.; Goel, A.; Li, X.; et al. Integrated microbiome and metabolome analysis reveals a novel interplay between commensal bacteria and metabolites in colorectal cancer. Theranostics 2019, 9, 4101–4114. [Google Scholar] [CrossRef]
- Sze, M.A.; Topçuoğlu, B.D.; Lesniak, N.A.; Ruffin, M.T.; Schloss, P.D. Fecal Short-Chain Fatty Acids Are Not Predictive of Colonic Tumor Status and Cannot Be Predicted Based on Bacterial Community Structure. mBio 2019, 10, e01454-19. [Google Scholar] [CrossRef] [Green Version]
- Song, M.; Garrett, W.S.; Chan, A.T. Nutrients, Foods, and Colorectal Cancer Prevention. Gastroenterology 2015, 148, 1244–1260. [Google Scholar] [CrossRef] [Green Version]
- O’Keefe, S.J.D.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E.H.; et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6, 1–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Leu, R.K.; Winter, J.M.; Christophersen, C.T.; Young, G.P.; Humphreys, K.J.; Hu, Y.; Miller, R.B.; Topping, D.L.; Bird, A.R.; Conlon, M.A.; et al. Butyrylated starch intake can prevent red meat-induced O6-methyl-2-deoxyguanosine adducts in human rectal tissue: A randomised clinical trial. Br. J. Nutr. 2015, 114, 220–230. [Google Scholar] [CrossRef] [Green Version]
- Park, M.; Kwon, J.; Shin, H.; Moon, S.M.; Kim, S.B.; Shin, U.S.; Han, Y.; Kim, Y. Butyrate enhances the efficacy of radiotherapy via FOXO3A in colorectal cancer patient-derived organoids. Int. J. Oncol. 2020, 57, 1307–1318. [Google Scholar] [CrossRef]
- Koliarakis, I.; Athanasakis, E.; Sgantzos, M.; Mariolis-Sapsakos, T.; Xynos, E.; Chrysos, E.; Souglakos, J.; Tsiaoussis, J. Intestinal Microbiota in Colorectal Cancer Surgery. Cancers 2020, 12, 3011. [Google Scholar] [CrossRef] [PubMed]
- Vargas, A.J.; Wertheim, B.C.; Gerner, E.W.; Thomson, C.A.; Rock, C.L.; Thompson, P.A. Dietary polyamine intake and risk of colorectal adenomatous polyps123. Am. J. Clin. Nutr. 2012, 96, 133–141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vargas, A.J.; Ashbeck, E.L.; Wertheim, B.C.; Wallace, R.B.; Neuhouser, M.L.; Thomson, C.A.; Thompson, P.A. Dietary polyamine intake and colorectal cancer risk in postmenopausal women. Am. J. Clin. Nutr. 2015, 102, 411–419. [Google Scholar] [CrossRef] [Green Version]
- Goossens, J.-F.; Bailly, C. Ursodeoxycholic acid and cancer: From chemoprevention to chemotherapy. Pharmacol. Ther. 2019, 203, 107396. [Google Scholar] [CrossRef]
- Pearson, T.; Caporaso, J.G.; Yellowhair, M.; Bokulich, N.A.; Padi, M.; Roe, D.J.; Wertheim, B.C.; Linhart, M.; Martinez, J.A.; Bilagody, C.; et al. Effects of ursodeoxycholic acid on the gut micro-biome and colorectal adenoma development. Cancer Med. 2019, 8, 617–628. [Google Scholar] [CrossRef] [Green Version]
- Alberts, D.S.; Martínez, M.E.; Hess, L.M.; Einspahr, J.G.; Green, S.B.; Bhattacharyya, A.K.; Guillen, J.; Krutzsch, M.; Batta, A.K.; Salen, G.; et al. Phase III Trial of Ursodeoxycholic Acid To Prevent Colorectal Adenoma Recurrence. J. Natl. Cancer Inst. 2005, 97, 846–853. [Google Scholar] [CrossRef] [Green Version]
- Thompson, P.A.; Wertheim, B.C.; Roe, D.J.; Ashbeck, E.L.; Jacobs, E.T.; Lance, P.; Martínez, M.E.; Alberts, D.S. Gender Modifies the Effect of Ursodeoxycholic Acid in a Randomized Controlled Trial in Colorectal Adenoma Patients. Cancer Prev. Res. 2009, 2, 1023–1030. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Strategies | Methods | Quantification | Metabolites Attributes | Advantages | Disadvantages | Prospects | |
|---|---|---|---|---|---|---|---|
| Untargeted and targeted | NMR | Yes | Polar or non-polar | Simple sample prepration, structural information, identify novel compounds | Low sensitivity, poor selectivity, poor for quantification | Machine learning and artificial interagency to assist metabolomics data processing, metabolite identification, and biomarker discovery | |
| MS | GC-MS | Yes | Volatile metabolites or the volatile derivatives of metabolites | The most common method for SCFAs, good spectrum libraries | Relatively complex sample preparation, standards or/and database dependence | ||
| LC-MS | Polar or non-polar | Softer ionization and lower temperature than GC-MS to detect larger/non-volatile and less stable metabolites | |||||
| DESI-MS | Broad metabolites particularly lipids | Rapid in situ assessment of metabolomic profiles | |||||
| MALDI-MS | Complex sample and broad metabolites | Rapid and tolerant of impurities | |||||
| NanoSI-MS | Stable-isotope labeled metabolites | Simultaneously identify and quantify metabolites in single cells | |||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, Y.; Nie, Y.; Yu, J.; Wong, C.C. Microbial Metabolites in Colorectal Cancer: Basic and Clinical Implications. Metabolites 2021, 11, 159. https://doi.org/10.3390/metabo11030159
Peng Y, Nie Y, Yu J, Wong CC. Microbial Metabolites in Colorectal Cancer: Basic and Clinical Implications. Metabolites. 2021; 11(3):159. https://doi.org/10.3390/metabo11030159
Chicago/Turabian StylePeng, Yao, Yuqiang Nie, Jun Yu, and Chi Chun Wong. 2021. "Microbial Metabolites in Colorectal Cancer: Basic and Clinical Implications" Metabolites 11, no. 3: 159. https://doi.org/10.3390/metabo11030159
APA StylePeng, Y., Nie, Y., Yu, J., & Wong, C. C. (2021). Microbial Metabolites in Colorectal Cancer: Basic and Clinical Implications. Metabolites, 11(3), 159. https://doi.org/10.3390/metabo11030159