
Mitochondrial Fission Governed by Drp1 Regulates Exogenous Fatty Acid Usage and Storage in Hela Cells

 , ,
, ,
Abstract
:1. Introduction
2. Results
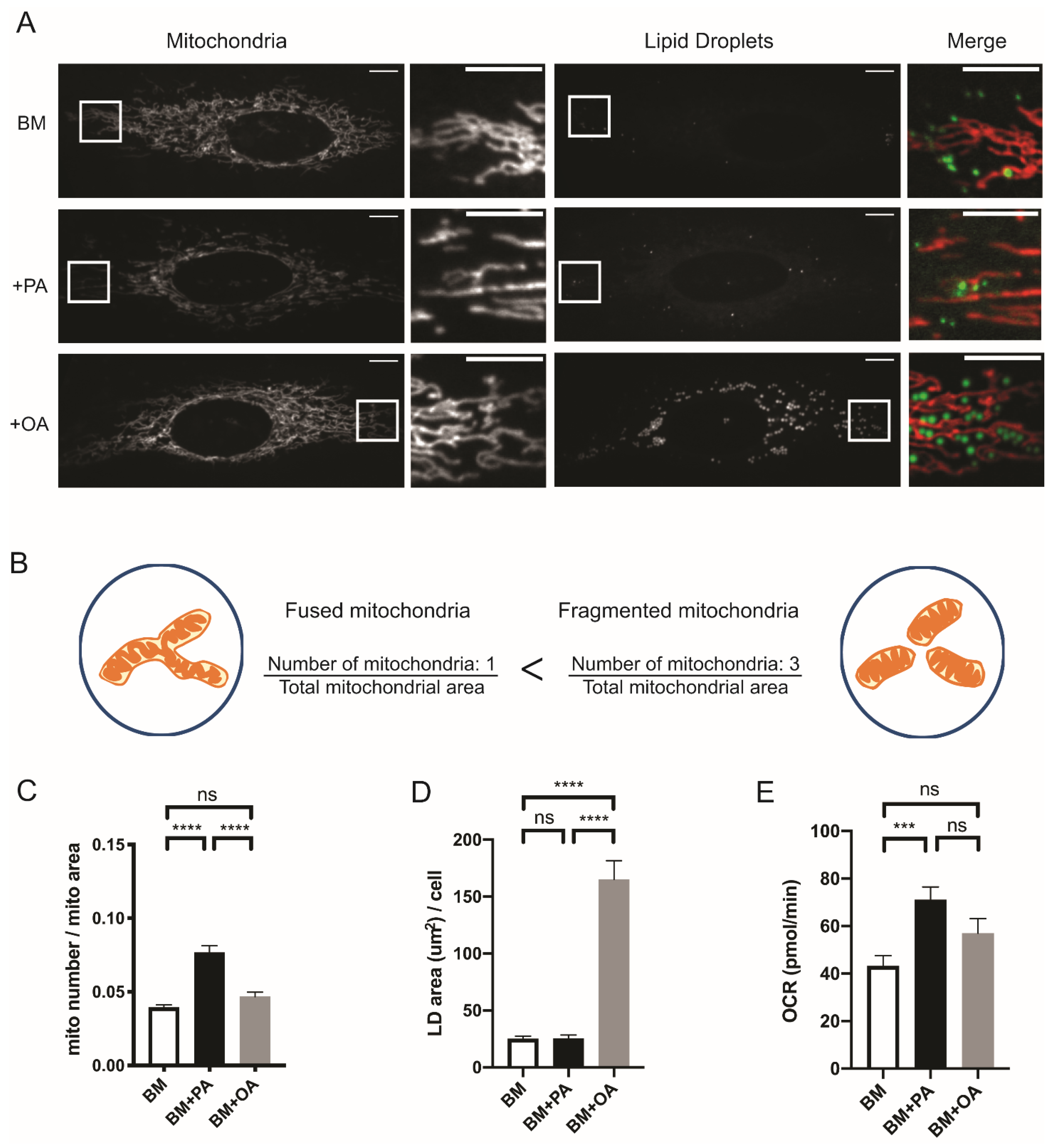
2.1. Mitochondrial Morphology Reflects the Exogenous Fatty Acid Usage and Storage
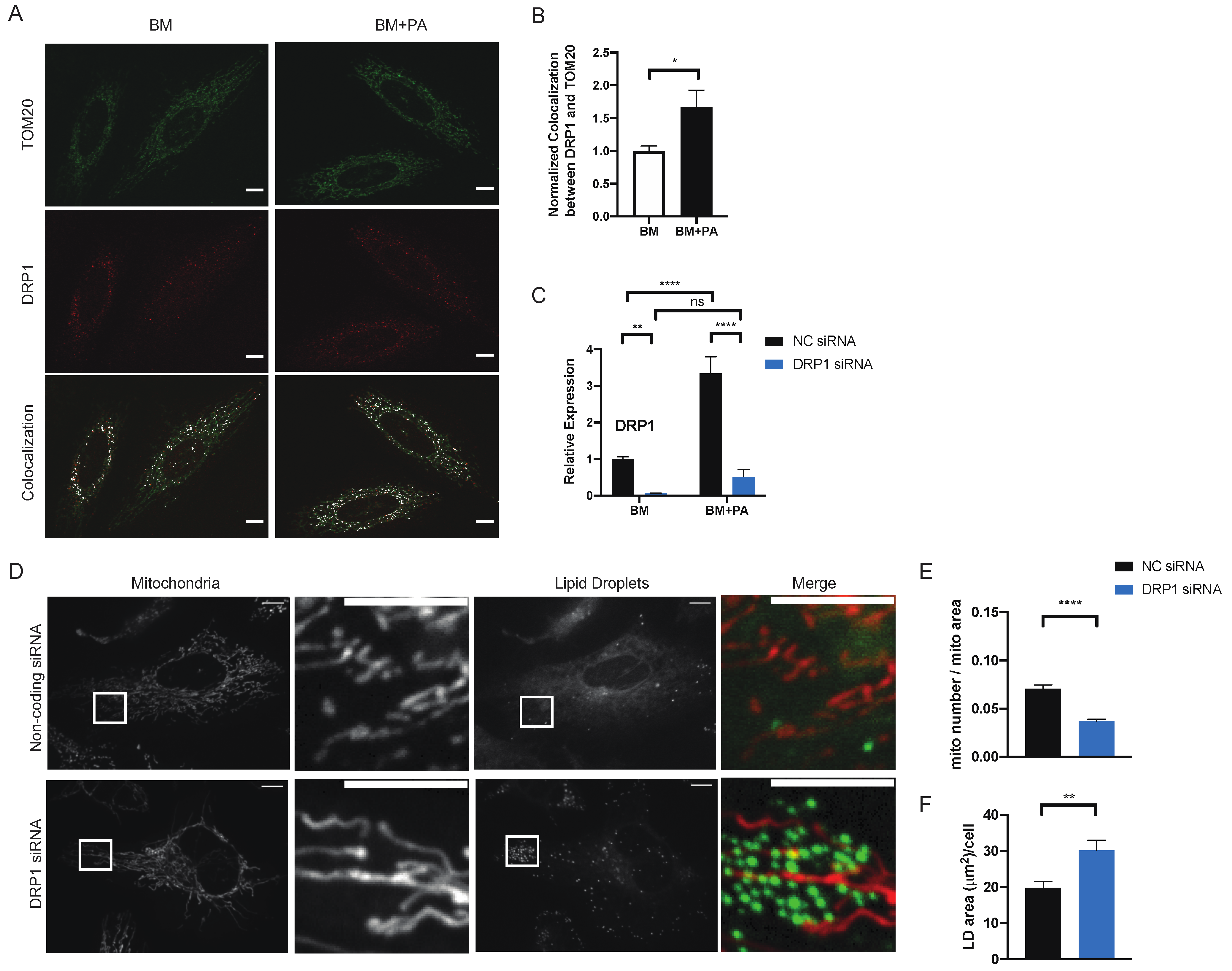
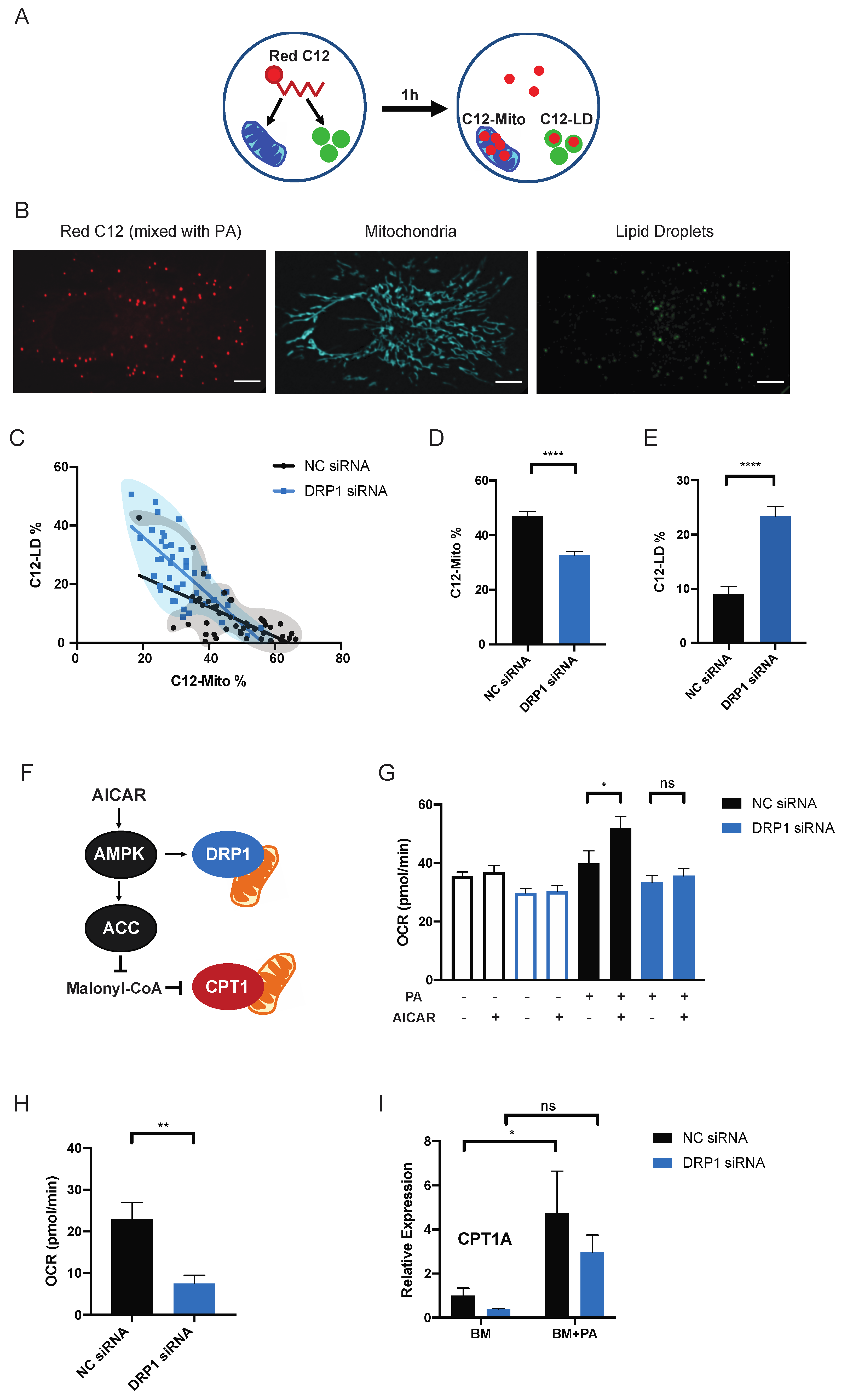
2.2. Mitochondrial Fission Directs the Distribution of Exogenous PA between Mitochondria and Lipid Droplets
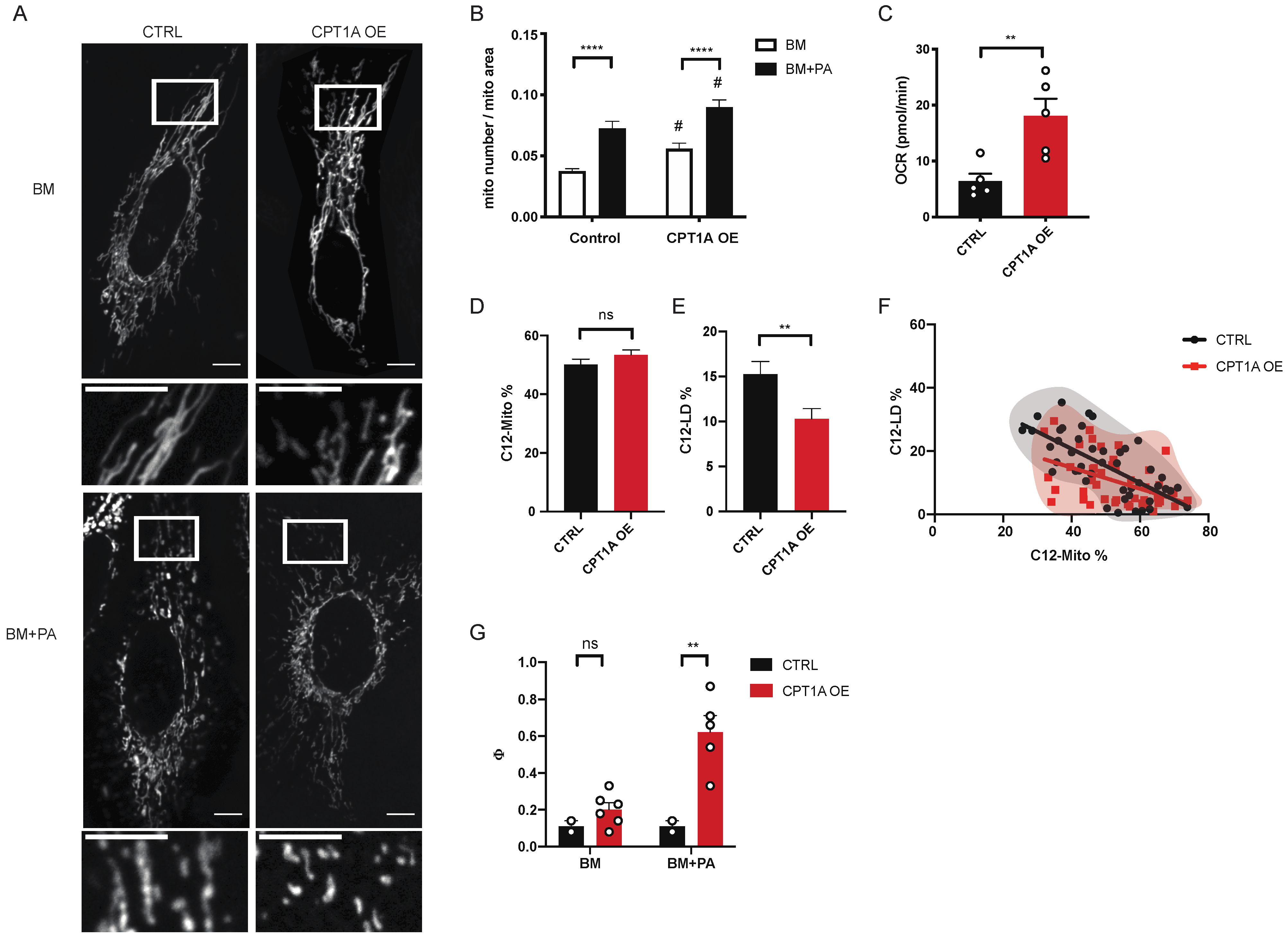
2.3. Mitochondrial Fission Works Independently of CPT1A on Fatty Acid Distribution
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Transfection
4.3. Imaging
4.4. Transmission Electron Microscopy
4.5. RT-qPCR
4.6. Mitochondrial Mass and ROS Measurement Using Flow Cytometry
4.7. Metabolism Assay
4.8. Image Analysis
4.9. Statistical Analyses
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ishihara, N.; Jofuku, A.; Eura, Y.; Mihara, K. Regulation of mitochondrial morphology by membrane potential, and DRP1-dependent division and FZO1-dependent fusion reaction in mammalian cells. Biochem. Biophys. Res. Commun. 2003, 301, 891–898. [Google Scholar] [CrossRef]
- Buck, M.D.; Sullivan, D.O.; Geltink, R.I.K.; Curtis, J.D.; Chang, C.-H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; Chen, Q.; et al. Mitochondrial Dynamics Controls T Cell Fate through Article Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Martínez-Diez, M.; Santamaría, G.; Ortega, A.D.; Cuezva, J.M. Biogenesis and Dynamics of Mitochondria during the Cell Cycle: Significance of 3′UTRs. PLoS ONE 2006, 1, e107. [Google Scholar] [CrossRef] [Green Version]
- Youle, R.J.; van der Bliek, A.M. Mitochondrial fission, fusion, and stress. Science 2012, 337, 1062–1065. [Google Scholar] [CrossRef] [Green Version]
- Molina, A.J.; Wikstrom, J.D.; Stiles, L.; Las, G.; Mohamed, H.; Elorza, A.; Walzer, G.; Twig, G.; Katz, S.; Corkey, B.E.; et al. Mitochondrial Networking Protects-Cells From Nutrient-Induced Apoptosis. Diabetes 2009, 58, 2303–2315. [Google Scholar] [CrossRef] [Green Version]
- Nasrallah, C.M.; Horvath, T.L. Mitochondrial dynamics in the central regulation of metabolism. Nat. Rev. Endocrinol. 2014, 10, 650–658. [Google Scholar] [CrossRef]
- Liesa, M.; Shirihai, O.S. Mitochondrial Dynamics in the Regulation of Nutrient Utilization and Energy Expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef] [Green Version]
- Rambold, A.S.; Cohen, S.; Lippincott-Schwartz, J. Fatty Acid Trafficking in Starved Cells: Regulation by Lipid Droplet Lipolysis, Autophagy, and Mitochondrial Fusion Dynamics. Dev. Cell 2015, 32, 678–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambold, A.S.; Kostelecky, B.; Elia, N.; Lippincott-Schwartz, J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc. Natl. Acad. Sci. USA 2011, 108, 10190–10195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carta, G.; Murru, E.; Banni, S.; Manca, C. Palmitic acid: Physiological role, metabolism and nutritional implications. Front. Physiol. 2017, 8, 902. [Google Scholar] [CrossRef] [Green Version]
- Alam, N.; Saggerson, E.D. Malonyl-CoA and the regulation of fatty acid oxidation in soleus muscle. Biochem. J. 1998, 334, 233–241. [Google Scholar] [CrossRef] [Green Version]
- McGarry, J.D.; Leatherman, G.F.; Foster, D.W. Carnitine palmitoyltransferase I. The site of inhibition of hepatic fatty acid oxidation by malonyl-CoA. J. Biol. Chem. 1978, 253, 4128–4136. [Google Scholar] [CrossRef]
- Toyama, E.Q.; Herzig, S.; Courchet, J.; Lewis, T.L.; Losón, O.C.; Hellberg, K.; Young, N.P.; Chen, H.; Polleux, F.; Chan, D.C.; et al. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 2016, 351, 275–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coppola, A.; Liu, Z.-W.; Andrews, Z.B.; Paradis, E.; Roy, M.-C.; Friedman, J.M.; Ricquier, D.; Richard, D.; Horvath, T.L.; Gao, X.-B.; et al. A Central Thermogenic-like Mechanism in Feeding Regulation: An Interplay between Arcuate Nucleus T3 and UCP2. Cell Metab. 2007, 5, 21–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrews, Z.B.; Liu, Z.-W.; Walllingford, N.; Erion, D.M.; Borok, E.; Friedman, J.M.; Tschöp, M.H.; Shanabrough, M.; Cline, G.; Shulman, G.I.; et al. UCP2 mediates ghrelin’s action on NPY/AgRP neurons by lowering free radicals. Nature 2008, 454, 846–851. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, M.O.; Liu, Z.W.; Horvath, T.L. Mitochondrial dynamics controlled by mitofusins regulate agrp neuronal activity and diet-induced obesity. Cell 2013, 155, 188–199. [Google Scholar] [CrossRef] [Green Version]
- Schneeberger, M.; Dietrich, M.O.; Sebastián, D.; Imbernón, M.; Castaño, C.; Garcia, A.; Esteban, Y.; Gonzalez-Franquesa, A.; Rodríguez, I.C.; Bortolozzi, A.; et al. Mitofusin 2 in POMC Neurons Connects ER Stress with Leptin Resistance and Energy Imbalance. Cell 2013, 155, 172–187. [Google Scholar] [CrossRef] [Green Version]
- Jin, S.; Yoon, N.A.; Liu, Z.-W.; Song, J.E.; Horvath, T.L.; Kim, J.D.; Diano, S. Drp1 is required for AgRP neuronal activity and feeding. eLife 2021, 10. [Google Scholar] [CrossRef]
- Cohen, S. Lipid Droplets as Organelles. Int. Rev. Cell Mol. Biol. 2018, 337, 83–110. [Google Scholar]
- Wang, H.; Sreenivasan, U.; Hu, H.; Saladino, A.; Polster, B.M.; Lund, L.M.; Gong, D.-W.; Stanley, W.C.; Sztalryd, C. Perilipin 5, a lipid droplet-associated protein, provides physical and metabolic linkage to mitochondria. J. Lipid Res. 2011, 52, 2159–2168. [Google Scholar] [CrossRef] [Green Version]
- Alves, T.C.; Pongratz, R.L.; Zhao, X.; Yarborough, O.; Sereda, S.; Shirihai, O.; Cline, G.W.; Mason, G.; Kibbey, R.G. Integrated, Step-Wise, Mass-Isotopomeric Flux Analysis of the TCA Cycle. Cell Metab. 2015, 22, 936–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benador, I.Y.; Veliova, M.; Mahdaviani, K.; Petcherski, A.; Wikstrom, J.D.; Assali, E.A.; Acín-Pérez, R.; Shum, M.; Oliveira, M.F.; Cinti, S.; et al. Mitochondria Bound to Lipid Droplets Have Unique Bioenergetics, Composition, and Dynamics that Support Lipid Droplet Expansion. Cell Metab. 2018, 27, 869–885.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.B.; Louie, S.M.; Daniele, J.R.; Tran, Q.; Dillin, A.; Zoncu, R.; Nomura, D.K.; Olzmann, J.A. DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell 2017, 42, 9–21.e5. [Google Scholar] [CrossRef] [Green Version]
- Pinkosky, S.L.; Scott, J.W.; Desjardins, E.M.; Smith, B.K.; Day, E.A.; Ford, R.J.; Langendorf, C.G.; Ling, N.X.Y.; Nero, T.L.; Loh, K.; et al. Long-chain fatty acyl-CoA esters regulate metabolism via allosteric control of AMPK β1 isoforms. Nat. Metab. 2020, 2, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, R.A.K.; Stephen, L.P.; Filippov, S.; Hanselman, J.C.; Cramer, C.T.; Newton, R.S. AMP-activated protein kinase: An emerging drug target to regulate imbalances in lipid and carbohydrate metabolism to treat cardio-metabolic diseases: Thematic Review Series: New Lipid and Lipoprotein Targets for the Treatment of Cardiometabolic Diseases. J. Lipid Res. 2012, 53, 2490–2514. [Google Scholar] [CrossRef] [Green Version]
- Koch, A.; Thiemann, M.; Grabenbauer, M.; Yoon, Y.; McNiven, M.A.; Schrader, M. Dynamin-like Protein 1 Is Involved in Peroxisomal Fission. J. Biol. Chem. 2003, 278, 8597–8605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adachi, Y.; Kato, T.; Yamada, T.; Murata, D.; Arai, K.; Stahelin, R.V.; Chan, D.C.; Iijima, M.; Sesaki, H. Drp1 Tubulates the ER in a GTPase-Independent Manner. Mol. Cell 2020, 80, 621–632.e6. [Google Scholar] [CrossRef]
- De Sousa, U.L.J.; Koss, M.D.; Fillies, M.; Gahl, A.; Scheeder, M.R.; Cardoso, M.C.; Leonhardt, H.; Geary, N.; Langhans, W.; Leonhardt, M. CPT1α over-expression increases long-chain fatty acid oxidation and reduces cell viability with incremental palmitic acid concentration in 293T cells. Biochem. Biophys. Res. Commun. 2005, 338, 757–761. [Google Scholar] [CrossRef]
- Lee, K.; Kerner, J.; Hoppel, C.L. Mitochondrial carnitine palmitoyltransferase 1a (CPT1a) is part of an outer membrane fatty acid transfer complex. J. Biol. Chem. 2011, 286, 25655–25662. [Google Scholar] [CrossRef] [Green Version]
- Jesinkey, S.R.; Madiraju, A.K.; Alves, T.C.; Yarborough, O.H.; Cardone, R.L.; Zhao, X.; Parsaei, Y.; Nasiri, A.R.; Butrico, G.; Liu, X.; et al. Mitochondrial GTP Links Nutrient Sensing to β Cell Health, Mitochondrial Morphology, and Insulin Secretion Independent of OxPhos. Cell Rep. 2019, 28, 759–772.e10. [Google Scholar] [CrossRef]
- Owen, O.E.; Kalhan, S.C.; Hanson, R.W. The key role of anaplerosis and cataplerosis for citric acid cycle function. J. Biol. Chem. 2002, 277, 30409–30412. [Google Scholar] [CrossRef] [Green Version]
- Aon, M.A.; Bhatt, N.; Cortassa, S. Mitochondrial and cellular mechanisms for managing lipid excess. Front. Physiol. 2014, 5, 282. [Google Scholar] [CrossRef] [Green Version]
- Garbarino, J.; Padamsee, M.; Wilcox, L.; Oelkers, P.M.; D’Ambrosio, D.; Ruggles, K.V.; Ramsey, N.; Jabado, O.; Turkish, A.; Sturley, S.L. Sterol and Diacylglycerol Acyltransferase Deficiency Triggers Fatty Acid-mediated Cell Death. J. Biol. Chem. 2009, 284, 30994–31005. [Google Scholar] [CrossRef] [Green Version]
- Finn, P.F.; Dice, J.F. Proteolytic and lipolytic responses to starvation. Nutrition 2006, 22, 830–844. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef] [Green Version]
- Kilian, N.; Goryaynov, A.; Lessard, M.D.; Hooker, G.; Toomre, D.; Rothman, J.E.; Bewersdorf, J. Assessing photodamage in live-cell STED microscopy. Nat. Methods 2018, 15, 755–756. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name. | Forward Primer | Reverse Primer |
|---|---|---|
| Actin | TGACGTGGACATCCGCAAAG | CTGGAAGGTGGACAGCGAGG |
| ACAA1 | GACAGGTCATCACGCTGCTCAA | CCAGGGTATTCAAAGACGGCAG |
| ACOX1 | GGCGCATACATGAAGGAGACCT | AGGTGAAAGCCTTCAGTCCAGC |
| CPT1A | AAATTACGTGAGCGACTGGTG | TGCTGCCTGAATGTGAGTTG |
| DGAT1 | GCTTCAGCAACTACCGTGGCAT | CCTTCAGGAACAGAGAAACCACC |
| DRP1 | GATGCCATAGTTGAAGTGGTGAC | CCACAAGCATCAGCAAAGTCTGG |
| EHHADH | CGGAGCATCGTGGAAAACAGCA | CCGAGTCTACAGCAATCACAGG |
| tRNA-Leu | CACCCAAGAACAGGGTTTGT | TGGCCATGGGTATGTTGTTA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Song, J.-E.; Alves, T.C.; Stutz, B.; Šestan-Peša, M.; Kilian, N.; Jin, S.; Diano, S.; Kibbey, R.G.; Horvath, T.L. Mitochondrial Fission Governed by Drp1 Regulates Exogenous Fatty Acid Usage and Storage in Hela Cells. Metabolites 2021, 11, 322. https://doi.org/10.3390/metabo11050322
Song J-E, Alves TC, Stutz B, Šestan-Peša M, Kilian N, Jin S, Diano S, Kibbey RG, Horvath TL. Mitochondrial Fission Governed by Drp1 Regulates Exogenous Fatty Acid Usage and Storage in Hela Cells. Metabolites. 2021; 11(5):322. https://doi.org/10.3390/metabo11050322
Chicago/Turabian StyleSong, Jae-Eun, Tiago C. Alves, Bernardo Stutz, Matija Šestan-Peša, Nicole Kilian, Sungho Jin, Sabrina Diano, Richard G. Kibbey, and Tamas L. Horvath. 2021. "Mitochondrial Fission Governed by Drp1 Regulates Exogenous Fatty Acid Usage and Storage in Hela Cells" Metabolites 11, no. 5: 322. https://doi.org/10.3390/metabo11050322
APA StyleSong, J. -E., Alves, T. C., Stutz, B., Šestan-Peša, M., Kilian, N., Jin, S., Diano, S., Kibbey, R. G., & Horvath, T. L. (2021). Mitochondrial Fission Governed by Drp1 Regulates Exogenous Fatty Acid Usage and Storage in Hela Cells. Metabolites, 11(5), 322. https://doi.org/10.3390/metabo11050322