
Phase I In Vitro Metabolic Profiling of the Synthetic Cannabinoid Receptor Agonists CUMYL-THPINACA and ADAMANTYL-THPINACA



Abstract
:1. Introduction
2. Results and Discussion
2.1. Metabolite Identification for CUMYL-THPINACA and ADAMANTYL-THPINACA after Incubation with pHLM and rCYP
2.2. In-Source Water Loss of Metabolites
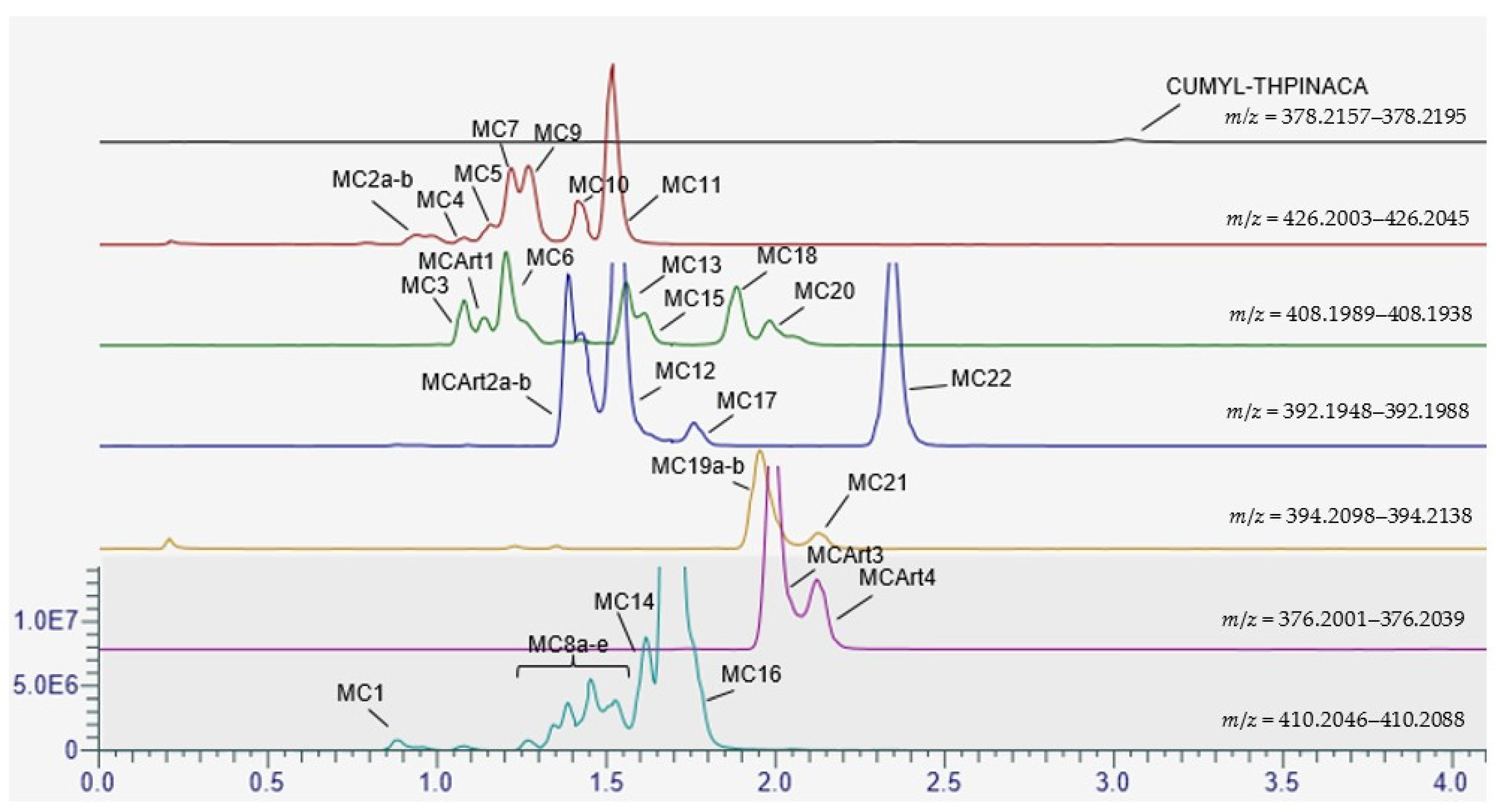
2.3. Metabolic Profiling of CUMYL-THPINACA
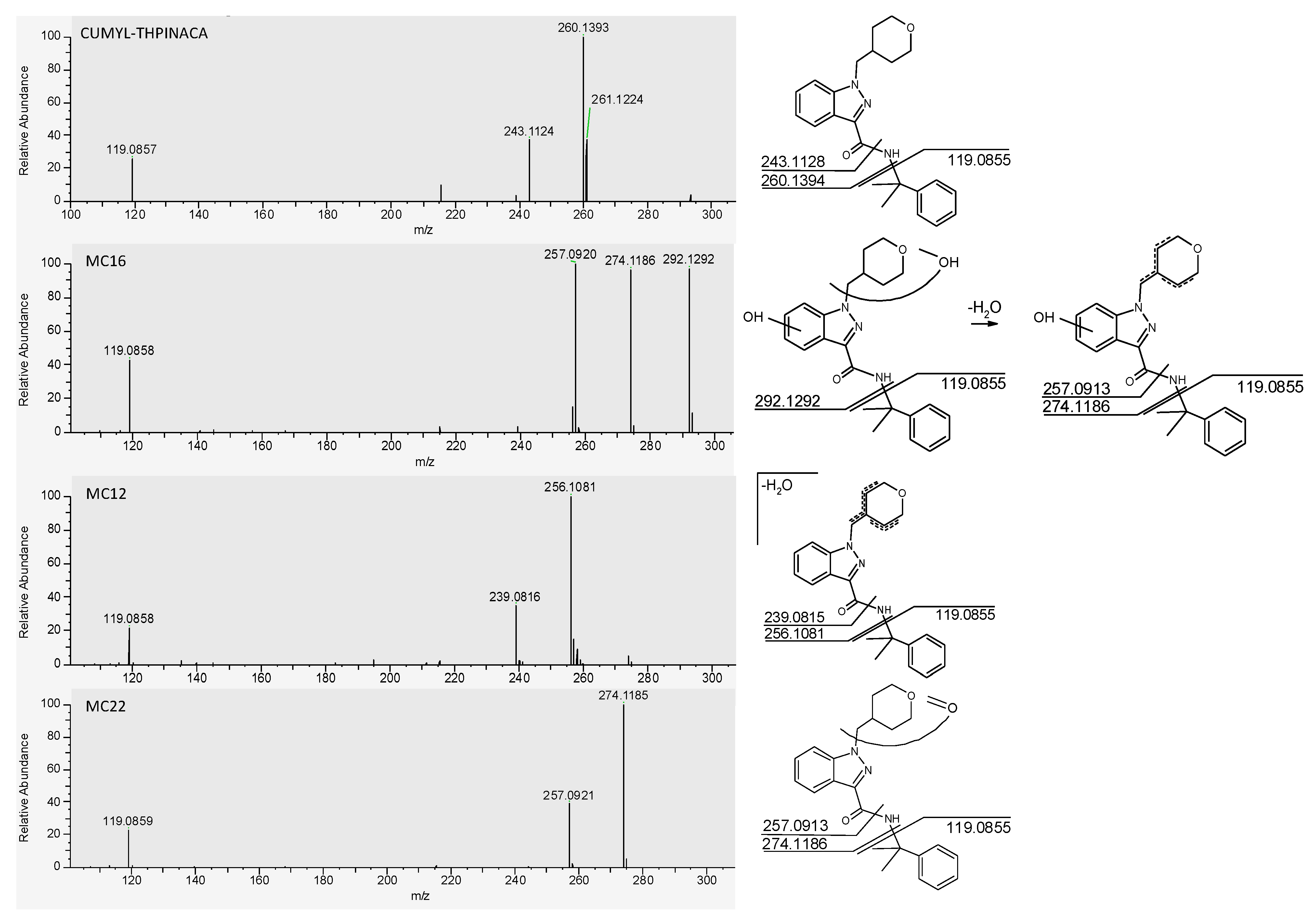
2.3.1. Mono-Hydroxylation
2.3.2. Di-Hydroxylation
2.3.3. Mono-Hydroxylation and Additional Desaturation and Carbonylation
2.3.4. Tri-Hydroxylation
2.3.5. Mono-Hydroxylation and Additional Desaturation and Carbonylation
2.3.6. Identification of the Primarily Involved CYP Isoenzymes
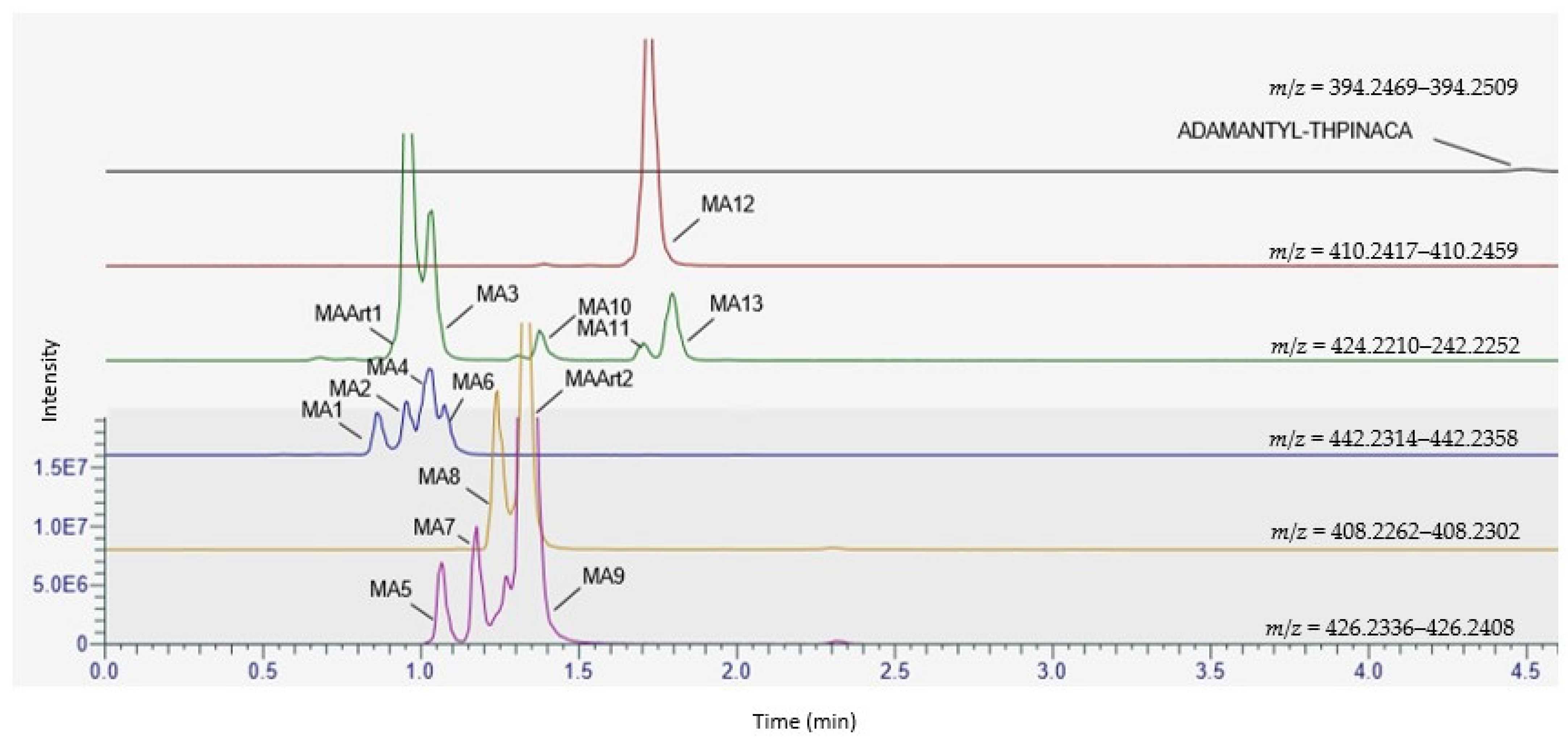
2.4. Metabolite Identification for ADAMANTYL-THPINACA
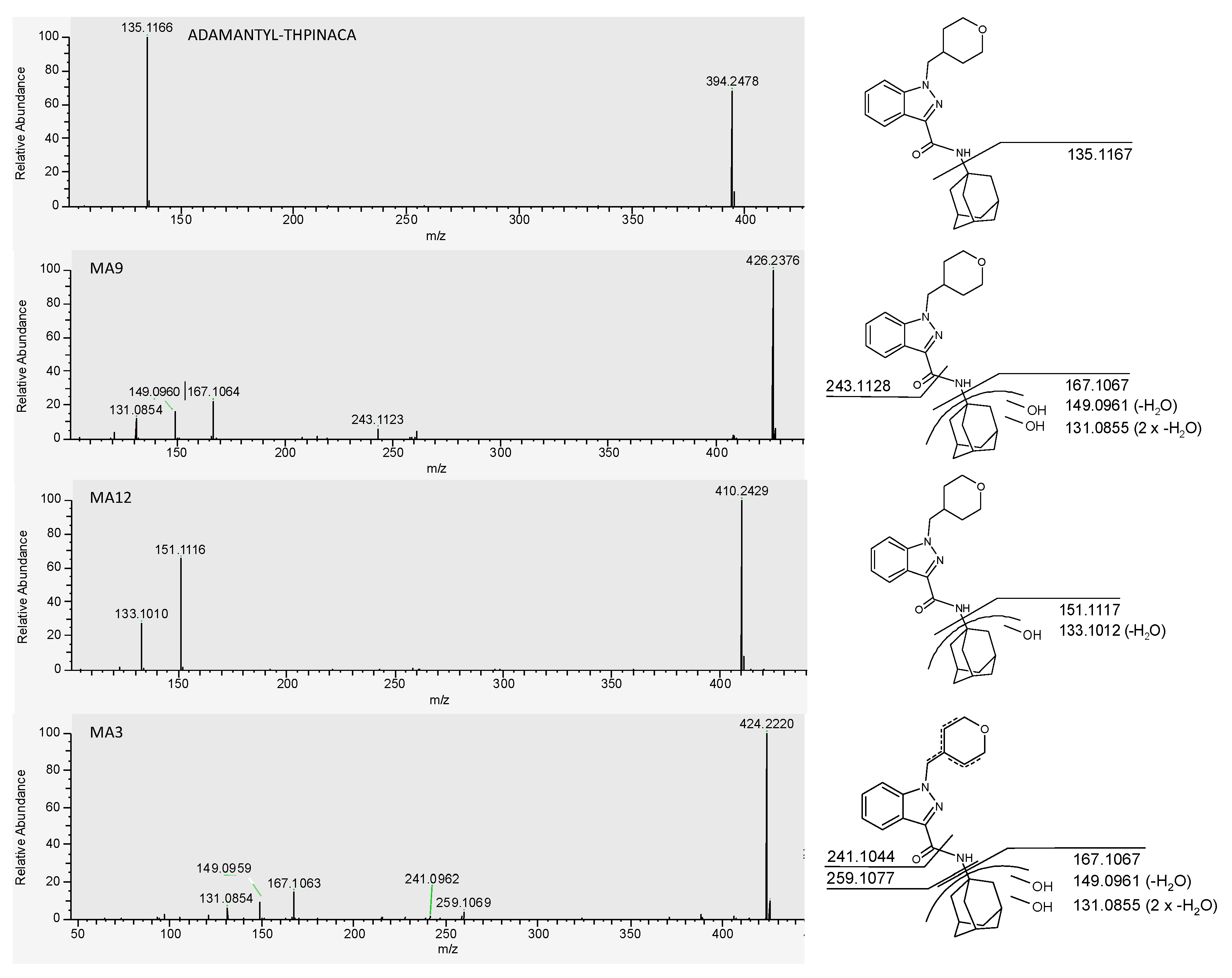
2.4.1. Mono-Hydroxylation
2.4.2. Di-Hydroxylation
2.4.3. Mono-Hydroxylation and Additional Desaturation
2.4.4. Tri-Hydroxylation
2.4.5. Di-Hydroxylation and Additional Desaturation, Mono-Hydroxylation and Additional Carbonylation
2.4.6. Identification of the Primarily Involved CYP Isoenzymes
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Microsomal Incubation with pHLM
3.3. Microsomal Incubation with rCYP
3.4. Sample Preparation
3.5. Derivatization Using Iodomethane
3.6. Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- European Monitoring Centre for Drugs and Drug Addiction. European Drug Report 2020: Trends and Developments; Publications Office of the EU: Luxembourg, 2020.
- Castaneto, M.S.; Gorelick, D.A.; Desrosiers, N.A.; Hartman, R.L.; Pirard, S.; Huestis, M.A. Synthetic cannabinoids: Epidemiology, pharmacodynamics, and clinical implications. Drug Alcohol Depend. 2014, 144, 12–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiley, J.L.; Marusich, J.A.; Huffman, J.W. Moving around the molecule: Relationship between chemical structure and in vivo activity of synthetic cannabinoids. Life Sci. 2014, 97, 55–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatch, M.B.; Forster, M. Cannabinoid-like effects of five novel carboxamide synthetic cannabinoids. NeuroToxicology 2019, 70, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Fantegrossi, W.E.; Moran, J.H.; Radominska-Pandya, A.; Prather, P.L. Distinct pharmacology and metabolism of K2 synthetic cannabinoids compared to Δ(9)-THC: Mechanism underlying greater toxicity? Life Sci. 2014, 97, 45–54. [Google Scholar] [CrossRef] [Green Version]
- Hutchison, R.D.; Ford, B.M.; Franks, L.N.; Wilson, C.D.; Yarbrough, A.L.; Fujiwara, R.; Su, M.K.; Fernandez, D.; James, L.P.; Moran, J.H.; et al. Atypical Pharmacodynamic Properties and Metabolic Profile of the Abused Synthetic Cannabinoid AB-PINACA: Potential Contribution to Pronounced Adverse Effects Relative to Δ(9)-THC. Front. Pharmacol. 2018, 9, 1084. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Angerer, V.; Jacobi, S.; Franz, F.; Auwärter, V.; Pietsch, J. Three fatalities associated with the synthetic cannabinoids 5F-ADB, 5F-PB-22, and AB-CHMINACA. Forensic Sci. Int. 2017, 281, e9–e15. [Google Scholar] [CrossRef] [PubMed]
- Harris, C.R.; Brown, A. Synthetic Cannabinoid Intoxication: A Case Series and Review. J. Emerg. Med. 2013, 44, 360–366. [Google Scholar] [CrossRef]
- Westin, A.A.; Frost, J.; Brede, W.R.; Gundersen, P.O.; Einvik, S.; Aarset, H.; Slørdal, L. Sudden Cardiac Death Following Use of the Synthetic Cannabinoid MDMB-CHMICA. J. Anal. Toxicol. 2016, 40, 86–87. [Google Scholar] [CrossRef] [Green Version]
- Hermanns-Clausen, M.; Kneisel, S.; Szabo, B.; Auwärter, V. Acute toxicity due to the confirmed consumption of synthetic cannabinoids: Clinical and laboratory findings. Addiction 2013, 108, 534–544. [Google Scholar] [CrossRef]
- Trecki, J.; Gerona, R.R.; Schwartz, M.D. Synthetic Cannabinoid-Related Illnesses and Deaths. N. Engl. J. Med. 2015, 373, 103–107. [Google Scholar] [CrossRef]
- Kleis, J.; Germerott, T.; Halter, S.; Héroux, V.; Roehrich, J.; Schwarz, C.S.; Hess, C. The synthetic cannabinoid 5F-MDMB-PICA: A case series. Forensic Sci. Int. 2020, 314, 110410. [Google Scholar] [CrossRef]
- Norman, C.; Walker, G.; McKirdy, B.; McDonald, C.; Fletcher, D.; Antonides, L.H.; Sutcliffe, O.B.; Nic Daéid, N.; McKenzie, C. Detection and quantitation of synthetic cannabinoid receptor agonists in infused papers from prisons in a constantly evolving illicit market. Drug Test. Anal. 2020, 12, 538–554. [Google Scholar] [CrossRef] [Green Version]
- Mogler, L.; Franz, F.; Rentsch, D.; Angerer, V.; Weinfurtner, G.; Longworth, M.; Banister, S.D.; Kassiou, M.; Moosmann, B.; Auwärter, V. Detection of the recently emerged synthetic cannabinoid 5F-MDMB-PICA in ‘legal high’ products and human urine samples. Drug Test. Anal. 2018, 10, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Norman, C.; Halter, S.; Haschimi, B.; Acreman, D.; Smith, J.; Krotulski, A.J.; Mohr, A.L.A.; Logan, B.K.; NicDaéid, N.; Auwärter, V.; et al. A transnational perspective on the evolution of the synthetic cannabinoid receptor agonists market: Comparing prison and general populations. Drug Test. Anal. 2021, 13, 841–852. [Google Scholar] [CrossRef] [PubMed]
- European Monitoring Centre for Drugs and Drug Addiction. EMCDDA Initial Report on the New Psychoactive Substance Methyl 3,3-Dimethyl-2-(1-(Pent-4-En-1-yl)-1H-Indazole-3-Carboxamido)Butanoate (MDMB-4en-PINACA), Initial Reports; Publications Office of the European Union: Luxembourg, 2020. Available online: https://www.emcdda.europa.eu/publications/initial-reports/mdmb-4en-pinaca_en (accessed on 1 December 2020).
- Scheidweiler, K.B.; Jarvis, M.J.Y.; Huestis, M.A. Nontargeted SWATH acquisition for identifying 47 synthetic cannabinoid metabolites in human urine by liquid chromatography-high-resolution tandem mass spectrometry. Anal. Bioanal. Chem. 2015, 407, 883–897. [Google Scholar] [CrossRef]
- Diao, X.; Huestis, M.A. Approaches, Challenges, and Advances in Metabolism of New Synthetic Cannabinoids and Identification of Optimal Urinary Marker Metabolites. Clin. Pharmacol. Ther. 2017, 101, 239–253. [Google Scholar] [CrossRef]
- Franz, F.; Angerer, V.; Moosmann, B.; Auwärter, V. Phase I metabolism of the highly potent synthetic cannabinoid MDMB-CHMICA and detection in human urine samples. Drug Test. Anal. 2017, 9, 744–753. [Google Scholar] [CrossRef]
- Diao, X.; Huestis, M.A. New Synthetic Cannabinoids Metabolism and Strategies to Best Identify Optimal Marker Metabolites. Front. Chem. 2019, 7, 109. [Google Scholar] [CrossRef] [PubMed]
- Bowden, M.; Williamson, J. Cannabinoid Compounds. WO2014/167530/A1, 11 April 2014. [Google Scholar]
- Åstrand, A.; Vikingsson, S.; Lindstedt, D.; Thelander, G.; Gréen, H.; Kronstrand, R.; Wohlfarth, A. Metabolism study for CUMYL-4CN-BINACA in human hepatocytes and authentic urine specimens: Free cyanide is formed during the main metabolic pathway. Drug Test. Anal. 2018, 10, 1270–1279. [Google Scholar] [CrossRef] [PubMed]
- Staeheli, S.N.; Poetzsch, M.; Veloso, V.P.; Bovens, M.; Bissig, C.; Steuer, A.E.; Kraemer, T. In vitro metabolism of the synthetic cannabinoids CUMYL-PINACA, 5F–CUMYL-PINACA, CUMYL-4CN-BINACA, 5F–CUMYL-P7AICA and CUMYL-4CN-B7AICA. Drug Test. Anal. 2018, 10, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Staeheli, S.N.; Steuer, A.E.; Kraemer, T. Identification of urinary metabolites of the synthetic cannabinoid 5F-CUMYL-P7AICA in human casework. Forensic Sci. Int. 2019, 294, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Mogler, L.; Wilde, M.; Huppertz, L.M.; Weinfurtner, G.; Franz, F.; Auwärter, V. Phase I metabolism of the recently emerged synthetic cannabinoid CUMYL-PEGACLONE and detection in human urine samples. Drug Test. Anal. 2018, 10, 886–891. [Google Scholar] [CrossRef] [PubMed]
- Mogler, L.; Halter, S.; Wilde, M.; Franz, F.; Auwärter, V. Human phase I metabolism of the novel synthetic cannabinoid 5F-CUMYL-PEGACLONE. Forensic Toxicol. 2019, 37, 154–163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asada, A.; Doi, T.; Tagami, T.; Takeda, A.; Satsuki, Y.; Kawaguchi, M.; Nakamura, A.; Sawabe, Y. Cannabimimetic activities of cumyl carboxamide-type synthetic cannabinoids. Forensic Toxicol. 2018, 36, 170–177. [Google Scholar] [CrossRef]
- Schoeder, C.T.; Hess, C.; Madea, B.; Meiler, J.; Müller, C.E.J.F.T. Pharmacological evaluation of new constituents of “Spice”: Synthetic cannabinoids based on indole, indazole, benzimidazole and carbazole scaffolds. Forensic Toxicol. 2018, 36, 385–403. [Google Scholar] [CrossRef] [Green Version]
- EMCDDA-Europol. Annual Report on the Implementation of Council Decision 2005/387/JHA. 2015. Available online: https://www.emcdda.europa.eu/system/files/publications/2880/TDAS16001ENN.pdf (accessed on 26 March 2021).
- Kovács, K.; Kereszty, É.; Berkecz, R.; Tiszlavicz, L.; Sija, É.; Körmöczi, T.; Jenei, N.; Révész-Schmehl, H.; Institóris, L. Fatal intoxication of a regular drug user following N-ethyl-hexedrone and ADB-FUBINACA consumption. J. Forensic Leg. Med. 2019, 65, 92–100. [Google Scholar] [CrossRef]
- Kadomura, N.; Ito, T.; Kawashima, H.; Matsuhisa, T.; Kinoshita, T.; Soda, M.; Kohyama, E.; Iwaki, T.; Nagai, H.; Kitaichi, K. In vitro metabolic profiles of adamantyl positional isomers of synthetic cannabinoids. Forensic Toxicol. 2021, 39, 26–44. [Google Scholar] [CrossRef]
- Broberg, M.N.; Knych, H.; Bondesson, U.; Pettersson, C.; Stanley, S.; Thevis, M.; Hedeland, M. Investigation of Equine In Vivo and In Vitro Derived Metabolites of the Selective Androgen Receptor Modulator (SARM) ACP-105 for Improved Doping Control. Metabolites 2021, 11, 85. [Google Scholar] [CrossRef]
- Holm, N.B.; Nielsen, L.M.; Linnet, K. CYP3A4 Mediates Oxidative Metabolism of the Synthetic Cannabinoid AKB-48. AAPS J. 2015, 17, 1237–1245. [Google Scholar] [CrossRef] [Green Version]
- Nielsen, L.M.; Holm, N.B.; Olsen, L.; Linnet, K. Cytochrome P450-mediated metabolism of the synthetic cannabinoids UR-144 and XLR-11. Drug Test. Anal. 2016, 8, 792–800. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef]
- Gaunitz, F.; Thomas, A.; Fietzke, M.; Franz, F.; Auwärter, V.; Thevis, M.; Mercer-Chalmers-Bender, K. Phase I metabolic profiling of the synthetic cannabinoids THJ-018 and THJ-2201 in human urine in comparison to human liver microsome and cytochrome P450 isoenzyme incubation. Int. J. Leg. Med. 2019, 133, 1049–1064. [Google Scholar] [CrossRef]
- Gaunitz, F.; Dahm, P.; Mogler, L.; Thomas, A.; Thevis, M.; Mercer-Chalmers-Bender, K.J.A.; Chemistry, B. In vitro metabolic profiling of synthetic cannabinoids by pooled human liver microsomes, cytochrome P450 isoenzymes, and Cunninghamella elegans and their detection in urine samples. Anal. Bioanal. Chem. 2019, 411, 3561–3579. [Google Scholar] [CrossRef]
- Swortwood, M.J.; Carlier, J.; Ellefsen, K.N.; Wohlfarth, A.; Diao, X.; Concheiro-Guisan, M.; Kronstrand, R.; Huestis, M.A. In vitro, in vivo and in silico metabolic profiling of α-pyrrolidinopentiothiophenone, a novel thiophene stimulant. Bioanalysis 2016, 8, 65–82. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mardal, M.; Dalsgaard, P.W.; Qi, B.; Mollerup, C.B.; Annaert, P.; Linnet, K. Metabolism of the synthetic cannabinoids AMB-CHMICA and 5C-AKB48 in pooled human hepatocytes and rat hepatocytes analyzed by UHPLC-(IMS)-HR-MSE. J. Chromatogr. B Analyt. Biomed. Life Sci. 2018, 1083, 189–197. [Google Scholar] [CrossRef]
- Carlier, J.; Diao, X.; Huestis, M.A. Synthetic cannabinoid BB-22 (QUCHIC): Human hepatocytes metabolism with liquid chromatography-high resolution mass spectrometry detection. J. Pharm. Biomed. Anal. 2018, 157, 27–35. [Google Scholar] [CrossRef] [PubMed]
- Fabregat-Safont, D.; Mardal, M.; Noble, C.; Cannaert, A.; Stove, C.P.; Sancho, J.V.; Linnet, K.; Hernández, F.; Ibáñez, M. Comprehensive investigation on synthetic cannabinoids: Metabolic behavior and potency testing, using 5F-APP-PICA and AMB-FUBINACA as model compounds. Drug Test. Anal. 2019, 11, 1358–1368. [Google Scholar] [CrossRef] [PubMed]
- Kevin, R.C.; Lefever, T.W.; Snyder, R.W.; Patel, P.R.; Fennell, T.R.; Wiley, J.L.; McGregor, I.S.; Thomas, B.F.J.F.T. In vitro and in vivo pharmacokinetics and metabolism of synthetic cannabinoids CUMYL-PICA and 5F-CUMYL-PICA. Forensic Toxicol. 2017, 35, 333–347. [Google Scholar] [CrossRef] [Green Version]
- Montesano, C.; Vincenti, F.; Fanti, F.; Marti, M.; Bilel, S.; Togna, A.R.; Gregori, A.; Di Rosa, F.; Sergi, M. Untargeted Metabolic Profiling of 4-Fluoro-Furanylfentanyl and Isobutyrylfentanyl in Mouse Hepatocytes and Urine by Means of LC-HRMS. Metabolites 2021, 11, 97. [Google Scholar] [CrossRef]
- Seitzer, P.M.; Searle, B.C. Incorporating In-Source Fragment Information Improves Metabolite Identification Accuracy in Untargeted LC-MS Data Sets. J. Proteome Res. 2019, 18, 791–796. [Google Scholar] [CrossRef]
- Anzenbacher, P.; Anzenbacherová, E. Cytochromes P450 and metabolism of xenobiotics. Cell. Mol. Life Sci. 2001, 58, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Hemmer, S.; Manier, S.K.; Fischmann, S.; Westphal, F.; Wagmann, L.; Meyer, M.R. Comparison of Three Untargeted Data Processing Workflows for Evaluating LC-HRMS Metabolomics Data. Metabolites 2020, 10, 378. [Google Scholar] [CrossRef] [PubMed]
- Gandhi, A.S.; Zhu, M.; Pang, S.; Wohlfarth, A.; Scheidweiler, K.B.; Liu, H.F.; Huestis, M.A. First characterization of AKB-48 metabolism, a novel synthetic cannabinoid, using human hepatocytes and high-resolution mass spectrometry. AAPS J. 2013, 15, 1091–1098. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID | Biotransformation | Formula | [M + H]+ Productions (m/z) | Mass Error (ppm) | Rt (min) | Area (n = 2) | Rank |
|---|---|---|---|---|---|---|---|
| MC1 | di-hydroxylation at cumyl | C23H28N3O4 | 410.2074 | 0.4 | 0.89 | 3.0 × 106 | 21 |
| 259.1077 | |||||||
| 151.0754 | |||||||
| MC2a–b | di-hydroxylation at cumyl, mono-hydroxylation at 4-methyl-tetrahydropyran | C23H28N3O5 | 426.2023 | 0.4 | a: 0.90 | 3.9 × 106 | 19 |
| 408.1918 | |||||||
| 259.1077 | b: 0.99 | ||||||
| 151.0754 | |||||||
| MC3 | mono-hydroxylation at cumyl, mono-hydroxylation and desaturation at 4-methyl-tetrahydropyran | C23H28N3O5 | 408.1918 | −0.1 | 1.08 | 8.00 × 106 | 14 |
| 256.1081 | |||||||
| 135.0804 | |||||||
| MC4 | di-hydroxylation at cumyl, mono-hydroxylation at 4-methyl-tetrahydropyran | C23H28N3O5 | 426.2023 | −0.2 | 1.08 | 1.1 × 106 | 22 |
| 408.1918 | |||||||
| 259.1077 | |||||||
| 151.0754 | |||||||
| MCArt1 | In-source water loss of MC5 | C23H28N3O5 | 408.1918 | 0.4 | 1.16 | 4.5 × 106 | - |
| 256.1081 | |||||||
| 135.0804 | |||||||
| MC5 | mono-hydroxylation at cumyl, di-hydroxylation at 4-methyl-tetrahydropyran | C23H28N3O5 | 426.2023 | 0.8 | 1.19 | 3.7 × 106 | 20 |
| 274.1186 | |||||||
| 256.1081 | |||||||
| 135.0804 | |||||||
| MC6 | di-hydroxylation at cumyl, desaturation at 4-methyl-tetrahydropyran | C23H28N3O5 | 408.1918 | −0.3 | 1.20 | 2.3 × 107 | 7 |
| 258.1237 | |||||||
| 151.0754 | |||||||
| MC7 | mono-hydroxylation at cumyl, mono-hydroxylation at indazole, mono-hydroxylation at 4-methyl-tetrahydropyran | C23H28N3O5 | 426.2023 | −0.6 | 1.23 | 1.6 × 107 | 10 |
| 292.1292 | |||||||
| 274.1186 | |||||||
| 257.0913 | |||||||
| 135.0804 | |||||||
| MC8a–e | mono-hydroxylation at cumyl and mono-hydroxylation at 4-methyl-tetrahydropyran | C23H28N3O4 | 410.2074 | −1.3 | a: 1.28 b: 1.35 c: 1.39 d: 1.46 e: 1.53 | 4.4 × 107 | 4 |
| 258.1237 | |||||||
| 241.0972 | |||||||
| 135.0804 | |||||||
| MC9 | di-hydroxylation at cumyl, mono-hydroxylation at 4-methyl-tetrahydropyran | C23H28N3O5 | 426.2023 | −0.6 | 1.27 | 1.9 × 107 | 9 |
| 258.1237 | |||||||
| 151.0754 | |||||||
| MCArt2a–b | In-source water loss MC8a–e | C23H26N3O3 | 392.1969 | −1.7 | a: 1.39 | 5.5 × 107 | - |
| 258.1237 | |||||||
| 241.0972 | b: 1.44 | ||||||
| 135.0804 | |||||||
| MC10 | di-hydroxylation at 4-methyl-tetrahydropyran, mono-hydroxylation at indazole | C23H28N3O5 | 426.2023 | −0.7 | 1.42 | 9.1 × 106 | 13 |
| 308.1241 | |||||||
| 290.1135 | |||||||
| 272.103 | |||||||
| 119.0855 | |||||||
| MC11 | tri-hydroxylation at 4-methyl-tetrahydropyran | C23H28N3O5 | 426.2023 | −0.5 | 1.51 | 3.8 × 107 | 5 |
| 308.1241 | |||||||
| 290.1135 | |||||||
| 272.103 | |||||||
| 254.0924 | |||||||
| 119.0855 | |||||||
| MC12 | mono-hydroxylation and desaturation at 4-methyl-tetrahydropyran | C23H26N3O3 | 392.1969 | −1.6 | 1.54 | 7.8 × 107 | 2 |
| 256.1081 | |||||||
| 239.0815 | |||||||
| MC13 | mono-hydroxylation at cumyl, carbonylation at 4-methyl-tetrahydropyran | C23H28N3O5 | 408.1918 | −1.2 | 1.56 | 1.1 × 107 | 12 |
| 274.1186 | |||||||
| 257.0913 | |||||||
| 135.0804 | |||||||
| MC14 | di-hydroxylation at cumyl | C23H28N3O4 | 410.2074 | −1.1 | 1.62 | 2.3 × 107 | 8 |
| 260.1394 | |||||||
| 151.0754 | |||||||
| MC15 | mono-hydroxylation at cumyl, carbonylation at 4-methyl-tetrahydropyran | C23H28N3O5 | 408.1918 | −0.1 | 1.62 | 6.3 × 106 | 16 |
| 274.1186 | |||||||
| 257.0913 | |||||||
| 135.0804 | |||||||
| MC16 | mono-hydroxylation at 4-methyl-tetrahydropyran, mono-hydroxylation at indazole | C23H28N3O4 | 410.2074 | −1.3 | 1.69 | 3.4 × 108 | 1 |
| 292.1292 | |||||||
| 274.1186 | |||||||
| 257.0913 | |||||||
| 119.0855 | |||||||
| MC17 | mono-hydroxylation and desaturation at 4-methyl-tetrahydropyran | C23H26N3O3 | 392.1969 | −0.9 | 1.81 | 5.6 × 106 | 17 |
| 256.1081 | |||||||
| 239.0815 | |||||||
| 119.0855 | |||||||
| MC18 | mono-hydroxylation and carbonylation at 4-methyl-tetrahydropyran | C23H28N3O5 | 408.1918 | −0.2 | 1.88 | 1.3 × 107 | 11 |
| 290.1135 | |||||||
| 273.087 | |||||||
| 272.103 | |||||||
| 119.0855 | |||||||
| MC19a–b | a: mono-hydroxylation at cumyl | C23H28N3O3 | 394.2118 | 1.4 | 1.99 | 3.0 × 107 | 6 |
| a: | 135.0804 | ||||||
| 260.1394 | |||||||
| 243.1128 | |||||||
| b: mono-hydroxylation at 4-methyl-tetrahydropyran | b: | 276.1343 | |||||
| 119.0855 | |||||||
| MCArt3 | In-source water loss MC19b | C23H25N3O2 | 376.2020 | −0.7 | 2.03 | 1.5 × 107 | - |
| 258.1237 | |||||||
| 119.0855 | |||||||
| MC20 | mono-hydroxylation and carbonylation at cumyl | C23H28N3O5 | 408.1918 | −0.1 | 1.98 | 7.0 × 106 | 15 |
| 260.1394 | |||||||
| 243.1128 | |||||||
| 149.1660 | |||||||
| MC21 | mono-hydroxylation at 4-methyl-tetrahydropyran | C23H28N3O3 | 394.2118 | 1.4 | 2.15 | 4.4 × 106 | 18 |
| 258.1237 | |||||||
| 119.0855 | |||||||
| MCArt4 | In-source water loss MC21 | C23H25N3O2 | 376.2020 | −0.5 | 2.17 | 4.7 × 106 | - |
| 258.1237 | |||||||
| 119.0855 | |||||||
| MC22 | carbonylation at 4-methyl-tetrahydropyran | C23H26N3O3 | 392.1969 | −1.3 | 2.39 | 5.0 × 107 | 3 |
| 274.1186 | |||||||
| 257.0913 | |||||||
| 119.0855 | |||||||
| CUMYL-THPINACA | C23H27N3O2 | 378.2176 | 0.1 | 3.07 | 8.9 × 105 | - | |
| 260.1394 | |||||||
| 243.1128 | |||||||
| 119.0855 | |||||||
| ID | 3A4 | 3A5 | 2D6 | 2C8 | 2C9 | 2C19 | 2B6 | 1A2 | 2E1 | 2A6 | Negative Control |
|---|---|---|---|---|---|---|---|---|---|---|---|
| n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | |
| MC1 | (+) | (+) | - | - | - | - | - | - | - | - | - |
| MC2a–b | + | (+) | - | - | - | - | - | - | - | - | - |
| MC3 | (+) | - | - | - | - | - | - | - | - | - | |
| MC4 | - | - | - | - | - | - | - | - | - | - | - |
| MCArt1 | (+) | (+) | - | - | - | - | - | - | - | - | - |
| MC5 | - | - | - | - | - | - | - | - | - | - | - |
| MC6 | (+) | (+) | - | - | - | - | - | - | - | - | - |
| MC7 | (+) | (+) | - | - | - | - | - | - | - | - | - |
| MC8a–e | + | + | (+) | (+) | (+) | (+) | - | - | - | - | - |
| MC9 | (+) | - | - | - | - | - | - | ||||
| MCArt2a–b | + | + | (+) | (+) | - | (+) | - | - | - | - | - |
| MC10 | + | + | - | - | - | - | - | - | - | - | - |
| MC11 | + | (+) | - | - | - | - | - | - | - | - | - |
| MC12 | ++ | + | - | - | - | - | - | - | - | - | - |
| MC13 | (+) | - | - | - | - | - | - | - | - | - | - |
| MC14 | + | + | (+) | (+) | - | (+) | - | - | - | - | - |
| MC15 | (+) | - | - | - | - | - | - | - | - | - | |
| MC16 | ++ | ++ | + | (+) | - | (+) | - | - | - | - | - |
| MC17 | + | (+) | - | - | - | - | - | - | - | - | - |
| MC18 | + | (+) | - | - | - | - | - | - | - | - | - |
| MC19a–b | +++ | ++ | ++ | + | (+) | + | (+) | (+) | - | - | - |
| MCArt3 | +++ | +++ | +++ | ++ | + | ++ | + | + | (+) * | (+) * | (+) * |
| MC20 | + | (+) | - | - | - | - | - | - | - | - | - |
| MC21 | +++ | +++ | ++ | ++ | + | ++ | (+) | (+) | - | - | - |
| MCArt4 | +++ | +++ | +++ | +++ | + | +++ | (+) * | (+) * | (+) * | (+) * | (+) * |
| MC22 | ++ | + | (+) | (+) | - | (+) | - | - | - | - | - |
| ID | Biotransformation | Formula | [M + H]+ Product ions (m/z) | Mass error (ppm) | Rt (min) | Area (n = 2) | Rank |
|---|---|---|---|---|---|---|---|
| MA1 | di-hydroxylation at adamantyl, mono-hydroxylation at 4-methyl-tetrahydropyran | C24H31N3O5 | 442.2336 | 1.4 | 0.87 | 9.2 × 106 | 11 |
| 424.2221 | |||||||
| 259.1077 | |||||||
| 167.1067 | |||||||
| 149.0961 | |||||||
| 131.0855 | |||||||
| MA2 | di-hydroxylation at adamantyl, mono-hydroxylation at 4-methyl-tetrahydropyran | C24H31N3O5 | 442.2336 | −0.7 | 0.97 | 1.1 × 107 | 10 |
| 424.2221 | |||||||
| 259.1077 | |||||||
| 167.1067 | |||||||
| 149.0961 | |||||||
| 131.0855 | |||||||
| MAArt1 | in-source water loss of MA2 | C24H29N3O4 | 424.2231 | −0.7 | 0.98 | 7.5 × 107 | |
| 259.1077 | |||||||
| 241.1044 | |||||||
| 167.1067 | |||||||
| 149.0961 | |||||||
| 131.0855 | |||||||
| MA3 | di-hydroxylation at adamantyl, desaturation at 4-methyl-tetrahydropyran | C24H29N3O4 | 424.2231 | −0.5 | 1.03 | 3.8 × 107 | 3 |
| 259.1077 | |||||||
| 241.1044 | |||||||
| 167.1067 | |||||||
| 149.0961 | |||||||
| 131.0855 | |||||||
| MA4 | tri-hydroxylation at adamantyl | C24H31N3O5 | 442. 2336 | −0.5 | |||
| 424.2231 | |||||||
| 260.1393 | |||||||
| 243.1128 | |||||||
| MA5 | di-hydroxylation at adamantyl | C24H31N3O3 | 426.2387 | 1.2 | 1.09 | 1.7 × 107 | 8 |
| 260.1394 | |||||||
| 243.1128 | |||||||
| 167.1067 | |||||||
| 149.0961 | |||||||
| 131.0855 | |||||||
| MA6 | mono-hydroxylation at adamantyl, mono-hydroxylation at indazole, mono-hydroxylation at 4-methyl-tetrahydropyran | C24H31N3O5 | 442.2336 | 0 | 1.1 | 1.3 × 107 | 9 |
| 424.2221 | |||||||
| 406.2114 | |||||||
| 151.1117 | |||||||
| 133.1012 | |||||||
| 274.1186 | |||||||
| 257.0921 | |||||||
| MA7 | mono-hydroxylation at adamantyl, mono-hydroxylation at 4-methyl-tetrahydropyran | C24H31N3O3 | 426.2387 | 0.5 | 1.2 | 2.4 × 107 | 6 |
| 151.1117 | |||||||
| 133.1012 | |||||||
| MA8 | Mono-hydroxylation at adamantyl, desaturation at 4-methyl-tetrahydropyran | C24H29N3O3 | 408.2282 | −0.2 | 1.27 | 3.2 × 107 | 4 |
| 151.1117 | |||||||
| 133.10118 | |||||||
| MA9 | di-hydroxylation at adamantyl | C24H31N3O3 | 426.2387 | −1.2 | 1.36 | 4.2 × 108 | 1 |
| 243.1128 | |||||||
| 167.1067 | |||||||
| 149.0961 | |||||||
| 131.0855 | |||||||
| MAArt2 | in-source water loss of MA9 | C24H29N3O3 | 408.2282 | −0.7 | 1.36 | 7.9 × 107 | |
| 260.1394 | |||||||
| 243.1128 | |||||||
| 149.0961 | |||||||
| 131.0855 | |||||||
| MA10 | Mono-hydroxylation at adamantyl, and carbonylation or mono-hydroxylation and desaturation at 4-methyl-tetrahydropyran | C24H29N3O4 | 424.2231 | 0 | 1.39 | 6.9 × 106 | 12 |
| 151.1117 | |||||||
| 133.1012 | |||||||
| MA11 | Di-hydroxylation at adamantyl, desaturation at 4-methyl-tetrahydropyran | C24H29N3O4 | 424.2231 | −0.9 | 1.73 | 4.1 × 106 | 13 |
| 259.1077 | |||||||
| 167.1067 | |||||||
| 149.0961 | |||||||
| 131.0855 | |||||||
| MA12 | mono-hydroxylation at adamantyl | C24H31N3O3 | 410.2438 | −0.5 | 1.75 | 7.6 × 107 | 2 |
| 151.1117 | |||||||
| 133.1012 | |||||||
| MA13 | carbonylation and mono-hydroxylation at adamantyl | C24H29N3O4 | 424.2231 | 0.2 | 1.8 | 1.8 × 107 | 7 |
| 406.2125 | |||||||
| 260.1394 | |||||||
| 243.1128 | |||||||
| 165.091 | |||||||
| MA1 | 119.0855 | ||||||
| ADAMANTYL-THPINACA | C24H31N3O3 | 394.2489 | 0.6 | 4.56 | 1.0 × 106 | - | |
| 135.1167 | |||||||
| ID | 3A4 | 3A5 | 2D6 | 2C8 | 2C9 | 2C19 | 2B6 | 1A2 | 2E1 | 2A6 | Negative Control |
|---|---|---|---|---|---|---|---|---|---|---|---|
| n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | n = 2 | |
| MA1 | + | (+) | - | - | - | - | - | - | - | - | - |
| MA2 | + | (+) | - | - | - | - | - | - | - | - | - |
| MAArt1 | ++ | (+) | - | - | - | - | - | - | - | - | - |
| MA3 | ++ | (+) | - | - | - | - | - | - | - | - | - |
| MA4 | + | (+) | - | - | - | - | - | - | - | - | - |
| MA5 | + | (+) | (+) | (+) | - | - | - | - | - | - | - |
| MA6 | (+) | (+) | - | - | - | - | - | - | - | - | - |
| MA7 | + | (+) | (+) | (+) | - | - | - | - | - | - | - |
| MA8 | + | ++ | + | (+) | - | - | - | - | - | - | - |
| MA9 | +++ | ++ | (+) | (+) | - | - | - | - | - | - | - |
| MAArt2 | ++ | + | (+) | (+) | - | - | - | - | - | - | - |
| MA10 | (+) | (+) | - | - | - | - | - | - | - | - | - |
| MA11 | (+) | (+) | - | - | - | - | - | - | - | - | - |
| MA12 | + | +++ | ++ | +++ | + | + | (+) * | (+) * | (+) * | (+) * | (+) * |
| MA13 | + | (+) | - | - | - | - | - | - | - | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monti, M.C.; Scheurer, E.; Mercer-Chalmers-Bender, K. Phase I In Vitro Metabolic Profiling of the Synthetic Cannabinoid Receptor Agonists CUMYL-THPINACA and ADAMANTYL-THPINACA. Metabolites 2021, 11, 470. https://doi.org/10.3390/metabo11080470
Monti MC, Scheurer E, Mercer-Chalmers-Bender K. Phase I In Vitro Metabolic Profiling of the Synthetic Cannabinoid Receptor Agonists CUMYL-THPINACA and ADAMANTYL-THPINACA. Metabolites. 2021; 11(8):470. https://doi.org/10.3390/metabo11080470
Chicago/Turabian StyleMonti, Manuela Carla, Eva Scheurer, and Katja Mercer-Chalmers-Bender. 2021. "Phase I In Vitro Metabolic Profiling of the Synthetic Cannabinoid Receptor Agonists CUMYL-THPINACA and ADAMANTYL-THPINACA" Metabolites 11, no. 8: 470. https://doi.org/10.3390/metabo11080470
APA StyleMonti, M. C., Scheurer, E., & Mercer-Chalmers-Bender, K. (2021). Phase I In Vitro Metabolic Profiling of the Synthetic Cannabinoid Receptor Agonists CUMYL-THPINACA and ADAMANTYL-THPINACA. Metabolites, 11(8), 470. https://doi.org/10.3390/metabo11080470