
Gut Microbiome and Metabolome Profiles Associated with High-Fat Diet in Mice



Abstract
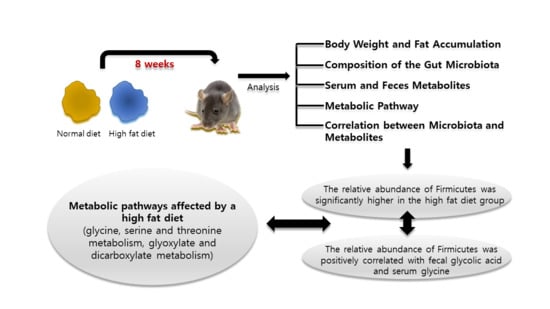
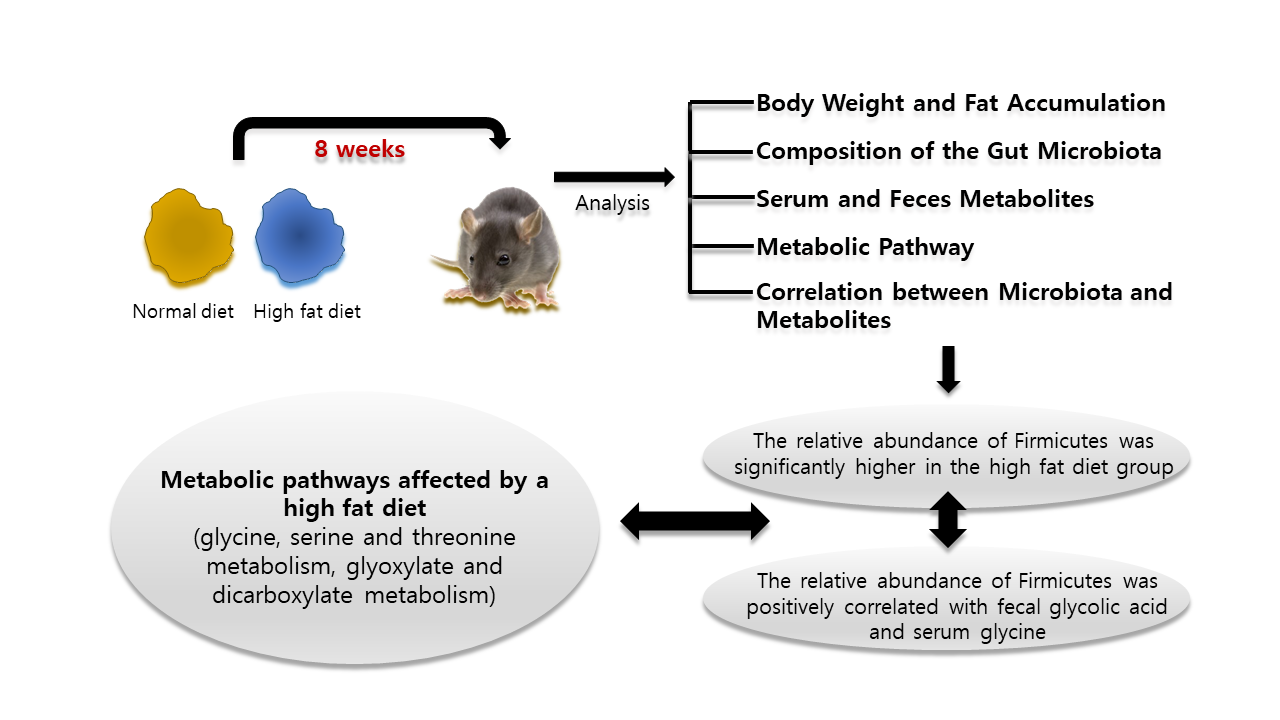
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
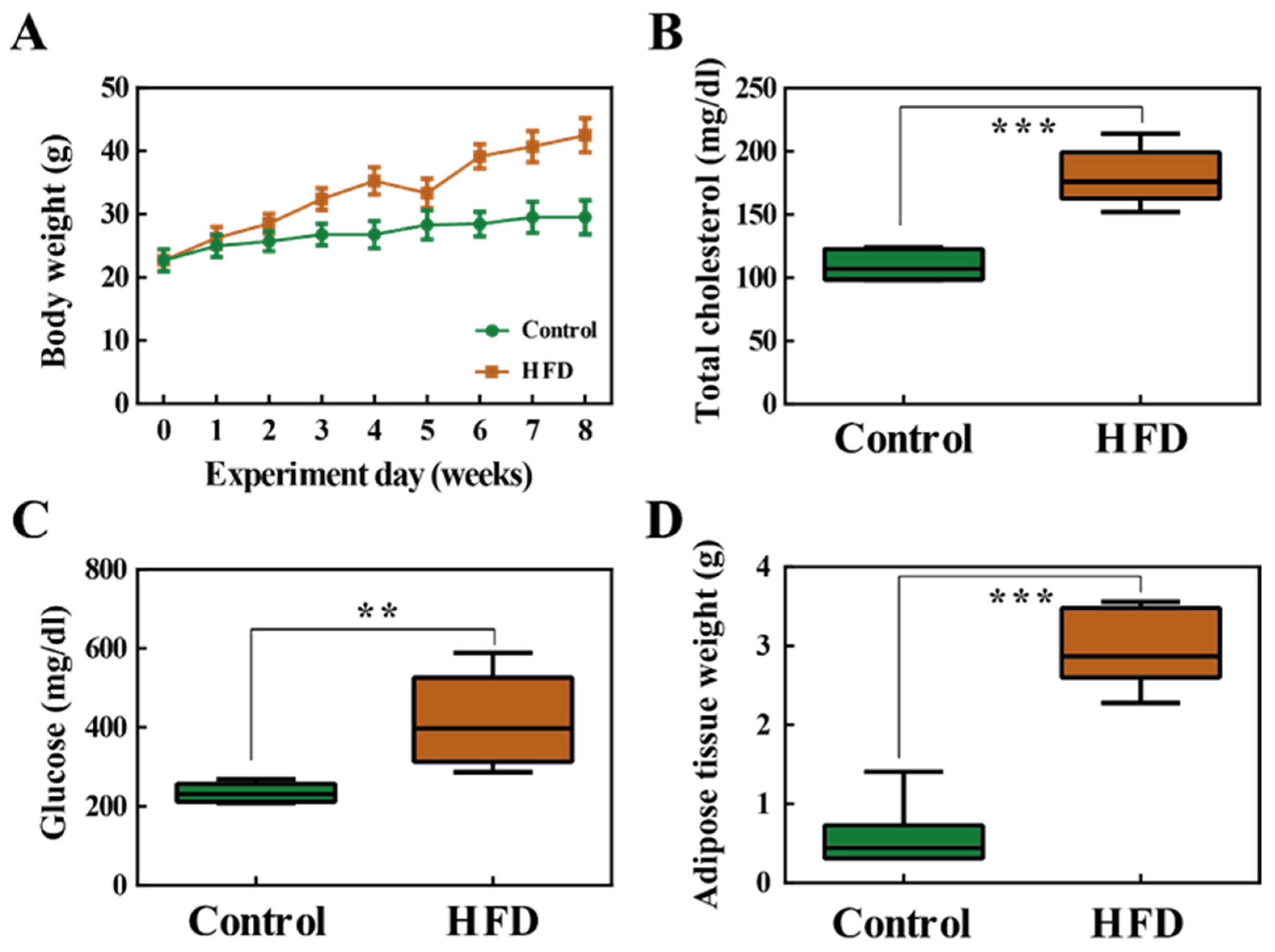
2.1. Body Weight and Fat Accumulation
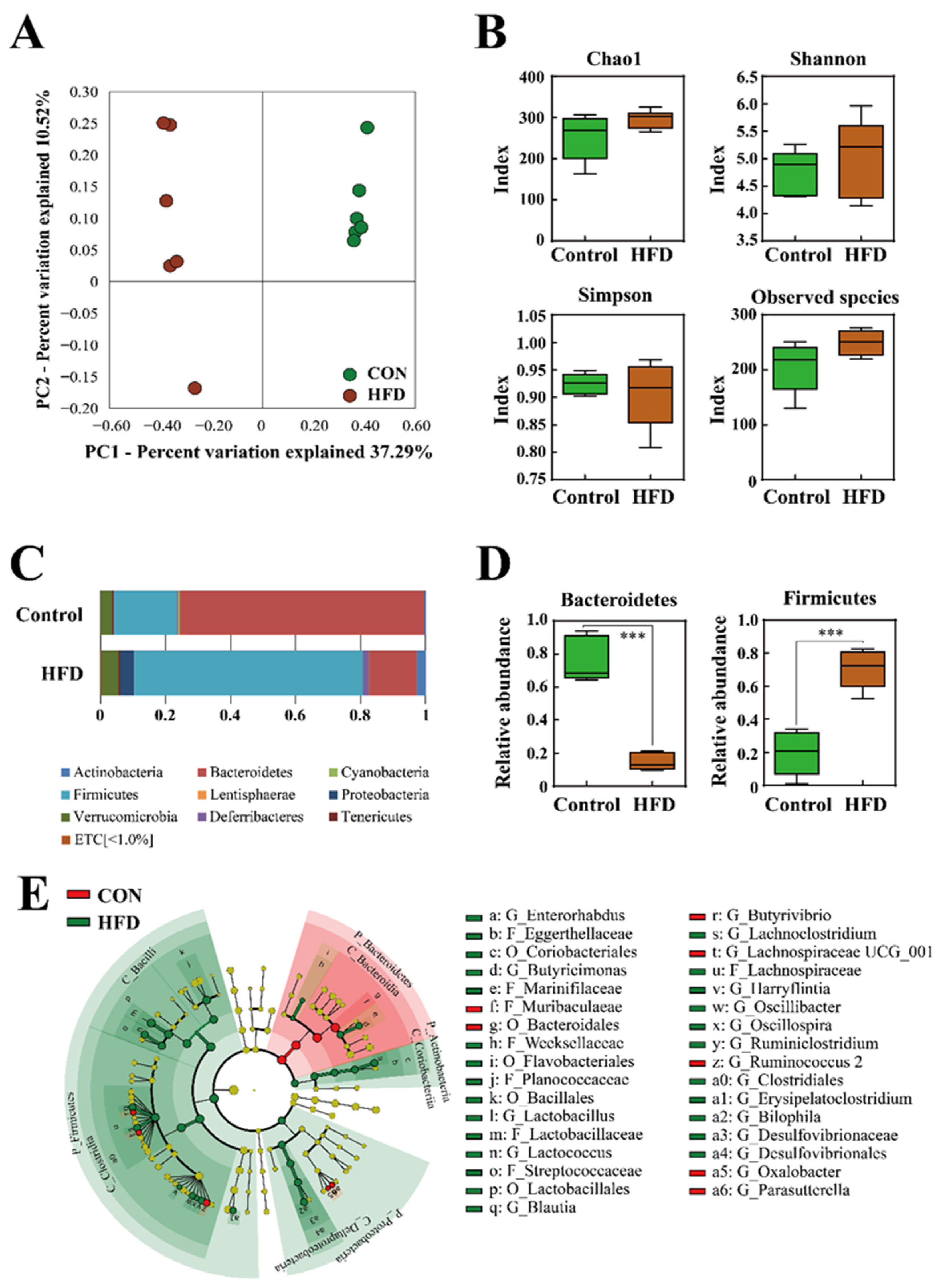
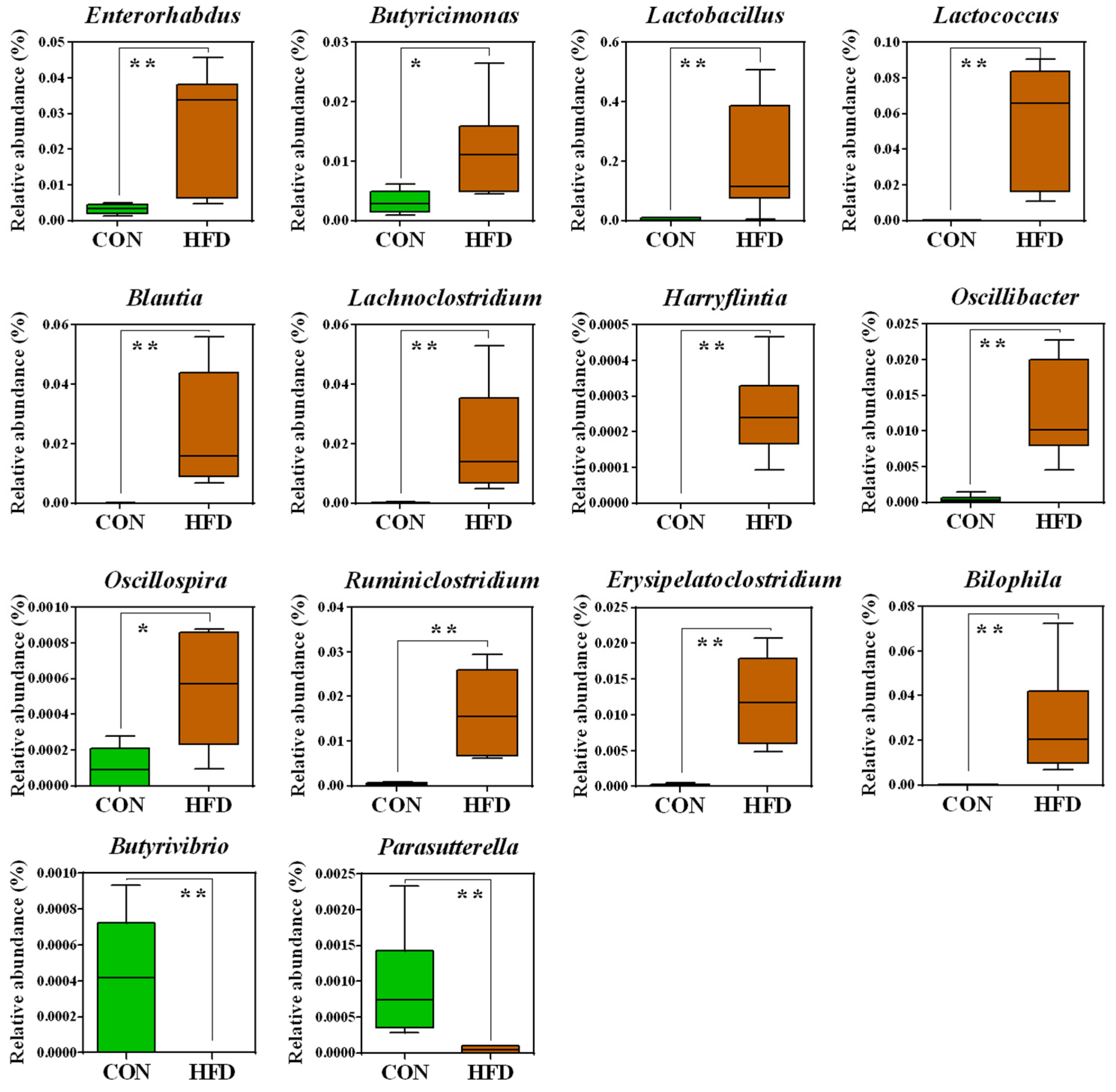
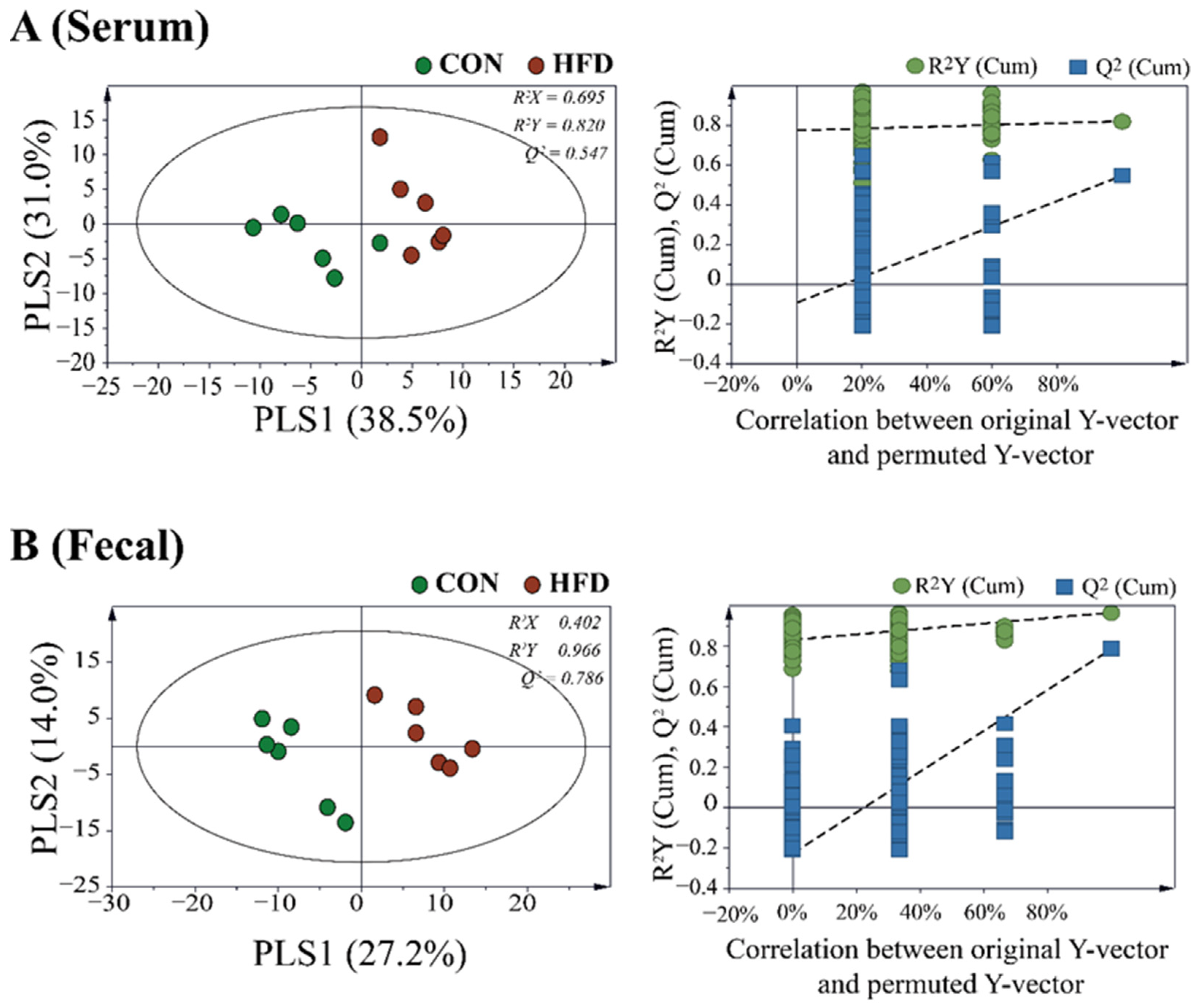
2.2. The Composition of the Gut Microbiota
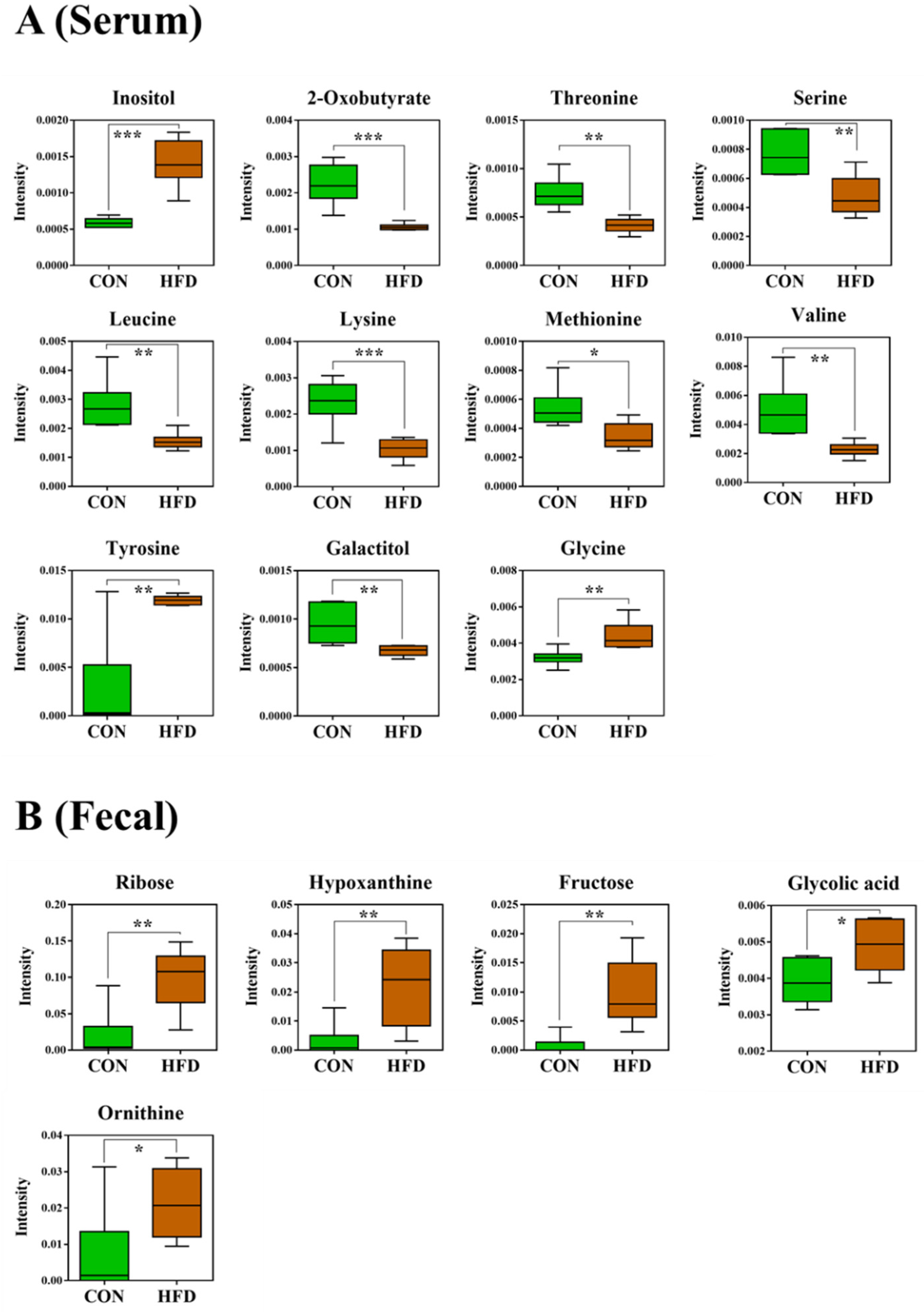
2.3. Serum and Feces Metabolites
2.4. Metabolic Pathway Analysis
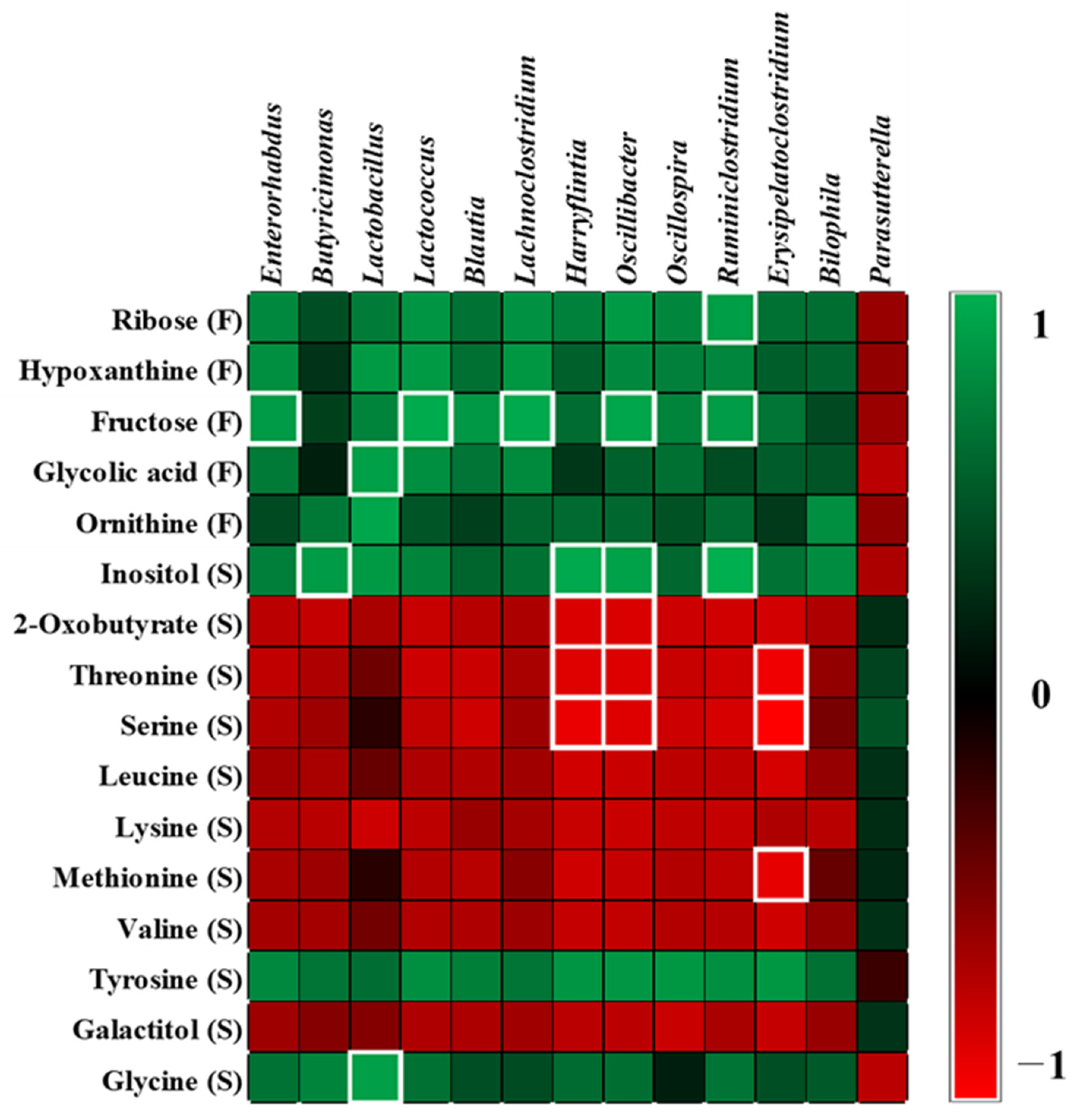
2.5. Correlation between Microbiota and Metabolites
3. Discussion
4. Materials and Methods
4.1. Animals and Treatment
4.2. DNA Extraction and 16S rRNA Gene Amplicon Sequencing
4.3. Sample Derivatization and GC-MS Analysis
4.4. Data Processing and Statistical Analysis
4.5. Metabolic Pathway Analysis
4.6. Correlation Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Permana, P.A.; Del Parigi, A.; Tataranni, P.A. Microarray gene expression profiling in obesity and insulin resistance. Nutrition 2004, 20, 134. [Google Scholar] [CrossRef]
- Kussmann, M.; Raymond, F.; Affolter, M. OMICS-driven biomarker discovery in nutrition and health. J. Biotechnol. 2006, 124, 758–787. [Google Scholar] [CrossRef]
- Goede, D.; Hauser, R. Increased prevalence of high Body Mass Index in patients presenting with pituitary tumours: Severe obesity in patients with macroprolactinoma. Swiss. Med. Wkly. 2006, 136, 254–258. [Google Scholar]
- Bartrina, J.A. Prevalence of obesity in developed countries: Current status and perspectives. Nutr. Hosp. 2002, 17, 34–41. [Google Scholar]
- Von Deneen, K.M.; Wei, Q.; Tian, J.; Liu, Y. Obesity in China: What are the causes? Curr. Pharm. Des. 2011, 17, 1132–1139. [Google Scholar] [CrossRef] [Green Version]
- Pickard, J.M.; Zeng, M.Y.; Caruso, R.; Núñez, G. Gut microbiota: Role in pathogen colonization, immune responses, and inflammatory disease. Immunol. Rev. 2017, 279, 70–89. [Google Scholar] [CrossRef]
- Dahiya, D.K.; Puniya, M.; Shandilya, U.K.; Dhewa, T.; Kumar, N.; Kumar, S.; Punjya, A.K.; Shukla, P. Gut microbiota modulation and its relationship with obesity using prebiotic fibers and probiotics: A review. Front. Microbiol. 2017, 8, 563. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Ley, R.E.; Turnbaugh, P.J.; Klein, S.; Gordon, J.I. Human gut microbes associated with obesity. Nature 2006, 444, 1022–1023. [Google Scholar] [CrossRef]
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103. [Google Scholar] [CrossRef]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvani, R.; Miccheli, A.; Landi, F.; Bossola, M.; Cesari, M.; Leeuwenburgh, C.; Sieber, C.C.; Bernabei, R.; Marzetti, E. Current nutritional recommendations and novel dietary strategies to manage sarcopenia. J. Frailty Aging 2013, 2, 38. [Google Scholar]
- Holmes, E.; Li, J.V.; Marchesi, J.R.; Nicholson, J.K. Gut microbiota composition and activity in relation to host metabolic phenotype and disease risk. Cell Metab. 2012, 16, 559–564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spratlin, J.L.; Serkova, N.J.; Eckhardt, S.G. Clinical applications of metabolomics in oncology: A review. Clin. Cancer Res. 2009, 15, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Connor, S.C.; Hansen, M.K.; Corner, A.; Smith, R.F.; Ryan, T.E. Integration of metabolomics and transcriptomics data to aid biomarker discovery in type 2 diabetes. Mol. Biosyst. 2010, 6, 909–921. [Google Scholar] [CrossRef]
- Dumas, M.E.; Barton, R.H.; Toye, A.; Cloarec, O.; Blancher, C.; Rothwell, A.; Fearnside, J.; Tatoud, R.; Blanc, V.; Lindon, J.C.; et al. Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin-resistant mice. Proc. Natl. Acad. Sci. USA 2006, 103, 12511–12516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [Green Version]
- Calvani, R.; Brasili, E.; Praticò, G.; Sciubba, F.; Roselli, M.; Finamore, A.; Marini, F.; Marzetti, E.; Miccheli, A. Application of NMR-based metabolomics to the study of gut microbiota in obesity. J. Clin. Gastroenterol. 2014, 48, S5–S7. [Google Scholar] [CrossRef]
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, 1–18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shearer, J.; Duggan, G.; Weljie, A.; Hittel, D.; Wasserman, D.; Vogel, H. Metabolomic profiling of dietary-induced insulin resistance in the high fat–fed C57BL/6J mouse. Diabetes Obes. Metab. 2008, 10, 950–958. [Google Scholar] [CrossRef]
- Rubio-Aliaga, I.; Roos, B.; Sailer, M.; McLoughlin, G.A.; Boekschoten, M.V.; van Erk, M.; Bachmair, E.M.; Van Schothorst, E.M.; Keijer, J.; Coort, S.L.; et al. Alterations in hepatic one-carbon metabolism and related pathways following a high-fat dietary intervention. Physiol. Genom. 2011, 43, 408–416. [Google Scholar] [CrossRef]
- He, Q.; Ren, P.; Kong, X.; Wu, Y.; Wu, G.; Li, P.; Hao, F.; Tang, H.; Blachier, F.; Yin, Y. Comparison of serum metabolite compositions between obese and lean growing pigs using an NMR-based metabonomic approach. J. Nutr. Biochem. 2012, 23, 133–139. [Google Scholar] [CrossRef]
- Liu, Y.; Li, T.R.R.; Xu, C.; Xu, T. Ribose accelerates gut motility and suppresses mouse body weight gaining. Int. J. Biol. Sci. 2016, 12, 701. [Google Scholar] [CrossRef] [Green Version]
- Rao, S.S.C.; Kavelock, R.; Beaty, J.; Ackerson, K.; Stumbo, P. Effects of fat and carbohydrate meals on colonic motor response. Gut 2000, 46, 205–211. [Google Scholar] [CrossRef]
- Sanguinetti, E.; Collado, M.C.; Marrachelli, V.G.; Monleon, D.; Selma-Royo, M.; Pardo-Tendero, M.M.; Burchielli, S.; Iozzo, P. Microbiome-metabolome signatures in mice genetically prone to develop dementia, fed a normal or fatty diet. Sci. Rep. 2018, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Bai, J.; Zhu, Y.; Dong, Y. Modulation of gut microbiota and gut-generated metabolites by bitter melon results in improvement in the metabolic status in high fat diet-induced obese rats. J. Funct. Foods 2018, 41, 127–134. [Google Scholar] [CrossRef]
- Lin, H.; An, Y.; Hao, F.; Wang, Y.; Tang, H. Correlations of fecal metabonomic and microbiomic changes induced by high-fat diet in the pre-obesity state. Sci. Rep. 2016, 6, 1–14. [Google Scholar]
- De Wit, D.; Derrien, M.; Bosch-Vermeulen, H.; Oosterink, E.; Keshtkar, S.; Duval, C.; de Vogel-van den Bosch, J.; Kleerebezem, M.; Müller, M.; van der Meer, R. Saturated fat stimulates obesity and hepatic steatosis and affects gut microbiota composition by an enhanced overflow of dietary fat to the distal intestine. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 303, G589–G599. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Shen, D.; Fang, Z.; Jie, Z.; Qiu, X.; Zhang, C.; Chen, Y.; Ji, L. Human gut microbiota changes reveal the progression of glucose intolerance. PLoS ONE 2013, 8, e71108. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.Y.; Ha, C.W.; Hoffmann, J.M.; Oscarsson, J.; Dinudom, A.; Mather, T.J.; Cook, D.I.; Hunt, N.H.; Caterson, I.D.; Holmes, A.J.; et al. Effects of dietary fat profile on gut permeability and microbiota and their relationships with metabolic changes in mice. Obesity 2015, 23, 1429–1439. [Google Scholar] [CrossRef]
- Schwiertz, A.; Taras, D.; Schäfer, K.; Beijer, S.; Bos, N.A.; Donus, C.; Hardt, P.D. Microbiota and SCFA in lean and overweight healthy subjects. Obesity 2010, 18, 190–195. [Google Scholar] [CrossRef] [PubMed]
- Murphy, E.; Cotter, P.; Healy, S.; Marques, T.M.; O’sullivan, O.; Fouhy, F.; Clarke, S.; O’toole, P.; Quigley, E.M.; Stanton, C.; et al. Composition and energy harvesting capacity of the gut microbiota: Relationship to diet, obesity and time in mouse models. Gut 2010, 59, 1635–1642. [Google Scholar] [CrossRef] [PubMed]
- Guspta, V.K.; Kim, M.; Bakshi, U.; Cunningham, K.Y.; Davis, J.M.; Lazaridis, K.N.; Nelson, H.; Sung, J. A predictive index for health status using species-level gut microbiome profiling. Nat. Commun. 2020, 11, 1–16. [Google Scholar]
- Rondina, M.T.; Weyrich, A.S.; Zimmerman, G.A. Platelets as cellular effectors of inflammation in vascular diseases. Circ. Res. 2013, 112, 1506–1519. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Quintel, A.; Alonso, M.; Campos, J.; Vizcaino, L.; Loidi, L.; Gude, F. Determinants of serum concentrations of lipopolysaccharide-binding protein (LBP) in the adult population: The role of obesity. PLoS ONE 2013, 8, e54600. [Google Scholar] [CrossRef] [PubMed]
- Gregor, M.F.; Hotamisligil, G.S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 2011, 29, 415–445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ignacio, A.; Fernandes, M.; Rodrigues, V.; Groppo, F.; Cardoso, A.; Avila-Campos, M.; Nakano, V. Correlation between body mass index and faecal microbiota from children. Clin. Microbiol. Infect. 2016, 22, 258.e1–258.e8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castaner, O.; Goday, A.; Park, Y.M.; Lee, S.H.; Magkos, F.; Shiow, S.A.T.E.; Schröder, H. The gut microbiome profile in obesity: A systematic review. Int. J. Pediatr. Endocrinol. 2018, 2018, 9. [Google Scholar] [CrossRef]
- Hu, S.; Xu, Y.; Gao, X.; Li, S.; Jiang, W.; Liu, Y.; Su, L.; Yang, H. Long-chain bases from sea cucumber alleviate obesity by modulating gut microbiota. Mar. Drugs 2019, 17, 455. [Google Scholar] [CrossRef] [Green Version]
- Biddle, A.S.; Tomb, J.F.; Fan, Z. Microbiome and blood analyte differences point to community and metabolic signatures in lean and obese horses. Front. Vet. Sci. 2018, 5, 225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Golubeva, A.V.; Crampton, S.; Desbonnet, L.; Edge, D.; O’Sullivan, O.; Lomasney, K.W.; Zhdanov, A.V.; Crispie, F.; Moloney, R.D.; Borre, Y.E.; et al. Prenatal stress-induced alterations in major physiological systems correlate with gut microbiota composition in adulthood. Psychoneuroendocrinology 2015, 60, 58–74. [Google Scholar] [CrossRef]
- Zhao, L.; Zhang, Q.; Ma, W.; Tian, F.; Shen, H.; Zhou, M. A combination of quercetin and resveratrol reduces obesity in high-fat diet-fed rats by modulation of gut microbiota. Food Funct. 2017, 8, 4644–4656. [Google Scholar] [CrossRef]
- He, N.; Wang, S.; Lv, Z.; Zhao, W.; Li, S. Low molecular weight chitosan oligosaccharides (LMW-COSs) prevent obesity-related metabolic abnormalities in association with the modification of gut microbiota in high-fat diet (HFD)-fed mice. Food Funct. 2020, 11, 9947–9959. [Google Scholar] [CrossRef]
- Liu, D.; Wen, B.; Zhu, K.; Luo, Y.; Li, J.; Li, Y.; Lin, H.; Huang, J.; Liu, Z. Antibiotics-induced perturbations in gut microbial diversity influence metabolic phenotypes in a murine model of high-fat diet-induced obesity. Appl. Microbiol. Biotechnol. 2019, 103, 5269–5283. [Google Scholar] [CrossRef]
- Adams, S.H. Emerging perspectives on essential amino acid metabolism in obesity and the insulin-resistant state. Adv. Nutr. 2011, 2, 445–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Felig, P.; Marliss, E.; Cahill, G.F., Jr. Plasma amino acid levels and insulin secretion in obesity. N. Engl. J. Med. 1969, 281, 811–816. [Google Scholar] [CrossRef]
- Liu, R.; Hong, J.; Xu, X.; Feng, Q.; Zhang, D.; Gu, Y.; Shi, J.; Zhao, S.; Liu, W.; Wang, X.; et al. Gut microbiome and serum metabolome alterations in obesity and after weight-loss intervention. Nat. Med. 2017, 23, 859. [Google Scholar] [CrossRef]
- Metges, C.C. Contribution of microbial amino acids to amino acid homeostasis of the host. J. Nutr. 2000, 130, 1857S–1864S. [Google Scholar] [CrossRef]
- Kim, J.Y.; Park, J.Y.; Kim, O.Y.; Ham, B.M.; Kim, H.J.; Kwon, D.Y.; Jang, Y.; Lee, J.H. Metabolic profiling of plasma in overweight/obese and lean men using ultra performance liquid chromatography and Q-TOF mass spectrometry (UPLC−Q-TOF MS). J. Proteome Res. 2010, 9, 4368–4375. [Google Scholar] [CrossRef]
- Newgard, C.B.; An, J.; Bain, J.R.; Muehlbauer, M.J.; Stevens, R.D.; Lien, L.F.; Haqq, A.M.; Shah, S.H.; Arlotto, M.; Slentz, C.A.; et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell. Metab. 2009, 9, 311–326. [Google Scholar] [CrossRef] [Green Version]
- She, P.; Van Horn, C.; Reid, T.; Hutson, S.M.; Cooney, R.N.; Lynch, C.J. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am. J. Physiol. Endocrinol. Metab. 2007, 293, e1552–e1563. [Google Scholar] [CrossRef] [Green Version]
- Duggan, G.E.; Hittel, D.S.; Sensen, C.W.; Weljie, A.M.; Vogel, H.J.; Shearer, J. Metabolomic response to exercise training in lean and diet-induced obese mice. J. Appl. Physiol. 2011, 110, 1311–1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Norvell, A.; McMahon, S.B. Cell biology. Rise of the rival. Science 2010, 327, 964–965. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ning, Z.; Mayne, J.; Yang, Y.; Deeke, S.A.; Walker, K.; Farnsworth, C.L.; Stokes, M.P.; Couture, J.F.; Mack, D.; et al. Widespread protein lysine acetylation in gut microbiome and its alterations in patients with Crohn’s disease. Nat. Commun. 2020, 11, 1–12. [Google Scholar] [CrossRef]
- Ma, Q.; Zhou, X.; Sun, Y.; Hu, L.; Zhu, J.; Shao, C.; Meng, Q.; Shan, A. Threonine, but not lysine and methionine, reduces fat accumulation by regulating lipid metabolism in obese mice. J. Agric. Food Chem. 2020, 68, 4876–4883. [Google Scholar] [CrossRef]
- Liu, Y.; Zheng, J.; Zhang, H.P.; Zhang, X.; Wang, L.; Wood, L.; Wang, G. Obesity-associated metabolic signatures correlate to clinical and inflammatory profiles of asthma: A pilot study. Allergy Asthma. Immunol. Res. 2018, 10, 628. [Google Scholar] [CrossRef]
- Maniscalco, M.; Paris, D.; Melck, D.J.; D’Amato, M.; Zedda, A.; Sofia, M.; Stellato, C.; Motta, A. Coexistence of obesity and asthma determines a distinct respiratory metabolic phenotype. J. Allergy Clin. Immunol. 2017, 139, 1536–1547.e1535. [Google Scholar] [CrossRef] [Green Version]
- Taylor, E.N.; Curhan, G.C. Body size and 24-hour urine composition. Am. J. Kidney Dis. 2006, 48, 905–915. [Google Scholar] [CrossRef]
- Walker, J.B. Metabolic Control of Creatine Biosynthesis: ii. restoration of transamidinase activity following creatine repression. J. Biol. Chem. 1961, 236, 493–498. [Google Scholar] [CrossRef]
- Konishi, F. The relationship of urinary 17-hydroxycorticosteroids to creatinine in obesity. Metabolism 1964, 13, 847–851. [Google Scholar] [CrossRef]
- Kazak, L.; Rahbani, J.F.; Samborska, B.; Lu, G.Z.; Jedrychowski, M.P.; Lajoie, M.; Zhang, S.; Ramsay, L.; Dou, F.Y.; Tenen, D.; et al. Ablation of adipocyte creatine transport impairs thermogenesis and causes diet-induced obesity. Nat. Metab. 2019, 1, 360–370. [Google Scholar] [CrossRef]
- Kazak, L.; Chouchani, E.T.; Lu, G.Z.; Jedrychowski, M.P.; Bare, C.J.; Mina, A.I.; Kumari, M.; Zhang, S.; Vuckovic, I.; Laznik-Bogoslavski, D.; et al. Genetic depletion of adipocyte creatine metabolism inhibits diet-induced thermogenesis and drives obesity. Cell. Metab. 2017, 26, 660–671.e3. [Google Scholar] [CrossRef] [Green Version]
- Parada, A.E.; Needham, D.M.; Fuhrman, J.A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 2016, 18, 1403–1414. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Park, S.E.; Seo, S.H.; Kim, E.J.; Byun, S.; Na, C.S.; Son, H.S. Changes of microbial community and metabolite in kimchi inoculated with different microbial community starters. Food. Chem. 2019, 274, 558–565. [Google Scholar] [CrossRef]
- Noble, W.S. How does multiple testing correction work? Nat. Biotechnol. 2009, 27, 1135–1137. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef] [Green Version]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Chong, J.; Wishart, D.B.; Xia, J. Using MetaboAnalyst 4.0 for comprehensive and integrative metabolomics data analysis. Curr. Protoc. Bioinform. 2019, 68, e86. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jo, J.-K.; Seo, S.-H.; Park, S.-E.; Kim, H.-W.; Kim, E.-J.; Kim, J.-S.; Pyo, J.-Y.; Cho, K.-M.; Kwon, S.-J.; Park, D.-H.; et al. Gut Microbiome and Metabolome Profiles Associated with High-Fat Diet in Mice. Metabolites 2021, 11, 482. https://doi.org/10.3390/metabo11080482
Jo J-K, Seo S-H, Park S-E, Kim H-W, Kim E-J, Kim J-S, Pyo J-Y, Cho K-M, Kwon S-J, Park D-H, et al. Gut Microbiome and Metabolome Profiles Associated with High-Fat Diet in Mice. Metabolites. 2021; 11(8):482. https://doi.org/10.3390/metabo11080482
Chicago/Turabian StyleJo, Jae-Kwon, Seung-Ho Seo, Seong-Eun Park, Hyun-Woo Kim, Eun-Ju Kim, Jeong-Sang Kim, Ju-Yeon Pyo, Kwang-Moon Cho, Sun-Jae Kwon, Dae-Hun Park, and et al. 2021. "Gut Microbiome and Metabolome Profiles Associated with High-Fat Diet in Mice" Metabolites 11, no. 8: 482. https://doi.org/10.3390/metabo11080482
APA StyleJo, J. -K., Seo, S. -H., Park, S. -E., Kim, H. -W., Kim, E. -J., Kim, J. -S., Pyo, J. -Y., Cho, K. -M., Kwon, S. -J., Park, D. -H., & Son, H. -S. (2021). Gut Microbiome and Metabolome Profiles Associated with High-Fat Diet in Mice. Metabolites, 11(8), 482. https://doi.org/10.3390/metabo11080482