
Antibiotic-Induced Dysbiosis of Microbiota Promotes Chicken Lipogenesis by Altering Metabolomics in the Cecum

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Antibiotic Treatment Promotes Lipogenesis of Chicken
2.2. Antibiotic Treatment Alters the Gut Microbiota
2.3. Antibiotic Treatment Alters Cecal Metabolomics
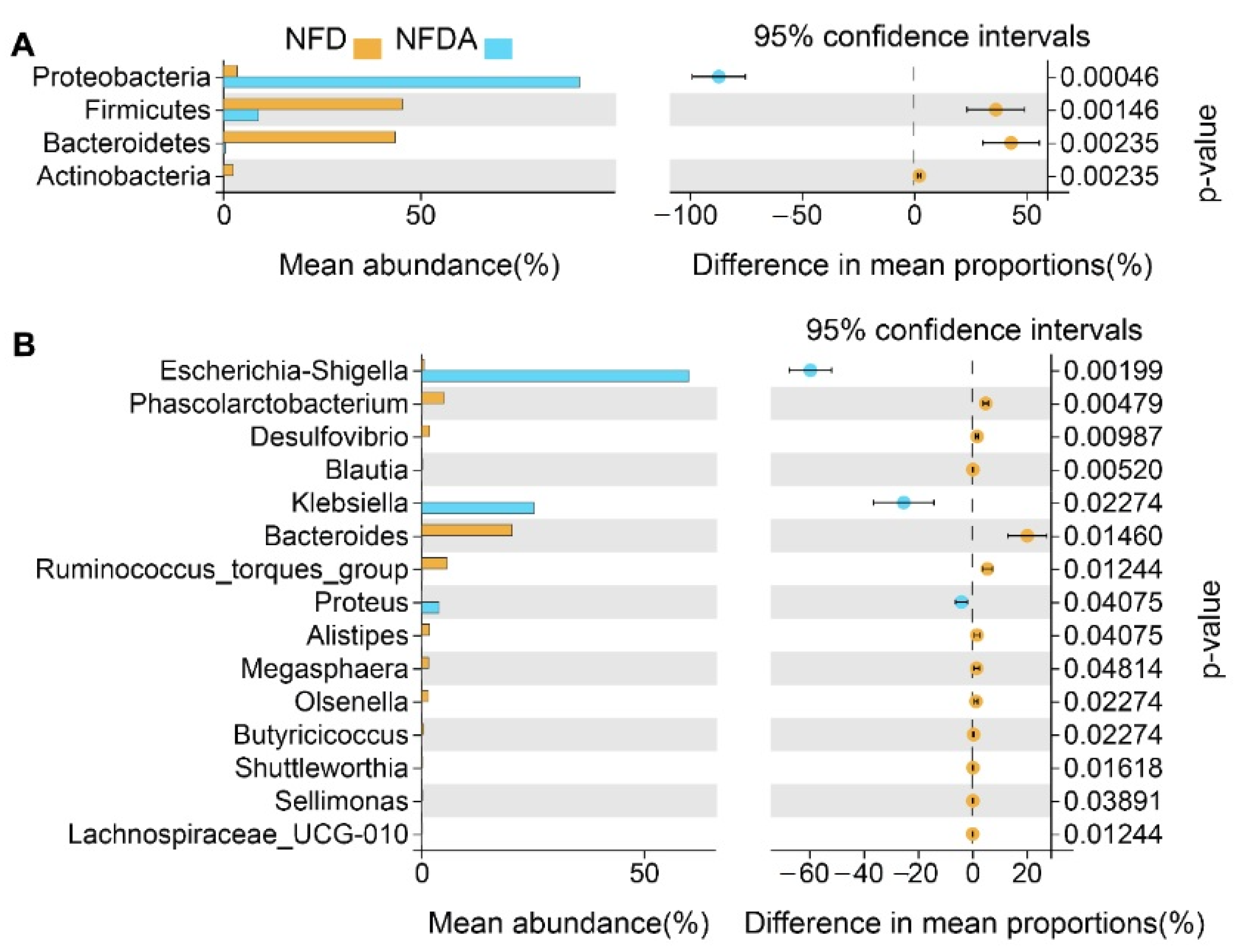
2.4. Identification of Differential Taxa
2.5. Prediction of the Function of Cecal Microbiota
2.6. Identification and Functional Annotation of Differential Metabolites
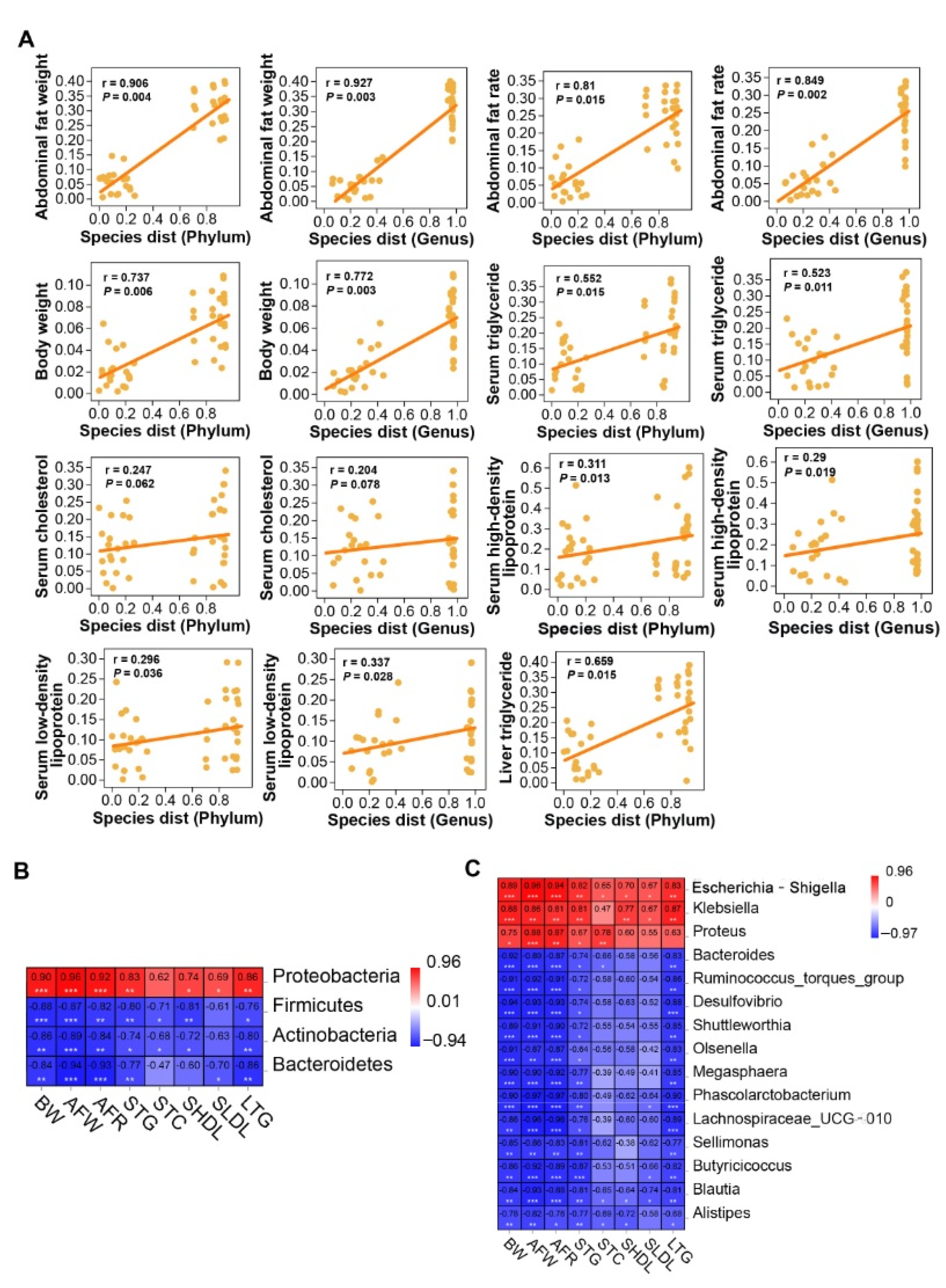
2.7. Cecal Microbiota Correlated to Lipogenesis of Chicken
2.8. Integrated Microbiome and Metabolomics Analysis
2.9. Metabolite Set Enrichment Analysis of Metabolites in the Networks
3. Discussion
4. Materials and Methods
4.1. Animal
4.2. Antibiotic Treatment
4.3. Sample Collection
4.4. Triglyceride Content Detection in Liver
4.5. Measurement of Serum LDL Oxidation, TC, and TG
4.6. 16S rRNA Sequencing
4.7. Metabolite Extraction and UHPLC-MS/MS Analysis
4.8. Metabolomics Data Analysis
4.9. Associations between Gut Microbiota and Lipogenesis
4.10. Integrated Microbiome and Metabolomics Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, Q.; Huang, J.J.; Yang, C.X.; Wang, Y.X.; Zhang, H.; Zhao, J.G.; Hui, L.I. CRISPR/Cas9 Mediated 1 Knockout and Its Impact on Chicken Preadipocytes Differentiation and Proliferation. Acta Vcterinaria Zootechhnica Sin. 2016, 47, 1775–1784. [Google Scholar] [CrossRef]
- Jiang, R. A Comparative Study on Lipid Metabolism and Related Gene Expression in Arbor Acres Broilers and Beijing-you Chickens. Chin. J. Anim. Nutr. 2010, 22, 1334–1441. [Google Scholar] [CrossRef]
- Jennen, D.G.; Vereijken, A.L.; Bovenhuis, H.; Crooijmans, R.P.; Veenendaal, A.; van der Poel, J.J.; Groenen, M.A. Detection and localization of quantitative trait loci affecting fatness in broilers. Poult. Sci. 2004, 83, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Min, L.I.; Dong, X.Y.; Hui, L.I.; Zhi-Qiang, D.U. Identification of RNA Editing Sites in Chicken Lines Divergently Selected for Abdominal Fat Content. Acta Vcterinaria Zootechhnica Sin. 2017, 48, 1611–1623. [Google Scholar] [CrossRef]
- Difford, G.; Lassen, J.; Lvendahl, P. Genes and microbes, the next step in dairy cattle breeding. In Proceedings of the Book of Abstracts of the 67th Annual Meeting European Federation of Animal Science, Belfast, UK, 29 August–2 September 2016. [Google Scholar]
- Wen, C.L.; Sun, C.J.; Yang, N. The concepts and research progress: From heritability to microbiability. Hereditas 2019, 41, 1023–1040. [Google Scholar] [CrossRef]
- Wen, C.; Yan, W.; Sun, C.; Ji, C.; Zhou, Q.; Zhang, D.; Zheng, J.; Yang, N. The gut microbiota is largely independent of host genetics in regulating fat deposition in chickens. ISME J. 2019, 13, 1422–1436. [Google Scholar] [CrossRef]
- Fröhlich, E.E.; Farzi, A.; Mayerhofer, R.; Reichmann, F.; Jačan, A.; Wagner, B.; Zinser, E.; Bordag, N.; Magnes, C.; Fröhlich, E.; et al. Cognitive impairment by antibiotic-induced gut dysbiosis: Analysis of gut microbiota-brain communication. Brain Behav. Immun. 2016, 56, 140–155. [Google Scholar] [CrossRef] [Green Version]
- Aguilera, M.; Cerdà-Cuéllar, M.; Martínez, V. Antibiotic-induced dysbiosis alters host-bacterial interactions and leads to colonic sensory and motor changes in mice. Gut Microbes 2015, 6, 10–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, H.; Gan, J.; Zeng, D.; Li, J.; Yu, H.; Zhao, H.; Yang, Y.; Tan, S.; Li, G.; Luo, C.; et al. Specific Microbial Taxa and Functional Capacity Contribute to Chicken Abdominal Fat Deposition. Front. Microbiol. 2021, 12, 643025. [Google Scholar] [CrossRef]
- Li, D.; Zhang, K.; Pan, Z.; Yu, M.; Lu, Y.; Wang, G.; Wu, J.; Zhang, J.; Zhang, K.; Du, W. Antibiotics promote abdominal fat accumulation in broilers. Anim. Sci. J. 2020, 91, e13326. [Google Scholar] [CrossRef]
- Zhu, Z.; Huang, J.; Li, X.; Xing, J.; Zhang, Z. Gut microbiota regulate tumor metastasis via circRNA/miRNA networks. Gut Microbes 2020, 12, 1–16. [Google Scholar] [CrossRef]
- Hu, X.; Mu, R.; Xu, M.; Yuan, X.; Jiang, P.; Guo, J.; Cao, Y.; Zhang, N.; Fu, Y. Gut microbiota mediate the protective effects on endometritis induced by Staphylococcus aureus in mice. Food Funct. 2020, 11, 3695–3705. [Google Scholar] [CrossRef]
- Ley, R.E.; Bäckhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [Green Version]
- Jeong, M.Y.; Jang, H.M.; Kim, D.H. High-fat diet causes psychiatric disorders in mice by increasing Proteobacteria population. Neurosc. Lett. 2019, 698, 51–57. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, A.; Sandhu, A.K.; Edirisinghe, I.; Burton-Freeman, B.M. Functional Deficits in Gut Microbiome of Young and Middle-Aged Adults with Prediabetes Apparent in Metabolising Bioactive (Poly)phenols. Nutrients 2020, 12, 3595. [Google Scholar] [CrossRef]
- Liu, D.; Huang, J.; Luo, Y.; Wen, B.; Wu, W.; Zeng, H.; Zhonghua, L. Fuzhuan Brick Tea Attenuates High-Fat Diet-Induced Obesity and Associated Metabolic Disorders by Shaping Gut Microbiota. J. Agr. Food Chem. 2019, 67, 13589–13604. [Google Scholar] [CrossRef]
- Garcia-Ribera, S.; Amat-Bou, M.; Climent, E.; Llobet, M.; Chenoll, E.; Corripio, R.; Ibáñez, L.; Ramon-Krauel, M.; Lerin, C. Specific Dietary Components and Gut Microbiota Composition are Associated with Obesity in Children and Adolescents with Prader-Willi Syndrome. Nutrients 2020, 12, 1063. [Google Scholar] [CrossRef] [PubMed]
- Guo, X. Detection of Firmicutes and Bacteroidetes in the Pig Gut and the Correlation between their Abundance and Fat Deposit. Ph.D. Thesis, Sichuan Agricultural University, Sichuan, China, 2009. [Google Scholar]
- Hou, Q.; Kwok, L.Y.; Zheng, Y.; Wang, L.; Guo, Z.; Zhang, J.; Huang, W.; Wang, Y.; Leng, L.; Li, H.; et al. Differential fecal microbiota are retained in broiler chicken lines divergently selected for fatness traits. Sci. Rep. 2016, 6, 37376. [Google Scholar] [CrossRef] [Green Version]
- Iwao, M.; Gotoh, K.; Arakawa, M.; Endo, M.; Honda, K.; Seike, M.; Murakami, K.; Shibata, H. Supplementation of branched-chain amino acids decreases fat accumulation in the liver through intestinal microbiota-mediated production of acetic acid. Sci. Rep. 2020, 10, 18768. [Google Scholar] [CrossRef]
- Hu, C.; Li, F.; Duan, Y.; Yin, Y.; Kong, X. Glutamic acid supplementation reduces body fat weight in finishing pigs when provided solely or in combination with arginine and it is associated with colonic propionate and butyrate concentrations. Food Funct. 2019, 10, 4693–4704. [Google Scholar] [CrossRef]
- Hu, C.J.; Jiang, Q.Y.; Zhang, T.; Yin, Y.L.; Li, F.N.; Deng, J.P.; Wu, G.Y.; Kong, X.F. Dietary supplementation with arginine and glutamic acid modifies growth performance, carcass traits, and meat quality in growing-finishing pigs. J. Anim. Sci. 2017, 95, 2680–2689. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Kong, X.; Hu, C.; Zhou, B.; Wang, C.; Shen, Q.W. Fatty Acid Content, Flavor Compounds, and Sensory Quality of Pork Loin as Affected by Dietary Supplementation with l-arginine and Glutamic Acid. J. Food Sci. 2019, 84, 3445–3453. [Google Scholar] [CrossRef]
- Lanska, D.J. The discovery of niacin, biotin, and pantothenic acid. Ann. Nutr. Metab. 2012, 61, 246–253. [Google Scholar] [CrossRef]
- Wang, B.; Zhang, X.; Yue, B.; Ge, W.; Zhang, M.; Ma, C.; Kong, M. Effects of pantothenic acid on growth performance, slaughter performance, lipid metabolism, and antioxidant function of Wulong geese aged one to four weeks. Anim. Nutr. 2016, 2, 312–317. [Google Scholar] [CrossRef]
- Beagle, W.S.; Begin, J.J. The effect of pantothenic acid on the diet of growing chicks on energy utilisation and body composition. Poult. Sci. 1976, 55, 950–957. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.; Zhang, S.; Li, H.; Yang, F.; Mushtaq, N.; Ullah, S.; Shi, Y.; An, C.; Xu, J. The influence of gut microbiota dysbiosis to the efficacy of 5-Fluorouracil treatment on colorectal cancer. Biomed. Pharmacother. 2018, 108, 184–193. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Pruesse, E.; Quast, C.; Knittel, K.; Fuchs, B.M.; Ludwig, W.; Peplies, J.; Glöckner, F.O. SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 2007, 35, 7188–7196. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2--approximately maximum-likelihood trees for large alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Lozupone, C.; Knight, R. UniFrac: A new phylogenetic method for comparing microbial communities. Appl. Environ. Microb. 2005, 71, 8228–8235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asshauer, K.P.; Wemheuer, B.; Daniel, R.; Meinicke, P. Tax4Fun: Predicting functional profiles from metagenomic 16S rRNA data. Bioinformatics 2015, 31, 2882–2884. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem. 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Zhao, X.; Zeng, Z.; Chen, A.; Lu, X.; Zhao, C.; Hu, C.; Zhou, L.; Liu, X.; Wang, X.; Hou, X.; et al. Comprehensive strategy to construct in-house database for accurate and batch identification of Small molecular metabolites. Anal. Chem. 2018, 90, 7635–7643. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Chong, J.; Zhou, G.; de Lima Morais, D.A.; Chang, L.; Barrette, M.; Gauthier, C.; Jacques, P.É.; Li, S.; Xia, J. MetaboAnalyst 5.0: Narrowing the gap between raw spectra and functional insights. Nucleic Acids Res. 2021, 49, W388–W396. [Google Scholar] [CrossRef] [PubMed]
- Bastian, M.; Heymann, S.; Jacomy, M. Gephi: An open source software for exploring and manipulating networks. Icwsm 2009, 8, 361–362. [Google Scholar] [CrossRef]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, T.; Ding, H.; Chen, L.; Lin, Y.; Gong, Y.; Pan, Z.; Zhang, G.; Xie, K.; Dai, G.; Wang, J. Antibiotic-Induced Dysbiosis of Microbiota Promotes Chicken Lipogenesis by Altering Metabolomics in the Cecum. Metabolites 2021, 11, 487. https://doi.org/10.3390/metabo11080487
Zhang T, Ding H, Chen L, Lin Y, Gong Y, Pan Z, Zhang G, Xie K, Dai G, Wang J. Antibiotic-Induced Dysbiosis of Microbiota Promotes Chicken Lipogenesis by Altering Metabolomics in the Cecum. Metabolites. 2021; 11(8):487. https://doi.org/10.3390/metabo11080487
Chicago/Turabian StyleZhang, Tao, Hao Ding, Lan Chen, Yueyue Lin, Yongshuang Gong, Zhiming Pan, Genxi Zhang, Kaizhou Xie, Guojun Dai, and Jinyu Wang. 2021. "Antibiotic-Induced Dysbiosis of Microbiota Promotes Chicken Lipogenesis by Altering Metabolomics in the Cecum" Metabolites 11, no. 8: 487. https://doi.org/10.3390/metabo11080487
APA StyleZhang, T., Ding, H., Chen, L., Lin, Y., Gong, Y., Pan, Z., Zhang, G., Xie, K., Dai, G., & Wang, J. (2021). Antibiotic-Induced Dysbiosis of Microbiota Promotes Chicken Lipogenesis by Altering Metabolomics in the Cecum. Metabolites, 11(8), 487. https://doi.org/10.3390/metabo11080487