
Metabolic Outcomes of Anaplerotic Dodecanedioic Acid Supplementation in Very Long Chain Acyl-CoA Dehydrogenase (VLCAD) Deficient Fibroblasts


Abstract
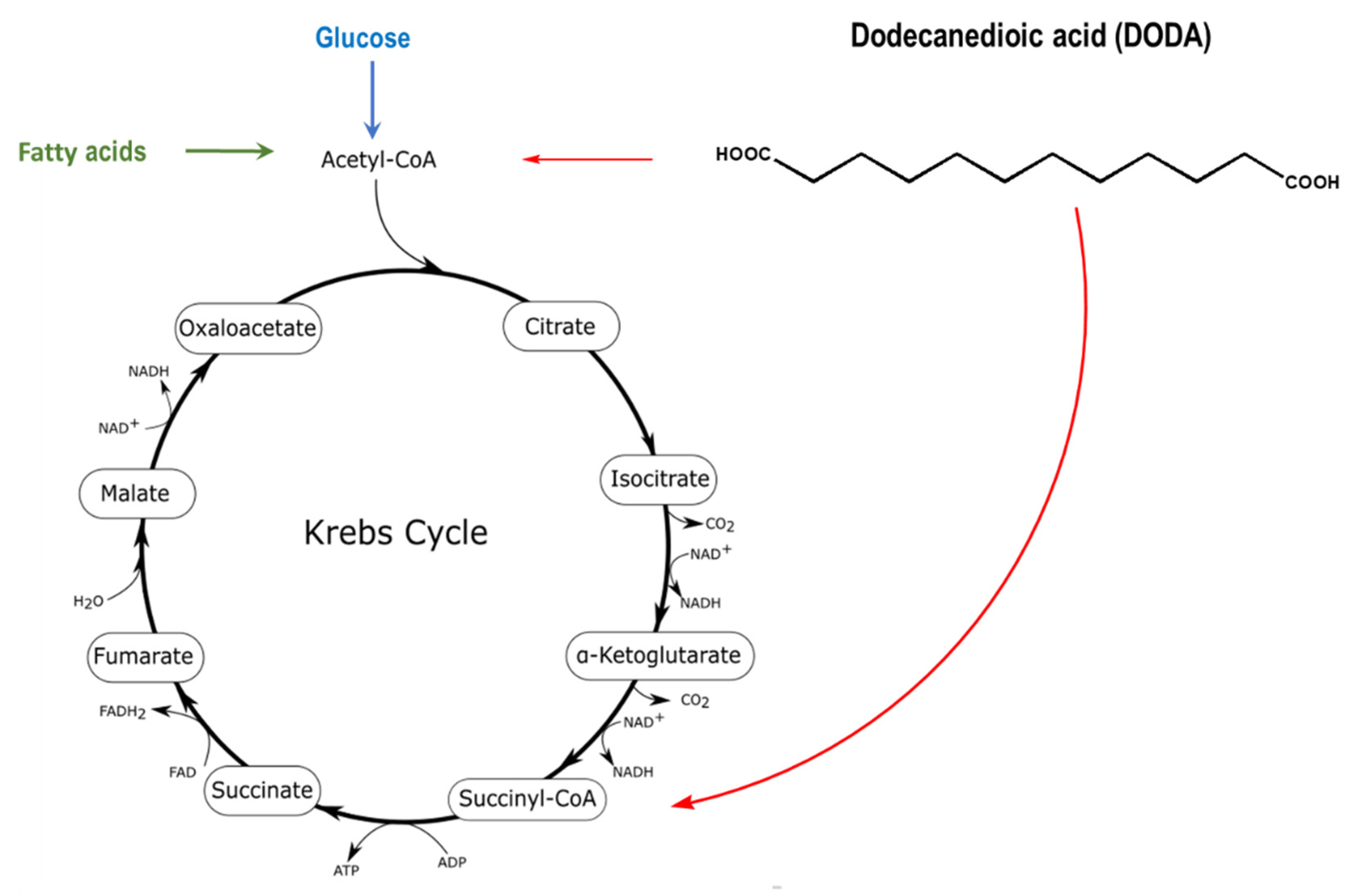
:1. Introduction
2. Results
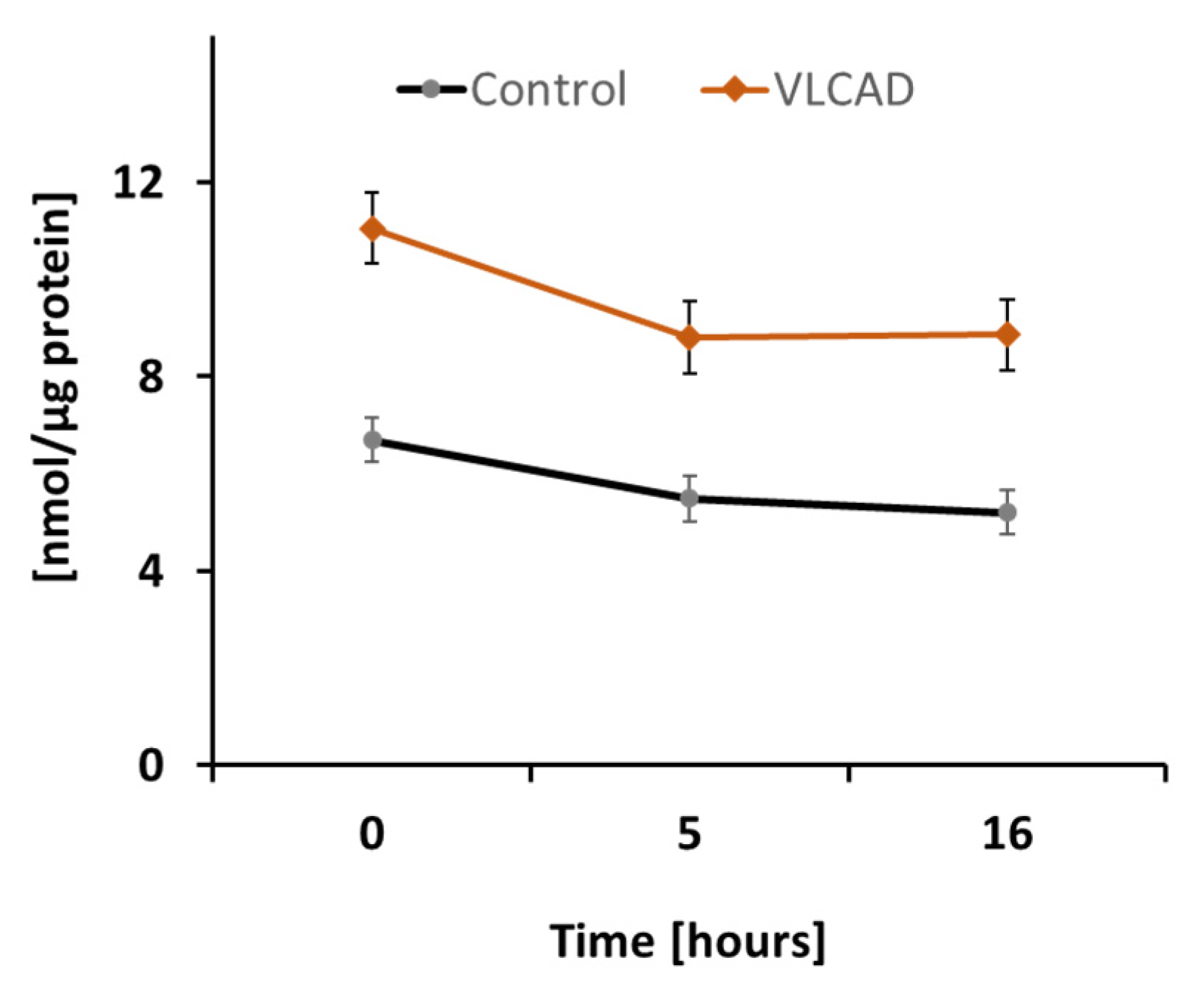
2.1. DODA Cellular Uptake
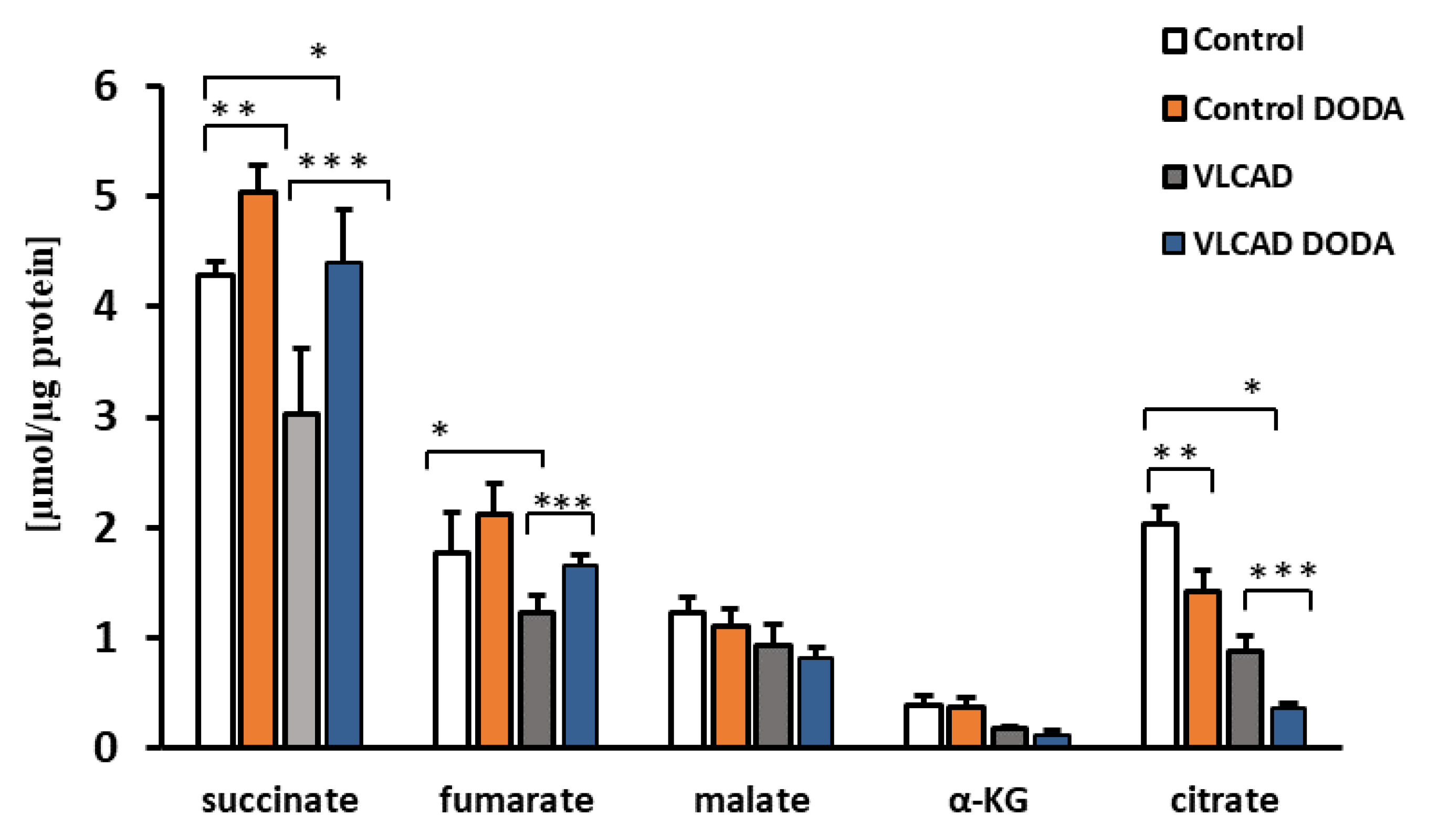
2.2. Krebs Cycle Intermediate
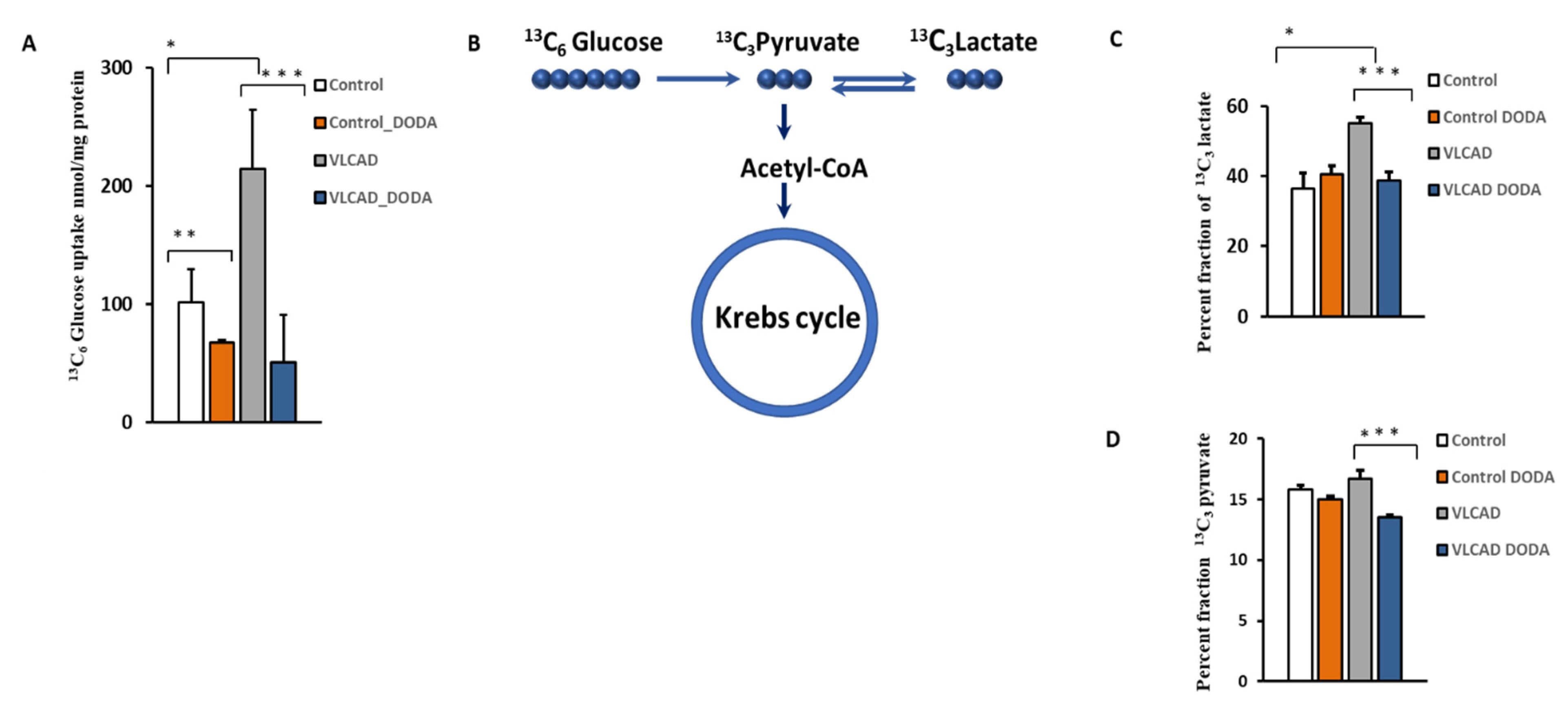
2.3. Glucose Uptake and Glycolytic Flux
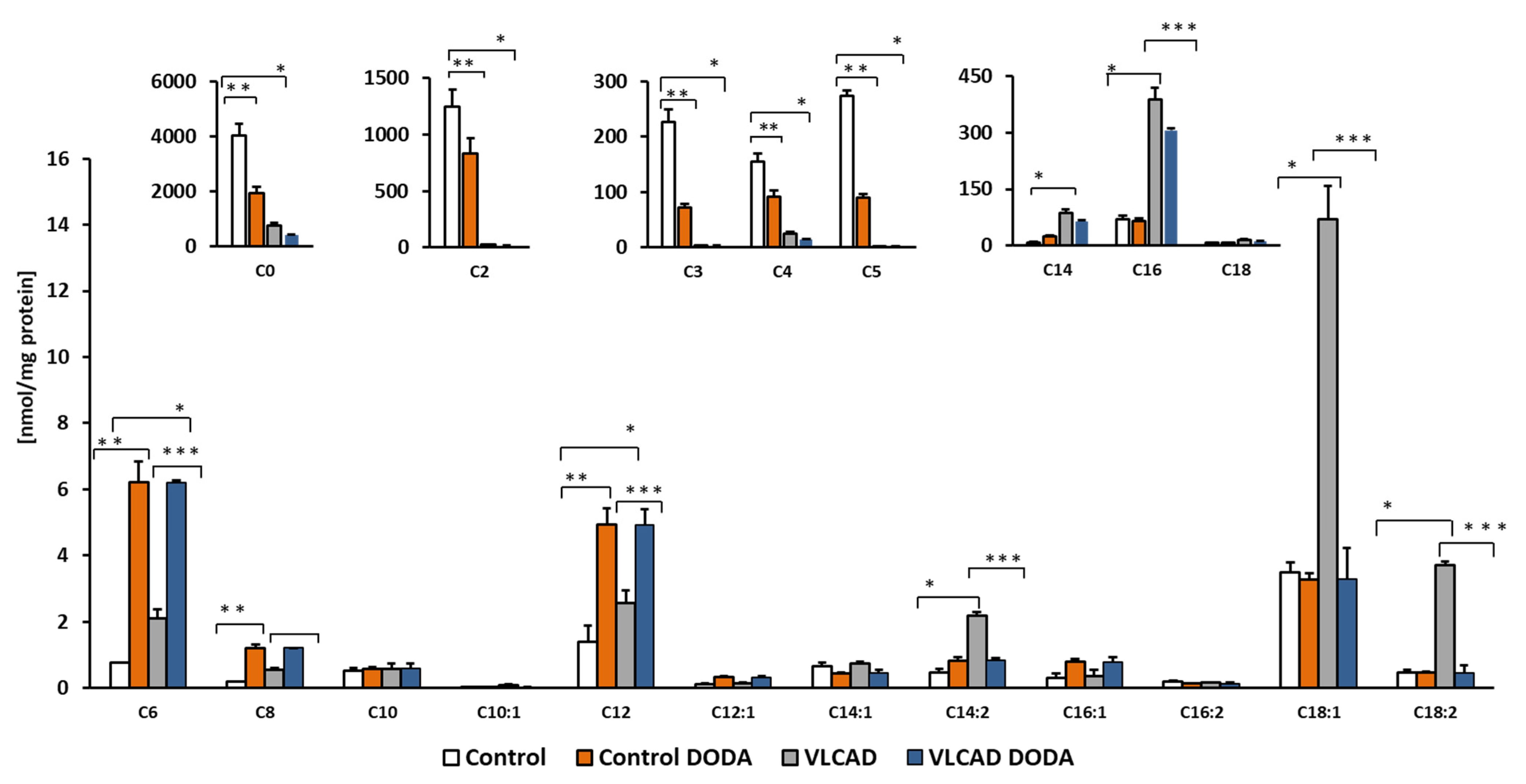
2.4. Acylcarnitines Profiles
3. Discussion
4. Materials and Methods
4.1. Cells
4.2. Dodecanedioic Acid Uptake Assay
4.3. Glucose Uptake Assay
4.4. Krebs Cycle Intermediates, Cellular Lactate, and Pyruvate
4.5. Acylcarnitine Analysis
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| DODA | Dodecanedioic acid |
| DA | Dicarboxylic acid |
| α-KG | α-Ketoglutarate |
| VLCFA | Very long-chain fatty acids |
| VLCADD | Very long-chain acyl-CoA dehydrogenase deficiency |
| LC-FAOD | Long-chain fatty acid oxidations disorders |
References
- Schiff, M.; Mohsen, A.-W.; Karunanidhi, A.; McCracken, E.; Yeasted, R.; Vockley, J. Molecular and cellular pathology of very-long-chain acyl-CoA dehydrogenase deficiency. Mol. Genet. Metab. 2013, 109, 21–27. [Google Scholar] [CrossRef]
- Pena, L.D.M.; van Calcar, S.C.; Hansen, J.; Edick, M.J.; Walsh Vockley, C.; Leslie, N.; Cameron, C.; Mohsen, A.W.; Berry, S.A.; Arnold, G.L.; et al. Outcomes and genotype-phenotype correlations in 52 individuals with VLCAD deficiency diagnosed by NBS and enrolled in the IBEM-IS database. Mol. Genet. Metab. 2016, 118, 272–281. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Saito, M.; Matsuoka, H.; Inagaki, N. A real-time method of imaging glucose uptake in single, living mammalian cells. Nat. Protoc. 2007, 2, 753–762. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J. Long-chain fatty acid oxidation disorders and current management strategies. Am. J. Manag. Care 2020, 26, S147–S154. [Google Scholar]
- Behrend, A.M.; Harding, C.O.; Shoemaker, J.D.; Matern, D.; Sahn, D.J.; Elliot, D.L.; Gillingham, M.B. Substrate oxidation and cardiac performance during exercise in disorders of long chain fatty acid oxidation. Mol. Genet. Metab. 2012, 105, 110–115. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Burton, B.; Berry, G.T.; Longo, N.; Phillips, J.; Sanchez-Valle, A.; Tanpaiboon, P.; Grunewald, S.; Murphy, E.; Humphrey, R.; et al. UX007 for the treatment of long chain-fatty acid oxidation disorders: Safety and efficacy in children and adults following 24 weeks of treatment. Mol. Genet. Metab. 2017, 120, 370–377. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Palmfeldt, J.; Gregersen, N.; Makhov, A.M.; Conway, J.F.; Wang, M.; McCalley, S.P.; Basu, S.; Alharbi, H.; St Croix, C.; et al. Mitochondrial fatty acid oxidation and the electron transport chain comprise a multifunctional mitochondrial protein complex running title: Structural architecture of mitochondrial energy metabolism. J. Biol. Chem. 2019, 294, 12380–12391. [Google Scholar] [CrossRef] [PubMed]
- Cecatto, C.; Amaral, A.U.; da Silva, J.C.; Wajner, A.; Schimit, M.D.O.V.; da Silva, L.H.R.; Wajner, S.M.; Zanatta, Â.; Castilho, R.F.; Wajner, M. Metabolite accumulation in VLCAD deficiency markedly disrupts mitochondrial bioenergetics and Ca2+ homeostasis in the heart. FEBS J. 2018, 285, 1437–1455. [Google Scholar] [CrossRef]
- Seminotti, B.; Leipnitz, G.; Karunanidhi, A.; Kochersperger, C.; Roginskaya, V.Y.; Basu, S.; Wang, Y.; Wipf, P.; Van Houten, B.; Mohsen, A.W.; et al. Mitochondrial energetics is impaired in very long-chain acyl-CoA dehydrogenase deficiency and can be rescued by treatment with mitochondria-targeted electron scavengers. Hum. Mol. Genet. 2019, 28, 928–941. [Google Scholar] [CrossRef]
- Diekman, E.F.; Visser, G.; Schmitz, J.P.J.; Nievelstein, R.A.J.; de Sain-van der Velden, M.; Wardrop, M.; Van der Pol, W.L.; Houten, S.M.; van Riel, N.A.W.; Takken, T.; et al. Altered Energetics of Exercise Explain Risk of Rhabdomyolysis in very long-chain acyl-CoA dehydrogenase deficiency. PLoS ONE 2016, 11, e0147818. [Google Scholar] [CrossRef]
- Tucci, S.; Flögel, U.; Hermann, S.; Sturm, M.; Schäfers, M.; Spiekerkoetter, U. Development and pathomechanisms of cardiomyopathy in very long-chain acyl-CoA dehydrogenase deficient (VLCAD−/−) mice. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 677–685. [Google Scholar] [CrossRef]
- Roe, C.R.; Sweetman, L.; Roe, D.S.; David, F.; Brunengraber, H. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J. Clin. Investig. 2002, 110, 259–269. [Google Scholar] [CrossRef]
- Roe, C.R.; Mochel, F. Anaplerotic diet therapy in inherited metabolic disease: Therapeutic potential. J. Inherit. Metab. Dis. 2006, 29, 332–340. [Google Scholar] [CrossRef]
- Brunengraber, H.; Roe, C.R. Anaplerotic molecules: Current and future. J. Inherit. Metab. Dis. 2006, 29, 327–331. [Google Scholar] [CrossRef]
- Roe, C.R.; Brunengraber, H. Anaplerotic treatment of long-chain fat oxidation disorders with triheptanoin: Review of 15 years experience. Mol. Genet. Metab. 2015, 116, 260–268. [Google Scholar] [CrossRef]
- Vockley, J.; Charrow, J.; Ganesh, J.; Eswara, M.; Diaz, G.A.; McCracken, E.; Conway, R.; Enns, G.M.; Starr, J.; Wang, R.; et al. Triheptanoin treatment in patients with pediatric cardiomyopathy associated with long chain-fatty acid oxidation disorders. Mol. Genet. Metab. 2016, 119, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Gillingham, M.B.; Heitner, S.B.; Martin, J.; Rose, S.; Goldstein, A.; El-Gharbawy, A.H.; Deward, S.; Lasarev, M.R.; Pollaro, J.; DeLany, J.P.; et al. Triheptanoin versus trioctanoin for long-chain fatty acid oxidation disorders: A double blinded, randomized controlled trial. J. Inherit. Metab. Dis. 2017, 40, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Sklirou, E.; Alodaib, A.N.; Dobrowolski, S.F.; Mohsen, A.-W.A.; Vockley, J. Physiological perspectives on the use of triheptanoin as anaplerotic therapy for long chain fatty acid oxidation disorders. Front. Genet. 2021, 11, 1–14. [Google Scholar] [CrossRef]
- Kinman, R.P.; Kasumov, T.; Jobbins, K.A.; Thomas, K.R.; Adams, J.E.; Brunengraber, L.N.; Kutz, G.; Brewer, W.U.; Roe, C.R.; Brunengraber, H. Parenteral and enteral metabolism of anaplerotic triheptanoin in normal rats. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E860–E866. [Google Scholar] [CrossRef] [PubMed]
- Bleeker, J.C.; Visser, G.; Clarke, K.; Ferdinandusse, S.; Haan, F.H.; Houtkooper, R.H.; IJlst, L.; Kok, I.L.; Langeveld, M.; Pol, W.L.; et al. Nutritional ketosis improves exercise metabolism in patients with very long-chain acyl-CoA dehydrogenase deficiency. J. Inherit. Metab. Dis. 2020, 43, 787–799. [Google Scholar] [CrossRef] [PubMed]
- Mingrone, G.; Castagneto, M. Medium-chain, even-numbered dicarboxylic acids as novel energy substrates: An update. Nutr. Rev. 2006, 64, 449–456. [Google Scholar] [CrossRef]
- Grego, A.V.; Mingrone, G. Dicarboxylic acids, an alternate fuel substrate in parenteral nutrition: An update. Clin. Nutr. 1995, 14, 143–148. [Google Scholar] [CrossRef]
- Mingrone, G.; De Gaetano, A.; Greco, A.V.; Capristo, E.; Benedetti, G.; Castagneto, M.; Gasbarrini, G. Dodecanedioic acid infusion induces a sparing effect on whole-body glucose uptake, mainly in non-insulin-dependent diabetes mellitus. Br. J. Nutr. 1997, 78, 723–735. [Google Scholar] [CrossRef]
- Vickers, A.E.M. Characterization of hepatic mitochondrial injury induced by fatty acid oxidation inhibitors. Toxicol. Pathol. 2009, 37, 78–88. [Google Scholar] [CrossRef] [PubMed]
- Wanders, R.J.A.; Komen, J.; Kemp, S. Fatty acid omega-oxidation as a rescue pathway for fatty acid oxidation disorders in humans. FEBS J. 2011, 278, 182–194. [Google Scholar] [CrossRef] [PubMed]
- Wood, J.C.; Magera, M.J.; Rinaldo, P.; Reed Seashore, M.; Strauss, A.W.; Friedman, A. Diagnosis of very long chain acyl-dehydrogenase deficiency from an infant’s newborn screening card. Pediatrics 2001, 108, e19. [Google Scholar] [CrossRef] [PubMed]
- Kølvraa, S.; Gregersen, N. In vitro studies on the oxidation of medium-chain dicarboxylic acids in rat liver. Biochim. Biophys. Acta (BBA) Lipids Lipid Metab. 1986, 876, 515–525. [Google Scholar] [CrossRef]
- Vamecq, J.; Draye, J.P.; Brison, J. Rat liver metabolism of dicarboxylic acids. Am. J. Physiol. 1989, 256, G680–G688. [Google Scholar] [CrossRef] [PubMed]
- Mingrone, G.; Greco, A.V.; Tataranni, A.; Raguso, C.; de Gaetano, A.; Castagneto, M. Pharmacokinetic profile of dodecanedioic acid, a proposed alternative fuel substrate. J. Parenter. Enter. Nutr. 1994, 18, 225–230. [Google Scholar] [CrossRef]
- Bharathi, S.S.; Zhang, Y.; Gong, Z.; Muzumdar, R.; Goetzman, E.S. Role of mitochondrial acyl-CoA dehydrogenases in the metabolism of dicarboxylic fatty acids. Biochem. Biophys. Res. Commun. 2020, 527, 162–166. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Bian, F.; Tomcik, K.; Kelleher, J.K.; Zhang, G.-F.; Brunengraber, H. Compartmentation of metabolism of the C12-, C9-, and C5-n-dicarboxylates in rat liver, investigated by mass isotopomer analysis. J. Biol. Chem. 2015, 290, 18671–18677. [Google Scholar] [CrossRef] [PubMed]
- Mingrone, G.; De Gaetano, A.; Greco, A.V.; Benedetti, G.; Capristo, E.; Castagneto, M.; Gasbarrini, G. Plasma clearance and oxidation of dodecanedioic acid in humans. J. Parenter. Enter. Nutr. 1996, 20, 38–42. [Google Scholar] [CrossRef] [PubMed]
- Bertuzzi, A.; Mingrone, G.; Gandolfi, A.; Greco, A.V.; Salinari, S. Disposition of dodecanedioic acid in humans. J. Pharmacol. Exp. Ther. 2000, 292, 846–852. [Google Scholar]
- Bertuzzi, A.; Mingrone, G.; Gandolfi, A.; Greco, A.V.; Salinari, S. Pharmacokinetic analysis of dodecanedioic acid in humans from bolus data. J. Parenter. Enter. Nutr. 1995, 19, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Marsden, D.; McCracken, E.; DeWard, S.; Barone, A.; Hsu, K.; Kakkis, E. Long-term major clinical outcomes in patients with long chain fatty acid oxidation disorders before and after transition to triheptanoin treatment—A retrospective chart review. Mol. Genet. Metab. 2015, 116, 53–60. [Google Scholar] [CrossRef]
- Mingrone, G.; Castagneto-Gissey, L.; Macé, K. Use of dicarboxylic acids in type 2 diabetes. Br. J. Clin. Pharmacol. 2013, 75, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Tserngg, K.-Y.; Jin, S.-J. Metabolic conversion of dicarboxylic acids to succinate in rat liver homogenates. A stable isotope tracer study. J. Biol. Chem. 1991, 266, 2924–2929. [Google Scholar] [CrossRef]
- Roe, C.R.; Yang, B.-Z.; Brunengraber, H.; Roe, D.S.; Wallace, M.; Garritson, B.K. Carnitine palmitoyltransferase II deficiency: Successful anaplerotic diet therapy. Neurology 2008, 71, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Longo, N.; Price, L.B.; Gappmaier, E.; Cantor, N.L.; Ernst, S.L.; Bailey, C.; Pasquali, M. Anaplerotic therapy in propionic acidemia. Mol. Genet. Metab. 2017, 122, 51–59. [Google Scholar] [CrossRef]
- Gaston, G.; Gangoiti, J.A.; Winn, S.; Chan, B.; Barshop, B.A.; Harding, C.O.; Gillingham, M.B. Cardiac tissue citric acid cycle intermediates in exercised very long-chain acyl-CoA dehydrogenase-deficient mice fed triheptanoin or medium-chain triglyceride. J. Inherit. Metab. Dis. 2020, 43, 1232–1242. [Google Scholar] [CrossRef]
- Smith, C.M.; Williamson, J.R. Inhibition of citrate synthase by succinyl-CoA and other metabolites. FEBS Lett. 1971, 18, 35–38. [Google Scholar] [CrossRef]
- Tucci, S.; Floegel, U.; Beermann, F.; Behringer, S.; Spiekerkoetter, U. Triheptanoin: Long-term effects in the very long-chain acyl-CoA dehydrogenase-deficient mouse. J. Lipid Res. 2017, 58, 196–207. [Google Scholar] [CrossRef]
- Houten, S.M.; Herrema, H.; Te Brinke, H.; Denis, S.; Ruiter, J.P.N.; Van Dijk, T.H.; Argmann, C.A.; Ottenhoff, R.; Mü Ller, M.; Groen, A.K.; et al. Impaired amino acid metabolism contributes to fasting-induced hypoglycemia in fatty acid oxidation defects. Hum. Mol. Genet. 2013, 22, 5249–5261. [Google Scholar] [CrossRef]
- Ventura, F.V.; Ruiter, J.P.N.; IJlst, L.; Tavares De Almeida, I.; Wanders, R.J.A. Lactic acidosis in long-chain fatty acid β-oxidation disorders. J. Inherit. Metab. Dis. 1998, 21, 645–654. [Google Scholar] [CrossRef]
- Yamada, K.A.; McHowat, J.; Yan, G.X.; Donahue, K.; Peirick, J.; Kleber, A.G.; Corr, P.B. Cellular uncoupling induced by accumulation of long-chain acylcarnitine during ischemia. Circ. Res. 1994, 74, 83–95. [Google Scholar] [CrossRef]
- Rutkowsky, J.M.; Knotts, T.A.; Ono-Moore, K.D.; McCoin, C.S.; Huang, S.; Schneider, D.; Singh, S.; Adams, S.H.; Hwang, D.H. Acylcarnitines activate proinflammatory signaling pathways. Am. J. Physiol. Endocrinol. Metab. 2014, 306, E1378–E1387. [Google Scholar] [CrossRef] [PubMed]
- Knabb, M.T.; Saffitz, J.E.; Corr, P.B.; Sobel, B.E. The dependence of electrophysiological derangements on accumulation of endogenous long-chain acyl carnitine in hypoxic neonatal rat myocytes. Circ. Res. 1986, 58, 230–240. [Google Scholar] [CrossRef] [PubMed]
- Dahash, B.A.; Sankararaman, S. Carnitine Deficiency; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Longo, N.; Frigeni, M.; Pasquali, M. Carnitine transport and fatty acid oxidation. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 2422–2435. [Google Scholar] [CrossRef]
- Mingrone, G.; De Gaetano, A.; Greco, A.V.; Capristo, E.; Benedetti, G.; Castagneto, M.; Gasbarrini, G. Comparison between dodecanedioic acid and long-chain triglycerides as an energy source in liquid formula diets. J. Parenter. Enter. Nutr. 1999, 23, 80–84. [Google Scholar] [CrossRef] [PubMed]
- McCann, M.R.; De la Rosa, M.V.G.; Rosania, G.R.; Stringer, K.A. L-carnitine and acylcarnitines: Mitochondrial biomarkers for precision medicine. Metabolites 2021, 11, 51. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, H.; Yamada, J.; Watanabe, T.; Suga, T. Compartmentation of dicarboxylic acid β-oxidation in rat liver: Importance of peroxisomes in the metabolism of dicarboxylic acids. Biochim. Biophys. Acta Gen. Subj. 1989, 990, 25–30. [Google Scholar] [CrossRef]
- Ferdinandusse, S.; Denis, S.; Van Roermund, C.W.T.; Wanders, R.J.A.; Dacremont, G. Identification of the peroxisomal beta-oxidation enzymes involved in the degradation of long-chain dicarboxylic acids. J. Lipid Res. 2004, 45, 1104–1111. [Google Scholar] [CrossRef] [PubMed]
- Westin, M.A.K.; Hunt, M.C.; Alexson, S.E.H. The identification of a succinyl-CoA thioesterase suggests a novel pathway for succinate production in peroxisomes. J. Biol. Chem. 2005, 280, 38125–38132. [Google Scholar] [CrossRef]
- Hardwick, J.P. Cytochrome P450 omega hydroxylase (CYP4) function in fatty acid metabolism and metabolic diseases. Biochem. Pharmacol. 2008, 75, 2263–2275. [Google Scholar] [CrossRef] [PubMed]
- Salinari, S.; Bertuzzi, A.; Gandolfi, A.; Greco, A.V.; Scarfone, A.; Manco, M.; Mingrone, G. Dodecanedioic acid overcomes metabolic inflexibility in type 2 diabetic subjects. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E1051–E1058. [Google Scholar] [CrossRef]
- House, A.; Fatica, E.; Shah, R.; Stergar, J.; Pearce, R.; Sandlers, Y. A protocol for metabolic characterization of human induced pluripotent derived cardiomyocytes (iPSCM). MethodsX 2019, 7, 100572. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Triheptanoin | DODA |
|---|---|
|
| n = 5 | Total Long Chain Acyl Carnitines (nmol/mg Protein) |
|---|---|
| Control | 91 |
| VLCAD | 511 |
| Control + DODA | 103 |
| VLCAD + DODA | 397 |
| Cell Type | 13C3 Lactate/13C3 Pyruvate |
|---|---|
| Control cells | 2.31 |
| VLCADD cells | 3.31 |
| Control cells/1 mM DODA | 2.70 |
| VLCADD cells/1 mM DODA | 2.86 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Radzikh, I.; Fatica, E.; Kodger, J.; Shah, R.; Pearce, R.; Sandlers, Y.I. Metabolic Outcomes of Anaplerotic Dodecanedioic Acid Supplementation in Very Long Chain Acyl-CoA Dehydrogenase (VLCAD) Deficient Fibroblasts. Metabolites 2021, 11, 538. https://doi.org/10.3390/metabo11080538
Radzikh I, Fatica E, Kodger J, Shah R, Pearce R, Sandlers YI. Metabolic Outcomes of Anaplerotic Dodecanedioic Acid Supplementation in Very Long Chain Acyl-CoA Dehydrogenase (VLCAD) Deficient Fibroblasts. Metabolites. 2021; 11(8):538. https://doi.org/10.3390/metabo11080538
Chicago/Turabian StyleRadzikh, Igor, Erica Fatica, Jillian Kodger, Rohan Shah, Ryan Pearce, and Yana I. Sandlers. 2021. "Metabolic Outcomes of Anaplerotic Dodecanedioic Acid Supplementation in Very Long Chain Acyl-CoA Dehydrogenase (VLCAD) Deficient Fibroblasts" Metabolites 11, no. 8: 538. https://doi.org/10.3390/metabo11080538
APA StyleRadzikh, I., Fatica, E., Kodger, J., Shah, R., Pearce, R., & Sandlers, Y. I. (2021). Metabolic Outcomes of Anaplerotic Dodecanedioic Acid Supplementation in Very Long Chain Acyl-CoA Dehydrogenase (VLCAD) Deficient Fibroblasts. Metabolites, 11(8), 538. https://doi.org/10.3390/metabo11080538