1. Introduction
3-hydroxy-3-methyl-glutaryl-CoA reductase (HMG-CoA reductase,
HMGCR) is a rate-limiting enzyme involved in the mevalonate pathway responsible for the synthesis of cholesterol, which is a vital precursor of different biologically active molecules, such as steroid hormones and lipoproteins [
1]. Statins are lipid-lowering drugs, which potently inhibit the activity of HMG-CoA reductase and effectively reduce the plasma concentration of LDL cholesterol. Since statins reduce the risk of cardiovascular events and overall mortality in the patients [
2], they are one of the most widely prescribed drugs in the world. For instance, in the USA, the number of adults taking statins increased from 21.8 million in 2002–2003 to 39.2 million in 2012–2013 [
3]. Apart from reducing plasma cholesterol levels, statins inhibit the synthesis of other components of the mevalonate pathway, such as major isoprenoids, farnesyl and geranylgeranyl pyrophosphate [
1,
4]. Aberrant synthesis of these compounds contributes to the development of different disorders, including cancer, cerebrovascular diseases, and Alzheimer’s disease [
4]. Statins’ anti-neoplastic, anti-inflammatory, and cardioprotective functions make them a potential therapeutic agent for these conditions [
5]. Despite being generally safe and well-tolerated, statin therapy is associated with an increased risk of new-onset type 2 diabetes (T2D) [
6]. Several pre-existing risk factors can stimulate the diabetogenic potential of the therapy, including therapy intensity, some genetic predisposition, sex, and age of the patients [
6,
7,
8,
9,
10]. Statins with high or moderate potency, such as atorvastatin and simvastatin, are reported to have a higher propensity to induce new-onset diabetes in patients compared to low potency statins, e.g., fluvastatin [
9]. Comparison between statins indicated that patients taking atorvastatin or simvastatin have increased risk of new-onset diabetes [
9], reduced insulin secretion, and insulin sensitivity [
11]. On the other hand, pravastatin is considered protective against diabetes development [
9,
12,
13]. This could be due to the lipophilicity of the drugs, as lipophilic statins (e.g., atorvastatin and simvastatin) can pass through the cell membrane of extrahepatic tissues, such as the pancreas, unlike hydrophilic statins (pravastatin) [
14], which could contribute to their diabetogenic effect.
Additionally, the rs12916
HMGCR polymorphism is associated with reduced hepatic
HMGCR expression and reduced LDL-cholesterol levels, as well as increased diabetes risk [
15]. Genetic inhibition of HMG-CoA reductase due to this polymorphism appears to resemble metabolic side effects of statin therapy. Therefore, it is of interest to understand whether the association of
HMGCR and its LDL-lowering polymorphism with certain plasma or adipose tissue metabolites could give a closer insight into the diabetogenic effects of statins.
Several possible mechanisms for the diabetogenic actions of statins have been proposed. Pancreatic β-cells are capable of de novo cholesterol synthesis. Inhibition of cholesterol production leads to deprivation of plasma membrane cholesterol, increased LDL receptor expression and increased uptake of plasma LDL in the β-cells [
16,
17]. Accumulating LDL in the β-cells from the bloodstream seems to induce inflammation and apoptosis of mice and human pancreatic β-cells and, hence, impair their function [
16,
18]. In vitro and animal studies on skeletal muscle cells have shown that simvastatin reduced glucose transporter 4 (GLUT4) translocation and impaired glucose uptake via interfering with insulin signaling cascade [
19,
20]. Animal studies have also demonstrated that the mevalonate pathway and its components seem to be highly important for the metabolic functions of adipose tissue [
21,
22]. Studies have also demonstrated that supra-therapeutic concentrations of statins (atorvastatin and simvastatin) inhibited 3T3-L1 adipocyte differentiation and GLUT4 translocation in adipocytes as well [
23,
24]. Interestingly, most studies have been performed in vitro using concentrations of statins in the range of 1–50 µM, which is more than 1000 times higher than the concentrations seen in human plasma [
25]. Similarly, the authors report that animal studies also use a much higher dose than the dose administered to the patients (1–100 mg/kg vs. 0.1–1 mg/kg, respectively) [
25]. Therefore, studies with relevant therapeutic concentrations are warranted. Finally, most of the studies use simvastatin lactone, which is an inactive prodrug [
25], therefore, its active metabolite, simvastatin hydroxy acid, also needs to be tested.
We hypothesize that inhibition of HMG-CoA reductase has a negative impact on plasma and adipose tissue metabolome and human adipose tissue metabolism, and pancreatic islet function. Therefore, our work aims to study the direct effects of pharmacological inhibition of HMG-CoA reductase with statins (simvastatin and its active metabolite simvastatin hydroxy acid) on human subcutaneous adipose tissue and isolated human pancreatic islet function. The concentration of the drugs was selected according to the reported therapeutic concentrations to perform clinically relevant experiments. Furthermore, we investigated the impact of a genetic HMGCR variant lowering LDL-cholesterol levels on the circulating metabolome.
3. Discussion
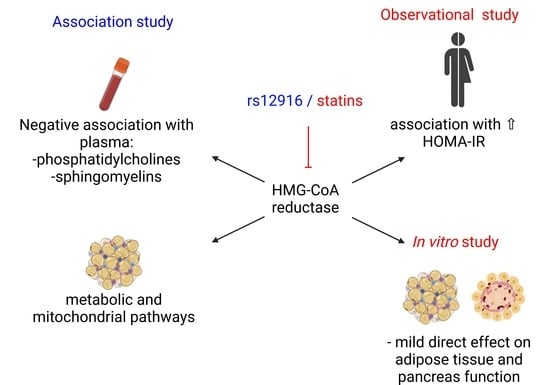
In this study, we observed that impaired HMG-CoA reductase activity caused by statin exposure or genetic variants are associated with diabetes and diabetes-related traits. We discovered that HMGCR expression is associated with several adipose tissue metabolic pathways, including insulin signaling and citrate cycle, and increased blood circulating levels of phosphatidylcholines and sphingomyelins, as well as insulin resistance. This led us to hypothesize that there might be direct effects of statins on adipose tissue metabolism or β-cell function. Direct treatment of human adipose tissue or β-cells ex vivo modestly reduced adipocyte glucose uptake and β-cell insulin secretion, albeit at supra-therapeutic concentrations.
Our analysis indicated the involvement of
HMGCR in a number of metabolic pathways in the adipose tissue. Among these, the most relevant to adipose tissue metabolism include insulin signaling and insulin resistance. Our data showed a positive association of
HMGCR with insulin signaling pathway, particularly, PI3K signaling, and negative association with NF-κB signaling, which suggests that reduced activity of
HMGCR could contribute to insulin resistance. A recent study in mice demonstrated that adipose tissue-specific deletion of
Hmgcr resulted in lipodystrophy and severe glucose intolerance and insulin resistance in the animals [
22]. We also observed that
HMGCR expression in the adipose tissue was also associated with several mitochondrial pathways, including oxidative phosphorylation, citrate cycle, and thermogenesis. Balaz and colleagues have reported the importance of the mevalonate pathway in adipocyte browning and thermogenesis [
21]. Furthermore, metabolic side effects of statins were linked to their ability to directly induce mitochondrial dysfunction as well as depletion of mevalonate pathway metabolites [
27]. We observed that
HMGCR expression in the adipose tissue did not differ between healthy individuals and T2D patients. However, the expression of the gene does not represent the activity of the enzyme, therefore, more detailed studies on HMG-CoA reductase are warranted. We compared the expression of
HMGCR in 31 statin-free individuals with
HMGCR expression in nine patients taking statins was significantly higher than in the adipose tissue of statin-free subjects. Statin therapy has been shown to induce compensatory upregulation in the expression and activity of HMG-CoA reductase in the liver in animal and human studies [
28,
29]. It appears that statins have similar effects in human adipose tissue as well. However, these results need to be interpreted with caution as the number of subjects in the two groups was limited and not balanced.
The T allele of the
HMGCR polymorphism rs12916 is associated with reduced hepatic
HMGCR expression and reduced LDL-cholesterol levels [
15]. We studied the association of this polymorphism with plasma metabolites and observed a strong negative correlation of the SNP with plasma sphingomyelins and phosphatidylcholines. The plasma concentrations of phosphatidylcholines and sphingomyelins were reduced in subjects with impaired HMG-CoA reductase activity. Reduced HMG-CoA reductase activity leads to diminished conversion of HMG-CoA to mevalonate [
1]. Mevalonate has been reported to increase sphingomyelin levels via inhibiting acid sphingomyelinase activity [
30], and therefore reduced sphingomyelin levels in subjects with reduced HMG-CoA reductase activity would have been expected. Inhibition of the HMG-CoA reductase has been shown to inhibit de novo synthesis of phosphatidylcholines via the cytidine diphosphate-choline pathway, which, in turn, could reduce its plasma lipid levels [
31]. Although most clinical studies suggested that sphingomyelins and phosphatidylcholines, such as C32:2, C34:1, and C34:2, are associated with reduced insulin sensitivity in humans [
32,
33], a metabolomic analysis in newly developed type 2 diabetes subjects shows that some phosphatidylcholines, are significantly associated with decreased risk of diabetes [
33,
34]. Interestingly, we observed a negative association of reduced
HMGCR activity with plasma sphingomyelins and phosphatidylcholines, suggesting that any disruption of phospholipid metabolism could affect metabolism. However, whether changes in phospholipids are a cause or consequence of insulin resistance requires further investigation.
Since pharmacological inhibition of HMG-CoA reductase with statins is associated with new-onset diabetes in patients, we compared insulin resistance between subjects taking statins and age- and BMI-matched control subjects. We tested the effect of statin treatment on systemic glucose tolerance in another independent cohort of 195 individuals, to verify whether the diabetogenic effect of statins might be due to pathophysiological mechanisms occurring in other tissues. After adjusting for multiple confounders, including BMI, age, sex, diabetes status, HbA1c, and LDL cholesterol levels, statins use was associated with an increased HOMA-IR. Similarly, diabetogenic effects of statins have been reported in patients previously [
6,
8,
35].
Since statin therapy was associated with increased systemic insulin resistance, we hypothesized that the diabetogenic effects of statin treatment could be mediated via a direct effect on adipose tissue and β-cell function. We observed that the drug or its active metabolite led to a 10% reduction in insulin-stimulated adipocyte glucose uptake only at the supra-therapeutic concentrations. Our data are consistent with the study on 3T3-L1 adipocytes treated with 100 ng/mL of simvastatin (ca 230 nM), which slightly reduce adipocyte glucose uptake [
23]. To the best of our knowledge, the effects of simvastatin hydroxy acid on adipocyte glucose uptake have not been previously studied. A similar trend in glucose uptake regulation was expected, as both drugs target the same enzyme. However, adipose tissue accounts for less than 10% of the whole-body glucose uptake [
35]. Therefore, the modest 10% reduction of the adipocyte glucose uptake is unlikely to lead to an increased risk of type 2 diabetes, as observed in patients taking statins. Neither simvastatin nor its active metabolite altered adipocyte lipolysis after 24 h incubation of the adipose tissue. Our data are in accordance with the study conducted by Henriksbo and colleagues, who reported no effects of atorvastatin on 3T3-L1 adipocyte lipolysis and glucose uptake [
35].
The presence of simvastatin and its active metabolite during the adipogenesis of SGBS cells did not alter their differentiation rate. Similar to our findings, Nakata and colleagues did not observe any changes in murine 3T3-L1 adipocyte maturation with simvastatin [
23]. We did not observe any significant effect on adipocyte GLUT4 expression after 14 days of differentiation at both mRNA and protein levels, while Nakata and co-workers reported that simvastatin reduced GLUT4 expression in 3T3-L1 cells after 10 days of culture [
23]. The discrepancy in our data could be due to the differences in the experimental setup, such as different cell lines or concentration differences, as Nakata and colleagues used a much higher concentration of the drug than in our setting.
We observed no significant effect of simvastatin and its active metabolite, SA, on the mRNA expression of adipokines in the adipose tissue. It has been shown that gene expression of adiponectin was not altered in the adipose tissue of animals treated with simvastatin for 2 weeks, while leptin mRNA expression was increased by the statins [
36]. Maeda and Horiuchi reported a reduction in the expression of leptin mRNA in 3T3-L1 adipocytes differentiated in the presence of simvastatin [
24]. Our data similarly showed a nominal reduction in
LEP and
ADIPOQ expression in SGBS cells. Similarly, reduced serum adiponectin concentration was reported in patients treated with simvastatin for 2 months [
37].
Simvastatin treatment nominally increased the expression of
IL1B in the adipose tissue. Our data are in accordance with the study by Henriksbo and colleagues, who reported that statins, including simvastatin, induce IL1β secretion via NLRP3 inflammasome activation in macrophages, which are the dominant leukocyte type in the adipose tissue [
38,
39]. Although statins have been reported to have anti-inflammatory effects by reducing circulating C-reactive protein and pro-inflammatory cytokines, they appear to induce adipose tissue inflammation. Interestingly, our data did not show any significant effect of simvastatin on the genes involved in the regulation of mitochondrial functions, namely
PPARGC1A,
TFAM, and
PDK4. We also did not observe any changes in
CAV1 expression, which could be used as a marker of cholesterol depletion in adipose tissue [
36]. This could be due to a relatively short-term incubation of adipose tissue and adipocytes with the drugs, while the patients taking statins are exposed to the drug for a much longer period. Additionally, the majority of in vitro studies use high concentrations of the drug [
25], while we selected the concentrations relevant to the plasma levels of the drugs.
Although our study demonstrated no direct effects of simvastatin and its active metabolite on adipose tissue metabolism, genetic or pharmacological inhibition of HMG-CoA reductase has been associated with weight gain, which itself is a risk factor for the development of diabetes [
15,
40] therefore, increased adiposity could be one of the contributors to diabetogenic effects of statins.
Similar to adipose tissue, we did not observe a significant effect of therapeutic concentration of simvastatin on the human pancreatic islets. When we increased the simvastatin concentration, the statin normalized palmitate-induced insulin hypersecretion, which has been suggested as a treatment strategy for early obesity intervention [
41]. In human islets exposed to a high statin concentration alone, GSIS was reduced [
42]. Thus, to what extent the observed positive effect of the higher statin concentration in β-cells of the palmitate-treated islets in the present study could be of clinical use remains to be decided. Statin-induced reduction in insulin secretion from β-cells happens because statins disrupt the production of endogenous cholesterol in β-cells, which is important to maintain the functionality of CaV channels and insulin secretion [
17]. Additionally, pancreatic β-cell-specific ablation of
HMGCR gene in mice resulted in a phenotype with severe hypoinsulinemia and hyperglycemia [
43]. This study revealed the importance of
HMGCR and mevalonate pathway for pancreatic β-cell development [
43].
Our study has several limitations. One of the limitations is the short incubation time compared to the duration of the treatment in the patients. HMG-CoA reductase is highly important for adipose tissue and pancreas functions [
22,
43]. Although 24 h incubation of MIN6 cells with atorvastatin (10 µM) has shown a reduction in cholesterol content in the cells [
44], measuring the cholesterol content or
HMGCR activity in human islets and adipose tissue is warranted. Unfortunately, it was not possible in our setting due to the limited amount of material and reduction in the number of donors because of the COVID-19 pandemic. To our best knowledge, there are no reports on the pancreatic β-cell function in individuals with rs12916 T-allele, which would be an important asset to understanding the role of HMG-CoA reductase in islet function. Additionally, glucose-stimulated insulin secretion in the human islets is also warranted, as we only measured GSIS after treatment of pancreatic islets with palmitate, which is a diabetogenic condition [
45]. Another limitation is a small sample size for both adipose tissue and pancreatic islet donors. Finally, for the observational study results, although we tried to adjust for several confounding factors, these data need to be interpreted with caution as the observational study performed in this work had a selection bias since we can assume that patients on statin treatment display a less favorable metabolic profile and are more prone to develop metabolic syndrome. In addition, it should be noted that there could be more parameters to consider, such as treatment duration, the dose of the medication, as well as the age of the patients since high doses of statins, older age, and female sex were associated with a higher risk of new-onset diabetes [
8,
46,
47].
Our study targeted the effects of statins on human cells and tissues. Our data show that genetic inhibition of HMG-CoA reductase is negatively correlated with plasma sphingomyelins and phosphatidylcholines, which appear to play a role in insulin resistance development. Our results suggest that HMGCR expression and activity in adipose tissue are associated with metabolic pathways; however, statin treatment has a minor direct effect on adipose tissue metabolism and β-cell insulin secretion. Other tissues, such as liver or skeletal muscle, might be of greater interest when investigating the diabetogenic effects of statins.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}