
Ultramicronized Palmitoylethanolamide Inhibits NLRP3 Inflammasome Expression and Pro-Inflammatory Response Activated by SARS-CoV-2 Spike Protein in Cultured Murine Alveolar Macrophages

 ,
,  , and
, and
Abstract
:
1. Introduction
2. Results
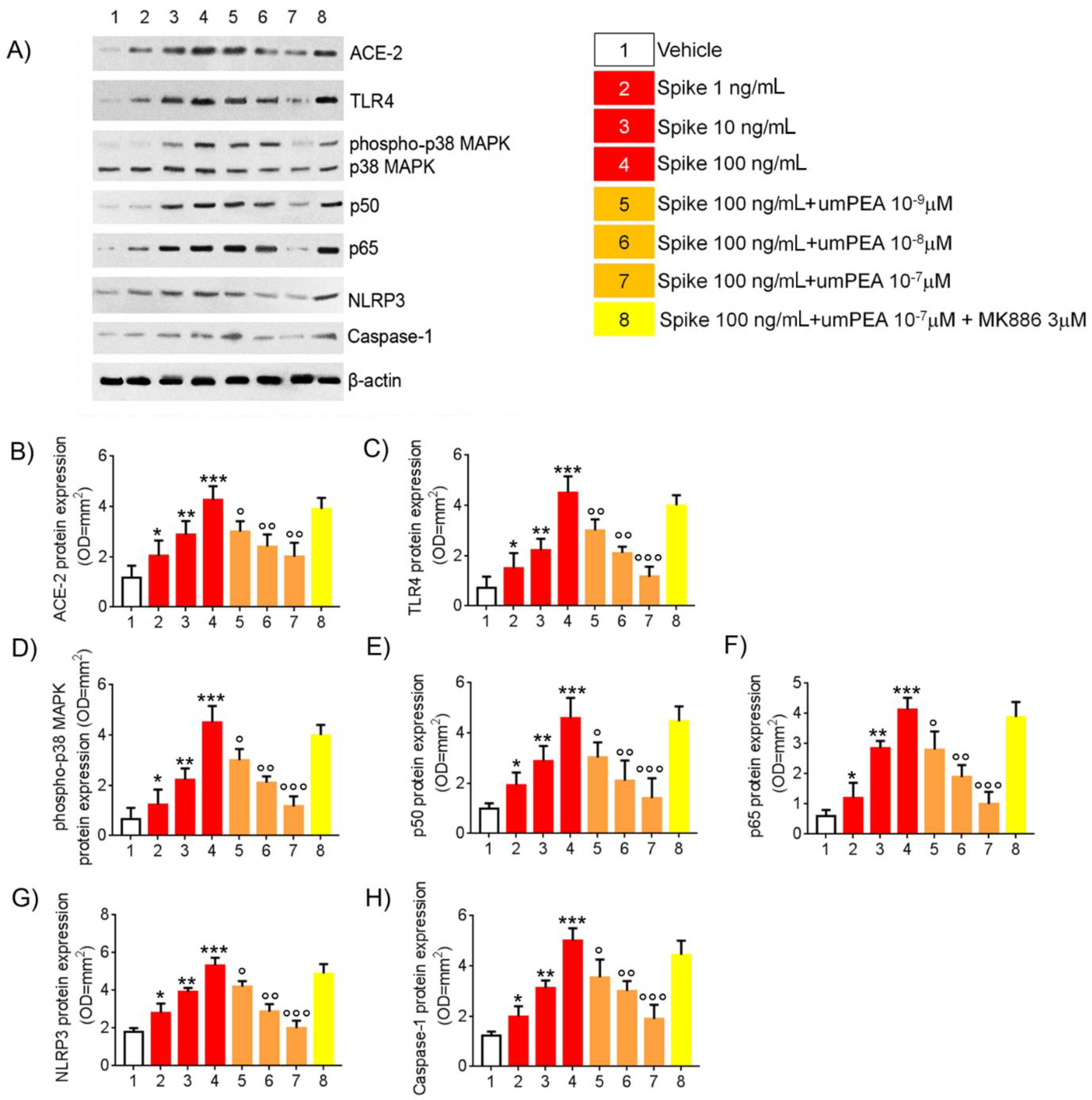
2.1. Western Blot Analysis Reveal Reduction of Pro-Inflammatory Proteins Expression Arbitrated by um-PEA in Alveolar Macrophages Challenged with SP
2.2. um-PEA Inhibited TNFα, IL-6, and IL-1β Release by SARS-CoV-2 SP Challenged Alveolar Macrophages
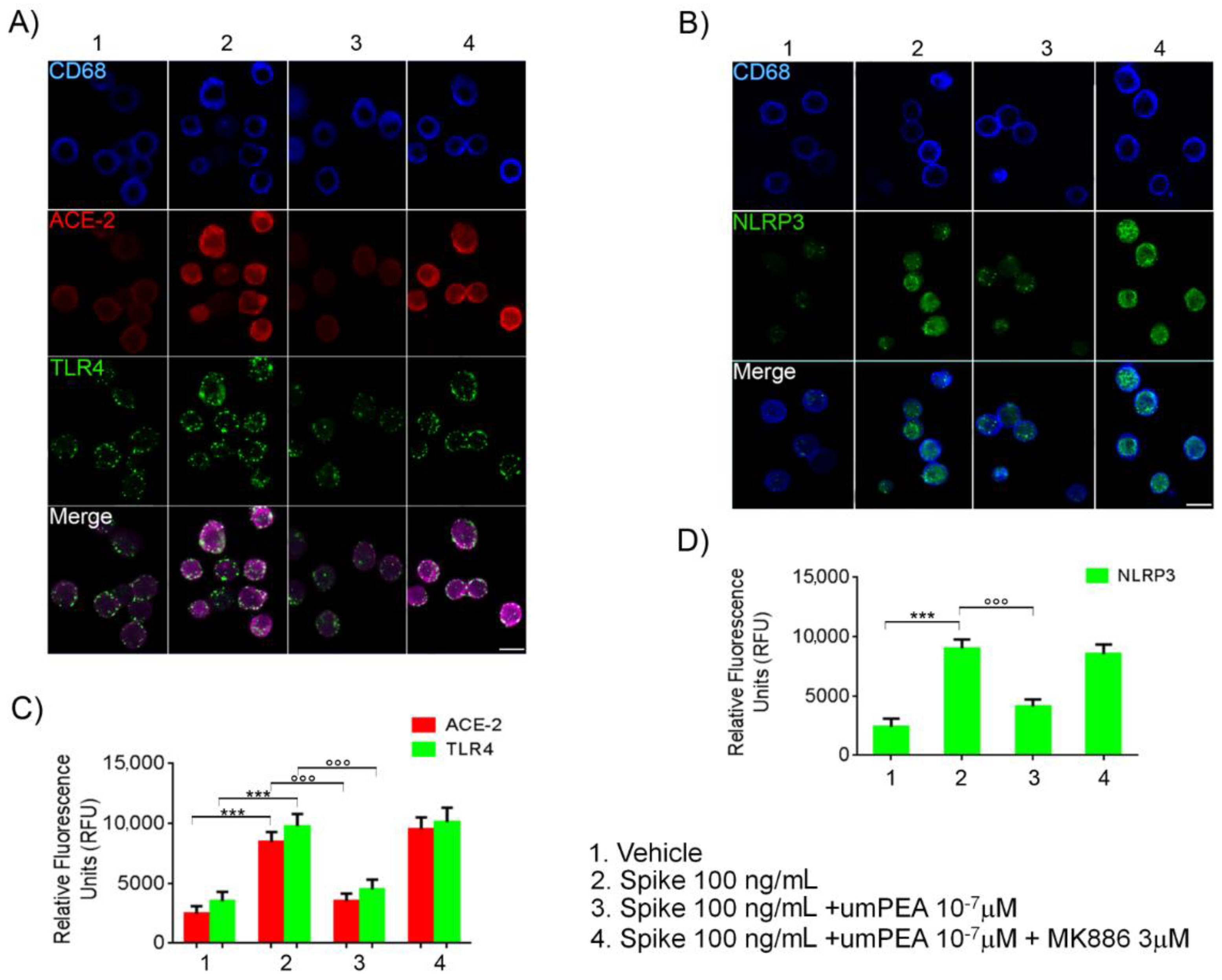
2.3. Immunofluorescent Analysis Confirmed um-PEA Downregulation of ACE-2, TLR4, and NLRP3 Proteins Expression in Alveolar Macrophages Exposed to SARS-CoV-2 SP
2.4. Um-PEA Did Not Show Cytotoxicity on WT Murine Alveolar Macrophages and Did Not Inhibit Pro-Inflammatory Markers Release in SARS-CoV-2 SP-Challenged PPAR-α -/- Murine Alveolar Macrophages
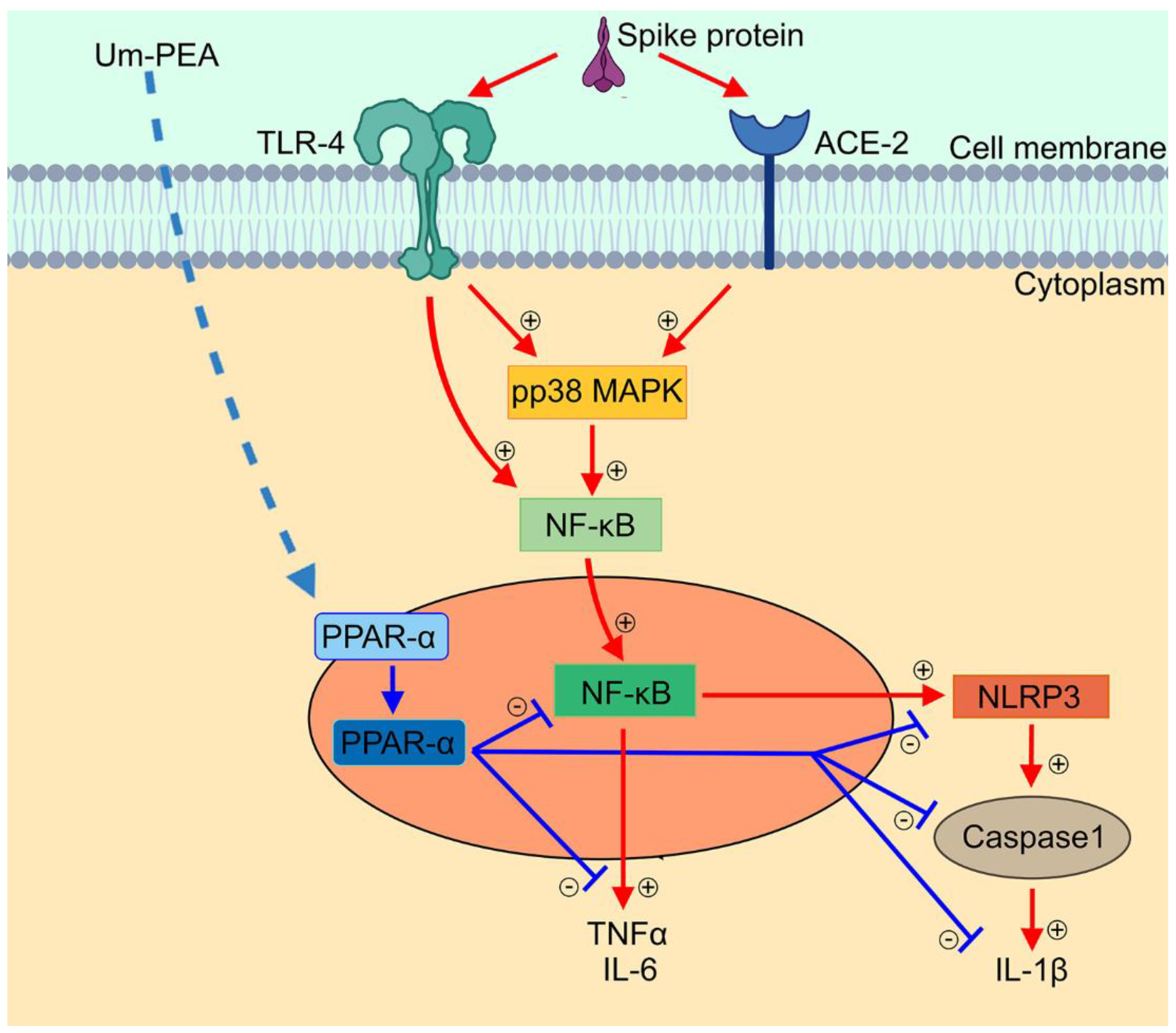
3. Discussion
4. Materials and Methods
4.1. Murine Alveolar Macrophages Isolation, Culture, and Treatments
4.2. Western Blot Analysis
4.3. Enzyme-Linked Immunosorbent Assay for TNFα, IL-6, and IL-1β
4.4. Immunofluorescence Analysis
4.5. Cytotoxicity Assay
4.6. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cao, Y.; Cai, K.; Xiong, L. Coronavirus disease 2019: A new severe acute respiratory syndrome from Wuhan in China. Acta Virol. 2020, 64, 245–250. [Google Scholar] [CrossRef]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, M.Y.; Zhao, R.; Gao, L.J.; Gao, X.F.; Wang, D.P.; Cao, J.M. SARS-CoV-2: Structure, Biology, and Structure-Based Therapeutics Development. Front. Cell. Infect. Microbiol. 2020, 10, 587269. [Google Scholar] [CrossRef]
- Lentz, S.; Roginski, M.A.; Montrief, T.; Ramzy, M.; Gottlieb, M.; Long, B. Initial emergency department mechanical ventilation strategies for COVID-19 hypoxemic respiratory failure and ARDS. Am. J. Emerg. Med. 2020, 38, 2194–2202. [Google Scholar] [CrossRef] [PubMed]
- Stanton, B.A.; Hampton, T.H.; Ashare, A. SARS-CoV-2 (COVID-19) and cystic fibrosis. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020, 319, L408–L415. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, T.; Frieman, M.B. The role of epidermal growth factor receptor (EGFR) signaling in SARS coronavirus-induced pulmonary fibrosis. Antivir. Res. 2017, 143, 142–150. [Google Scholar] [CrossRef]
- Chen, X.; Tang, J.; Shuai, W.; Meng, J.; Feng, J.; Han, Z. Macrophage polarization and its role in the pathogenesis of acute lung injury/acute respiratory distress syndrome. Inflamm. Res. 2020, 69, 883–895. [Google Scholar] [CrossRef]
- Abassi, Z.; Knaney, Y.; Karram, T.; Heyman, S.N. The Lung Macrophage in SARS-CoV-2 Infection: A Friend or a Foe? Front. Immunol. 2020, 11, 1312. [Google Scholar] [CrossRef]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef] [PubMed]
- Quagliariello, V.; Bonelli, A.; Caronna, A.; Lombari, M.C.; Conforti, G.; Libutti, M.; Iaffaioli, R.V.; Berretta, M.; Botti, G.; Maurea, N. SARS-CoV-2 infection: NLRP3 inflammasome as plausible target to prevent cardiopulmonary complications? Eur. Rev. Med. Pharmacol. Sci. 2020, 24, 9169–9171. [Google Scholar]
- Rodrigues, T.S.; de Sá, K.S.G.; Ishimoto, A.Y.; Becerra, A.; Oliveira, S.; Almeida, L.; Gonçalves, A.V.; Perucello, D.B.; Andrade, W.A.; Castro, R.; et al. Inflammasomes are activated in response to SARS-CoV-2 infection and are associated with COVID-19 severity in patients. J. Exp. Med. 2021, 218, e20201707. [Google Scholar] [CrossRef]
- Cabral, G.A.; Ferreira, G.A.; Jamerson, M.J. Endocannabinoids and the Immune System in Health and Disease. Handb. Exp. Pharmacol. 2015, 231, 185–211. [Google Scholar] [PubMed]
- Rankin, L.; Fowler, C.J. The Basal Pharmacology of Palmitoylethanolamide. Int. J. Mol. Sci. 2020, 21, 7942. [Google Scholar] [CrossRef]
- Esposito, G.; Capoccia, E.; Turco, F.; Palumbo, I.; Lu, J.; Steardo, A.; Cuomo, R.; Sarnelli, G.; Steardo, L. Palmitoylethanolamide improves colon inflammation through an enteric glia/toll like receptor 4-dependent PPAR-α activation. Gut 2014, 63, 1300–1312. [Google Scholar] [CrossRef]
- Sarnelli, G.; Gigli, S.; Capoccia, E.; Iuvone, T.; Cirillo, C.; Seguella, L.; Nobile, N.; D’Alessandro, A.; Pesce, M.; Steardo, L.; et al. Palmitoylethanolamide Exerts Antiproliferative Effect and Downregulates VEGF Signaling in Caco-2 Human Colon Carcinoma Cell Line Through a Selective PPAR-α-Dependent Inhibition of Akt/mTOR Pathway. Phytother. Res. 2016, 30, 963–970. [Google Scholar] [CrossRef]
- Rinne, P.; Guillamat-Prats, R.; Rami, M.; Bindila, L.; Ring, L.; Lyytikäinen, L.P.; Raitoharju, E.; Oksala, N.; Lehtimäki, T.; Weber, C.; et al. Palmitoylethanolamide Promotes a Proresolving Macrophage Phenotype and Attenuates Atherosclerotic Plaque Formation. Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2562–2575. [Google Scholar] [CrossRef] [PubMed]
- Lackovic, V.; Borecký, L.; Kresáková, J. Effect of impulsin treatment of interferon production and antiviral resistance of mice. Arch. Immunol. Ther. Exp. 1977, 25, 655–661. [Google Scholar]
- Pesce, M.; Seguella, L.; Cassarano, S.; Aurino, L.; Sanseverino, W.; Lu, J.; Corpetti, C.; Del Re, A.; Vincenzi, M.; Sarnelli, G.; et al. Phytotherapics in COVID19: Why palmitoylethanolamide? Phytother. Res. 2020, 35, 2514–2522. [Google Scholar] [CrossRef]
- Petrosino, S.; Di Marzo, V. The pharmacology of palmitoylethanolamide and first data on the therapeutic efficacy of some of its new formulations. Br. J. Pharmacol. 2017, 174, 1349–1365. [Google Scholar] [CrossRef] [PubMed]
- Noce, A.; Albanese, M.; Marrone, G.; Di Lauro, M.; Pietroboni Zaitseva, A.; Palazzetti, D.; Guerriero, C.; Paolino, A.; Pizzenti, G.; Di Daniele, F.; et al. Ultramicronized Palmitoylethanolamide (um-PEA): A New Possible Adjuvant Treatment in COVID-19 patients. Pharmaceuticals 2021, 14, 336. [Google Scholar] [CrossRef]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van den Berg, D.F.; Te Velde, A.A. Severe COVID-19: NLRP3 Inflammasome Dysregulated. Front. Immunol. 2020, 11, 1580. [Google Scholar] [CrossRef] [PubMed]
- Zhao, C.; Zhao, W. NLRP3 Inflammasome-A Key Player in Antiviral Responses. Front. Immunol. 2020, 11, 211. [Google Scholar] [CrossRef] [Green Version]
- Brandão, S.C.S.; Ramos, J.D.O.X.; Dompieri, L.T.; Godoi ET, A.M.; Figueiredo, J.L.; Sarinho, E.S.C. Is Toll-like receptor 4 involved in the severity of COVID-19 pathology in patients with cardiometabolic comorbidities? Cytokine Growth Factor Rev. 2021, 58, 102–110. [Google Scholar] [CrossRef] [PubMed]
- Choudhury, A.; Mukherjee, S. In silico studies on the comparative characterization of the interactions of SARS-CoV-2 spike glycoprotein with ACE-2 receptor homologs and human TLRs. J. Med. Virol. 2020, 92, 2105–2113. [Google Scholar]
- Zhao, Y.; Kuang, M.; Li, J.; Zhu, L.; Jia, Z.; Guo, X.; Hu, Y.; Kong, J.; Yin, H.; Wang, X.; et al. Publisher Correction: SARS-CoV-2 spike protein interacts with and activates TLR4. Cell Res. 2021, 31, 825. [Google Scholar] [CrossRef]
- Mitchell, S.; Vargas, J.; Hoffmann, A. Signaling via the NFκB system. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Luo, B.; Huang, F.; Liu, Y.; Liang, Y.; Wei, Z.; Ke, H.; Zeng, Z.; Huang, W.; He, Y. NLRP3 Inflammasome as a Molecular Marker in Diabetic Cardiomyopathy. Front. Physiol. 2017, 8, 519. [Google Scholar] [CrossRef] [Green Version]
- Veiras, L.C.; Cao, D.; Saito, S.; Peng, Z.; Bernstein, E.A.; Shen, J.Z.Y.; Koronyo-Hamaoui, M.; Okwan-Duodu, D.; Giani, J.F.; Khan, Z.; et al. Overexpression of ACE in Myeloid Cells Increases Immune Effectiveness and Leads to a New Way of Considering Inflammation in Acute and Chronic Diseases. Curr. Hypertens. Rep. 2020, 22, 4. [Google Scholar]
- Bernstein, K.E.; Koronyo, Y.; Salumbides, B.C.; Sheyn, J.; Pelissier, L.; Lopes, D.H.; Shah, K.H.; Bernstein, E.A.; Fuchs, D.T.; Yu, J.J.; et al. Angiotensin-converting enzyme overexpression in myelomonocytes prevents Alzheimer’s-like cognitive decline. J. Clin. Investig. 2014, 124, 1000–1012. [Google Scholar] [CrossRef] [Green Version]
- Khan, Z.; Shen, X.Z.; Bernstein, E.A.; Giani, J.F.; Eriguchi, M.; Zhao, T.V.; Gonzalez-Villalobos, R.A.; Fuchs, S.; Liu, G.Y.; Bernstein, K.E. Angiotensin-converting enzyme enhances the oxidative response and bactericidal activity of neutrophils. Blood 2017, 130, 328–339. [Google Scholar] [CrossRef] [PubMed]
- Okwan-Duodu, D.; Datta, V.; Shen, X.Z.; Goodridge, H.S.; Bernstein, E.A.; Fuchs, S.; Liu, G.Y.; Bernstein, K.E. Angiotensin-converting enzyme overexpression in mouse myelomonocytic cells augments resistance to Listeria and methicillin-resistant Staphylococcus aureus. J. Biol. Chem. 2010, 285, 39051–39060. [Google Scholar] [CrossRef] [Green Version]
- Okwan-Duodu, D.; Weiss, D.; Peng, Z.; Veiras, L.C.; Cao, D.Y.; Saito, S.; Khan, Z.; Bernstein, E.A.; Giani, J.F.; Taylor, W.R.; et al. Overexpression of myeloid angiotensin-converting enzyme (ACE) reduces atherosclerosis. Biochem. Biophys. Res. Commun. 2019, 520, 573–579. [Google Scholar] [CrossRef]
- Shen, X.Z.; Li, P.; Weiss, D.; Fuchs, S.; Xiao, H.D.; Adams, J.A.; Williams, I.R.; Capecchi, M.R.; Taylor, W.R.; Bernstein, K.E. Mice with enhanced macrophage angiotensin-converting enzyme are resistant to melanoma. Am. J. Pathol. 2007, 170, 2122–2134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korbecki, J.; Bobiński, R.; Dutka, M. Self-regulation of the inflammatory response by peroxisome proliferator-activated receptors. Inflamm. Res. 2019, 68, 443–458. [Google Scholar] [CrossRef] [Green Version]
- Fan, E.K.Y.; Fan, J. Regulation of alveolar macrophage death in acute lung inflammation. Respir. Res. 2018, 19, 50. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; He, R.; Zhang, L.; Hao, B.; Jiang, W.; Wang, W.; Geng, Q. Inflammatory Caspases Drive Pyroptosis in Acute Lung Injury. Front. Pharmacol. 2021, 12, 631256. [Google Scholar] [CrossRef] [PubMed]
- Broug-Holub, E.; Toews, G.B.; van Iwaarden, J.F.; Strieter, R.M.; Kunkel, S.L.; Paine, R., 3rd; Standiford, T.J. Alveolar macrophages are required for protective pulmonary defenses in murine Klebsiella pneumonia: Elimination of alveolar macrophages increases neutrophil recruitment but decreases bacterial clearance and survival. Infect. Immun. 1997, 65, 1139–1146. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.; Wu, Q.; Tang, H.; Chen, J.; Wu, Q.; Yuan, X.; Xiong, S.; Ye, Y.; Lv, H. NLRP3 Regulated CXCL12 Expression in Acute Neutrophilic Lung Injury. J. Inflamm. Res. 2020, 13, 377–386. [Google Scholar] [CrossRef]
- Repasy, T.; Martinez, N.; Lee, J.; West, K.; Li, W.; Kornfeld, H. Bacillary replication and macrophage necrosis are determinants of neutrophil recruitment in tuberculosis. Microbes Infect. 2015, 17, 564–574. [Google Scholar] [CrossRef] [Green Version]
- Tian, X.; Sun, H.; Casbon, A.J.; Lim, E.; Francis, K.P.; Hellman, J.; Prakash, A. NLRP3 Inflammasome Mediates Dormant Neutrophil Recruitment following Sterile Lung Injury and Protects against Subsequent Bacterial Pneumonia in Mice. Front. Immunol. 2017, 8, 1337. [Google Scholar] [CrossRef]
- Reusch, N.; De Domenico, E.; Bonaguro, L.; Schulte-Schrepping, J.; Baßler, K.; Schultze, J.L.; Aschenbrenner, A.C. Neutrophils in COVID-19. Front. Immunol. 2021, 12, 652470. [Google Scholar] [CrossRef] [PubMed]
- Awadasseid, A.; Wu, Y.; Tanaka, Y.; Zhang, W. Current advances in the development of SARS-CoV-2 vaccines. Int. J. Biol. Sci. 2021, 17, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.H.; Huang, H.Y.; Tu, Y.F.; Lai, W.Y.; Wang, C.L.; Sun, J.R.; Chien, Y.; Lin, T.W.; Lin, Y.Y.; Chien, C.S.; et al. Highlight of severe acute respiratory syndrome coronavirus-2 vaccine development against COVID-19 pandemic. J. Chin. Med. Assoc. 2021, 84, 9–13. [Google Scholar] [CrossRef]
- Ding, X.; Jin, S.; Tong, Y.; Jiang, X.; Chen, Z.; Mei, S.; Zhang, L.; Billiar, T.R.; Li, Q. TLR4 signaling induces TLR3 up-regulation in alveolar macrophages during acute lung injury. Sci. Rep. 2017, 7, 34278. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antibody | Host | Clonality | Dilution | Brand | Antibody |
|---|---|---|---|---|---|
| Anti-TLR-4 | Rabbit | Polyclonal | 1:300 v/v | Bioss Antibodies, Boston, MA, USA | Anti-TLR-4 |
| Anti-NF-kappaB p65 subunit | Rabbit | Polyclonal | 1:5000 v/v | Santa Cruz Biotechnology, Dallas, TX, USA | Anti-NF-kappaB p65 subunit |
| Anti-NF-kappaB p50 subunit | Mouse | Monoclonal | 1:1000 v/v | Santa Cruz Biotechnology, Dallas, TX, USA | Anti-NF-kappaB p50 subunit |
| Anti-phospho-p38 MAPK | Rabbit | Polyclonal | 1:100 v/v | Santa Cruz Biotechnology, Dallas, TX, USA | Anti-phospho-p38 MAPK |
| Anti-phospho-p38 MAPK | Mouse | Monoclonal | 1:100 v/v | Santa Cruz Biotechnology, Dallas, TX, USA | Anti-phospho-p38 MAPK |
| Anti-Caspase-1 | Mouse | Monoclonal | 1:100 v/v | Santa Cruz Biotechnology, Dallas, TX, USA | Anti-Caspase-1 |
| Anti-β actin | Mouse | Monoclonal | 1:5000 v/v | Proteintech Manchester, UK | Anti-β actin |
| Antibody | Host | Clonality | Dilution | Brand |
|---|---|---|---|---|
| Anti-TLR-4 | Rabbit | Polyclonal | 1:50 v/v | Bioss Antibodies, Boston, MA, USA |
| Anti-ACE-2 | Mouse | Monoclonal | 1:50 v/v | Santa Cruz Biotechnology, Dallas, TX, USA |
| Anti-CD68 | Goat | Monoclonal | 1:200 v/v | AbCam, Cambridge, UK |
| Anti-NLRP3 | Rabbit | Monoclonal | 1:100 v/v | Thermo Fisher Scientific, Waltham, MA, USA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Del Re, A.; Corpetti, C.; Pesce, M.; Seguella, L.; Steardo, L.; Palenca, I.; Rurgo, S.; De Conno, B.; Sarnelli, G.; Esposito, G. Ultramicronized Palmitoylethanolamide Inhibits NLRP3 Inflammasome Expression and Pro-Inflammatory Response Activated by SARS-CoV-2 Spike Protein in Cultured Murine Alveolar Macrophages. Metabolites 2021, 11, 592. https://doi.org/10.3390/metabo11090592
Del Re A, Corpetti C, Pesce M, Seguella L, Steardo L, Palenca I, Rurgo S, De Conno B, Sarnelli G, Esposito G. Ultramicronized Palmitoylethanolamide Inhibits NLRP3 Inflammasome Expression and Pro-Inflammatory Response Activated by SARS-CoV-2 Spike Protein in Cultured Murine Alveolar Macrophages. Metabolites. 2021; 11(9):592. https://doi.org/10.3390/metabo11090592
Chicago/Turabian StyleDel Re, Alessandro, Chiara Corpetti, Marcella Pesce, Luisa Seguella, Luca Steardo, Irene Palenca, Sara Rurgo, Barbara De Conno, Giovanni Sarnelli, and Giuseppe Esposito. 2021. "Ultramicronized Palmitoylethanolamide Inhibits NLRP3 Inflammasome Expression and Pro-Inflammatory Response Activated by SARS-CoV-2 Spike Protein in Cultured Murine Alveolar Macrophages" Metabolites 11, no. 9: 592. https://doi.org/10.3390/metabo11090592
APA StyleDel Re, A., Corpetti, C., Pesce, M., Seguella, L., Steardo, L., Palenca, I., Rurgo, S., De Conno, B., Sarnelli, G., & Esposito, G. (2021). Ultramicronized Palmitoylethanolamide Inhibits NLRP3 Inflammasome Expression and Pro-Inflammatory Response Activated by SARS-CoV-2 Spike Protein in Cultured Murine Alveolar Macrophages. Metabolites, 11(9), 592. https://doi.org/10.3390/metabo11090592