
A Multidrug Approach to Modulate the Mitochondrial Metabolism Impairment and Relative Oxidative Stress in Fanconi Anemia Complementation Group A

 , , ,
, , , 
Abstract
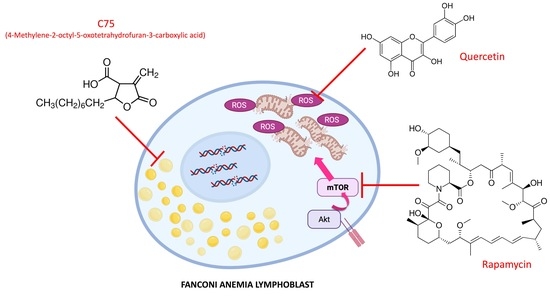
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
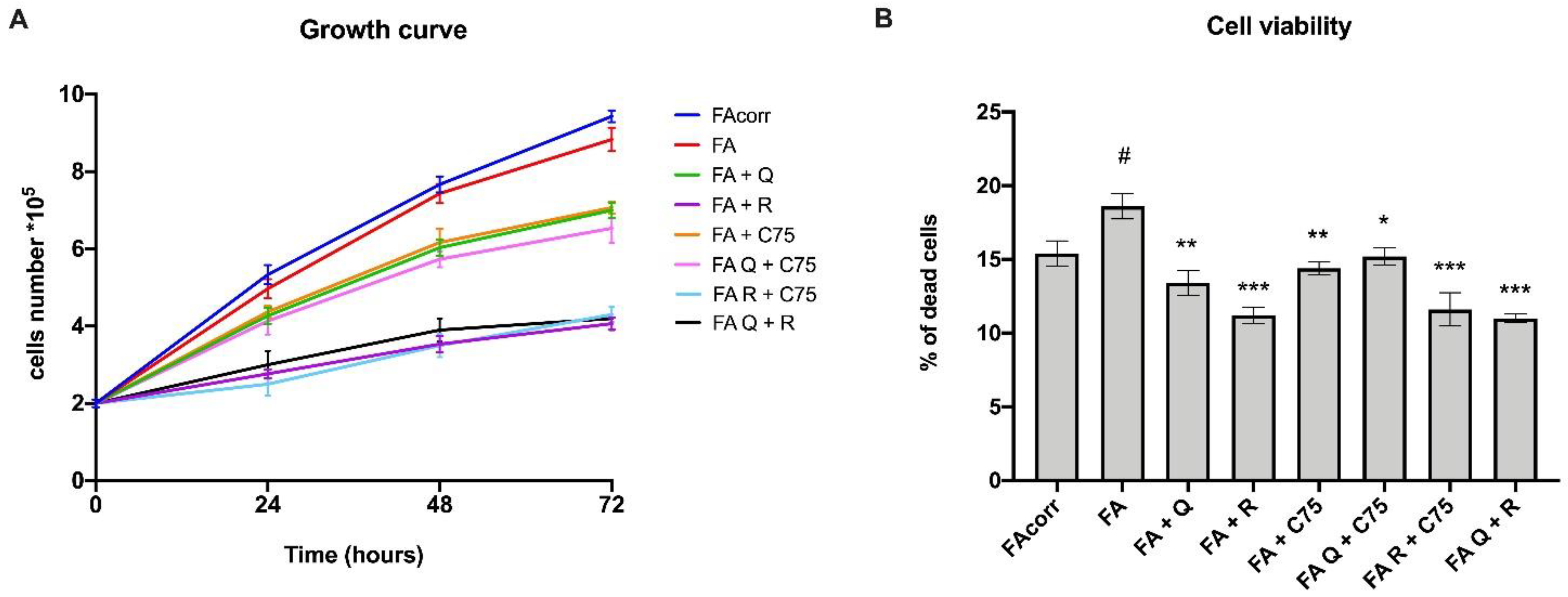
2.1. Quercetin, C75, Rapamycin, and Their Combinations Slow Downs Cellular Growth and Limit the Cell Death
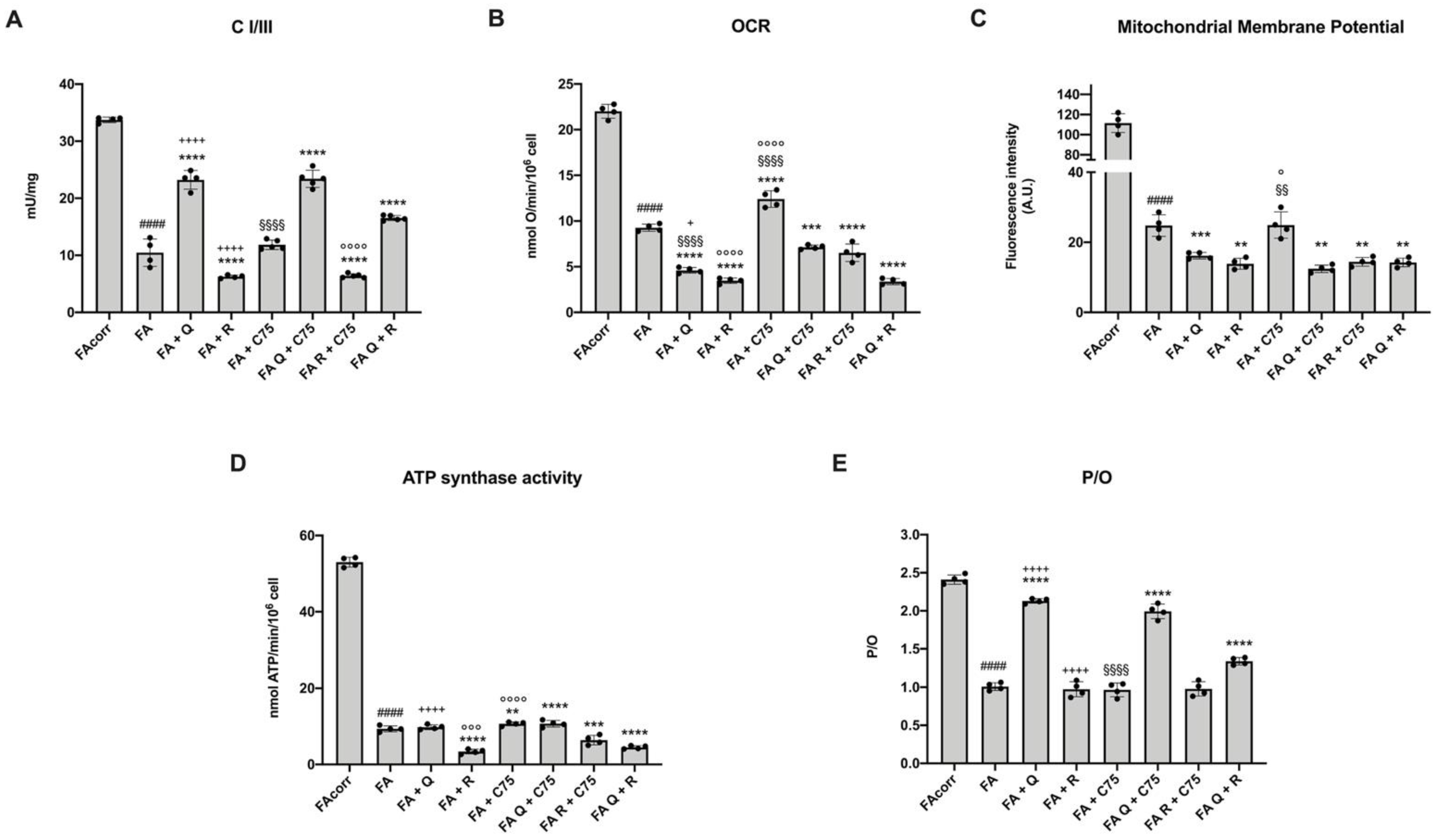
2.2. Quercetin, C75, Rapamycin, and Their Combinations Modulate the Mitochondrial Aerobic Metabolism in FA Cells
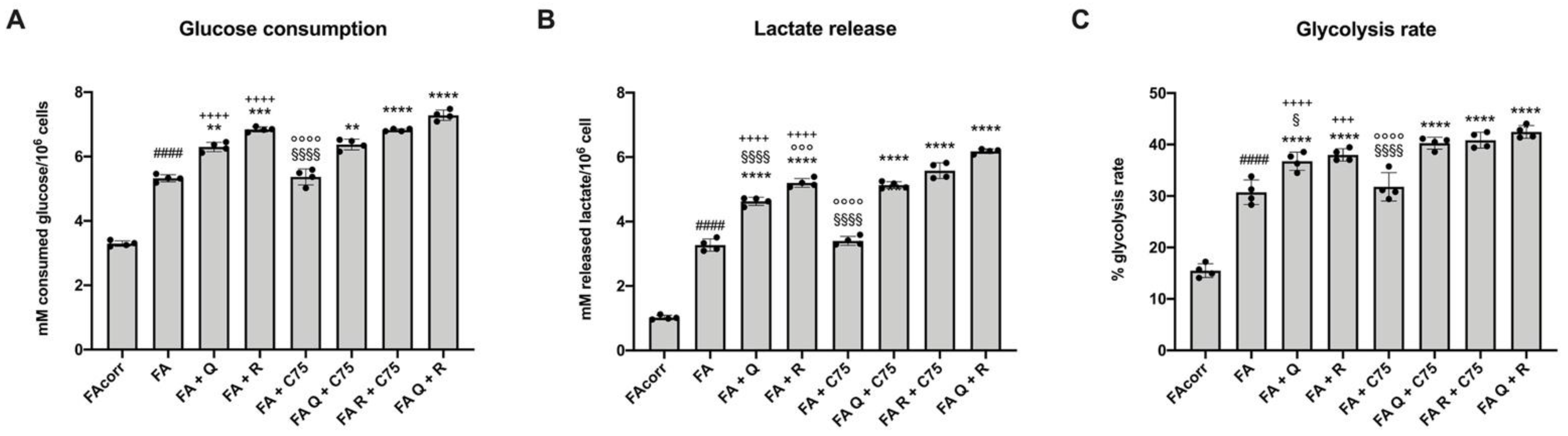
2.3. Quercetin, and Rapamycin and Their Combinations, but Not C75, Modulate the Anaerobic Glycolysis in FA Cells
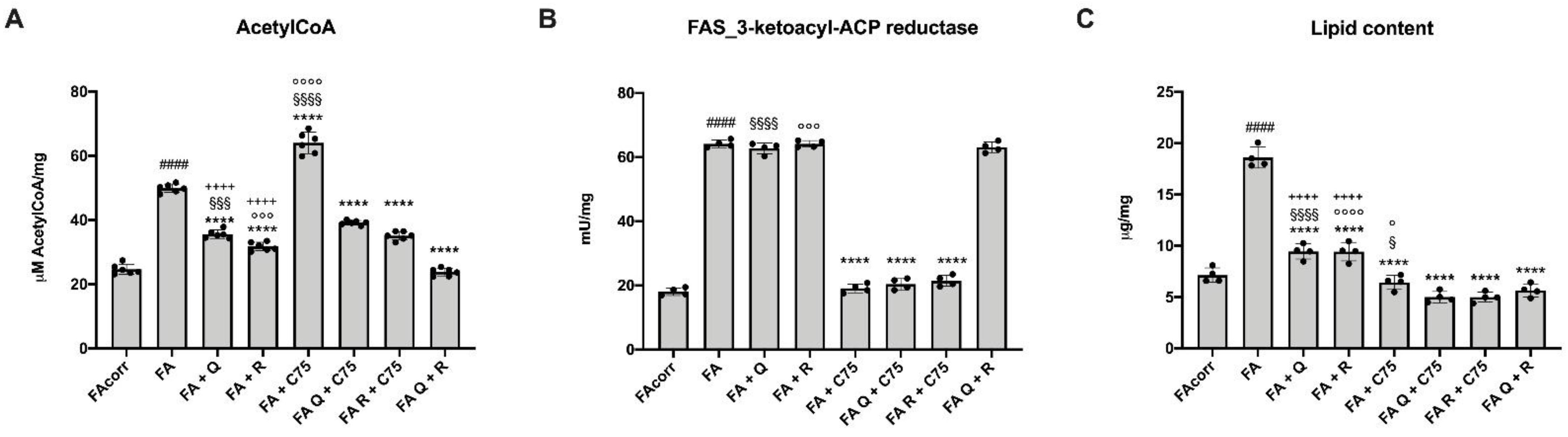
2.4. Quercetin, C75, Rapamycin and Their Combination Modulated the Lipid Content in FA Cells
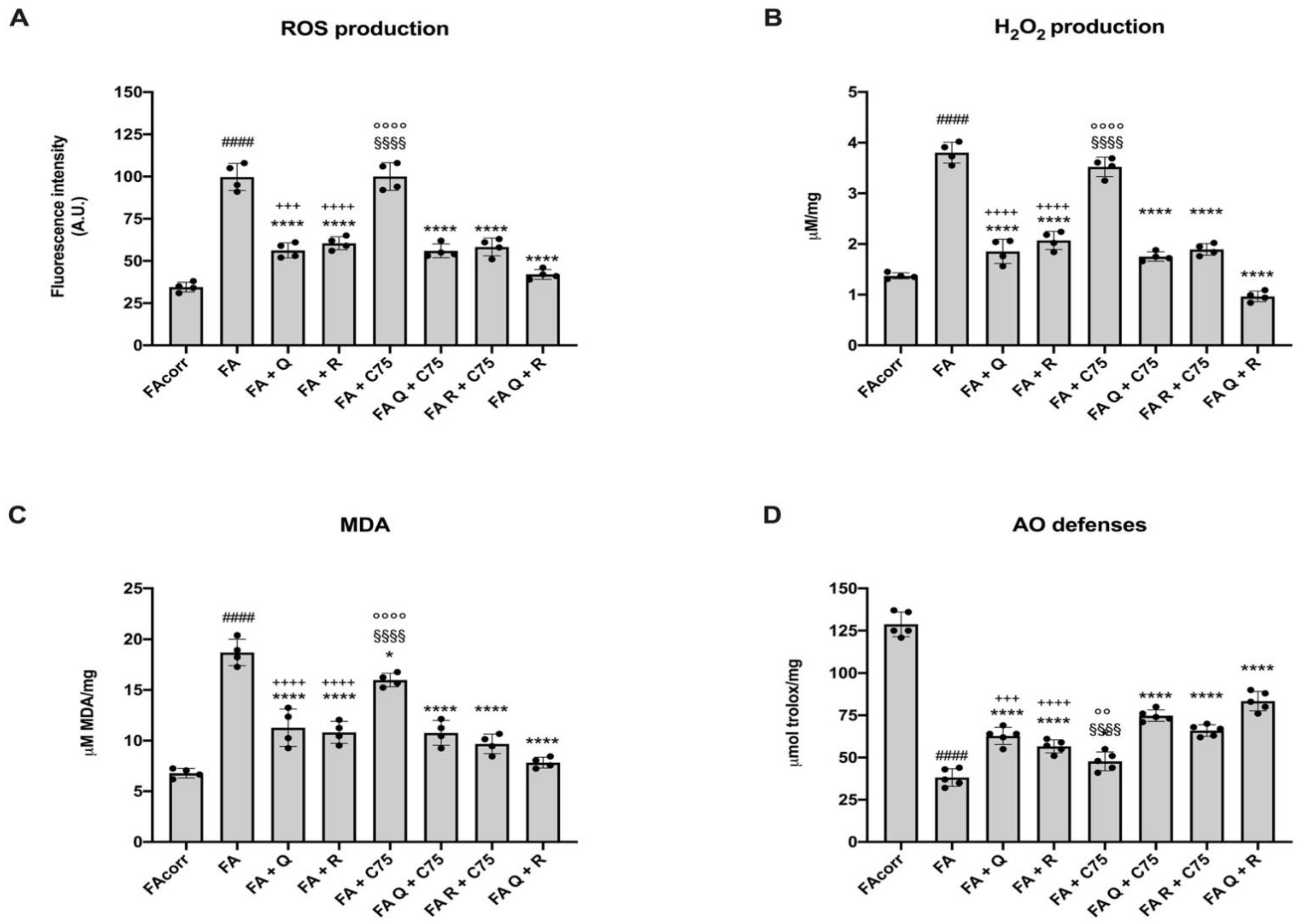
2.5. Quercetin, C75, Rapamycin, and Their Combination Decrease Oxidative Stress Production, the Relative Oxidative Damages, and Increase the Antioxidant Defenses in FA Cells
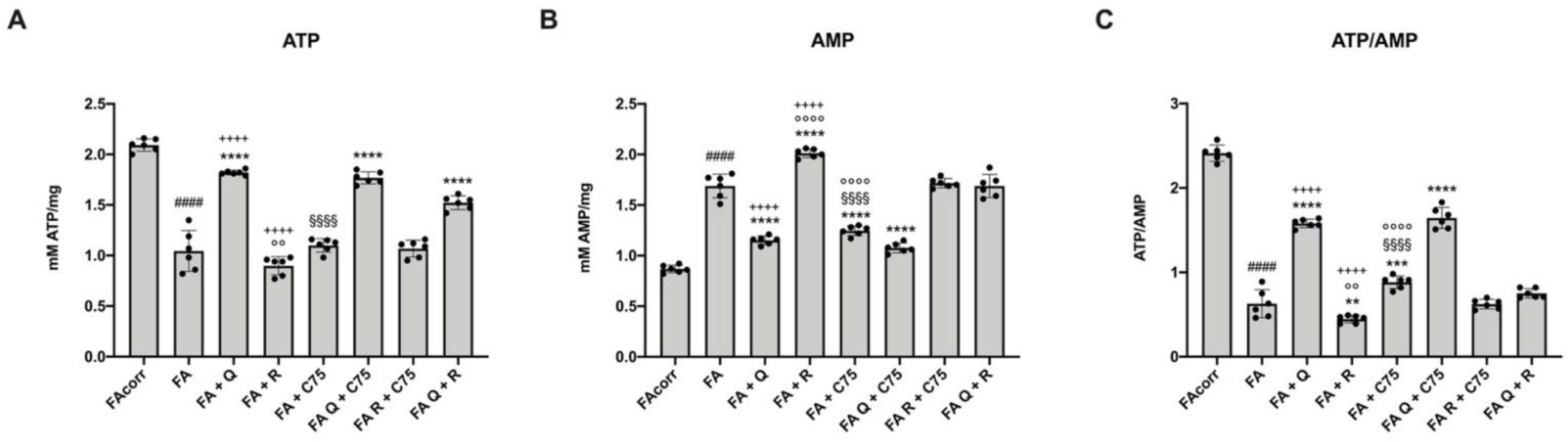
2.6. The Treatment with Quercetin and C75, but Not Rapamycin, Improve the Energy Status in FA Cells
2.7. All Treatments Reduce the Damages and DNA Double-Strand Breaks Induced by Hydroxy Urea
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Treatments
4.2. Flow-Cytometric Assays for Cell Viability
4.3. Assay of the Electron Transport between Complexes I and III
4.4. Oxygen Consumption Rate Assay
4.5. Mitochondrial Trans Membrane Potential by Flow Cytometry
4.6. Bioluminescent Luciferase Fo-F1 ATP Synthase Assay
4.7. Glucose Consumption and Lactate Release Assay
4.8. Evaluation of ATP/AMP Ratio
4.9. Evaluation of Acetyl-CoA Concentration
4.10. 3-ketoacyl-ACP Reductase Assay
4.11. Evaluation of Lipid Content
4.12. Oxidative Stress, Lipid Peroxidation and Antioxidant Defenses Evaluation
4.13. Western Blot Analysis
4.14. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Castella, M.; Pujol, R.; Callén, E.; Trujillo, J.P.; Casado, J.A.; Gille, H.; Lach, F.P.; Auerbach, A.D.; Schindler, D.; Benítez, J.; et al. Origin, functional role, and clinical impact of Fanconi anemia FANCA mutations. Blood 2011, 117, 3759–3769. [Google Scholar] [CrossRef]
- Dufour, C. How I manage patients with Fanconi anaemia. Br. J. Haematol. 2017, 178, 32–47. [Google Scholar] [CrossRef] [Green Version]
- de Winter, J.P.; Joenje, H. The genetic and molecular basis of Fanconi anemia. Mutat. Res. 2009, 668, 11–19. [Google Scholar] [CrossRef]
- Moldovan, G.-L.; D’Andrea, A.D. How the fanconi anemia pathway guards the genome. Annu. Rev. Genet. 2009, 43, 223–249. [Google Scholar] [CrossRef] [Green Version]
- Palovcak, A.; Liu, W.; Yuan, F.; Zhang, Y. Maintenance of genome stability by Fanconi anemia proteins. Cell Biosci. 2017, 7, 8. [Google Scholar] [CrossRef] [Green Version]
- Grompe, M. Fanconi anemia and DNA repair. Hum. Mol. Genet. 2001, 10, 2253–2259. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faivre, L.; Guardiola, P.; Lewis, C.; Dokal, I.; Ebell, W.; Zatterale, A.; Altay, C.; Poole, J.; Stones, D.; Kwee, M.L.; et al. Association of complementation group and mutation type with clinical outcome in Fanconi anemia. Blood 2000, 96, 4064–4070. [Google Scholar]
- Morgan, N.V.; Tipping, A.J.; Joenje, H.; Mathew, C.G. High frequency of large intragenic deletions in the Fanconi anemia group A gene. Am. J. Hum. Genet. 1999, 65, 1330–1341. [Google Scholar] [CrossRef] [Green Version]
- Bottega, R.; Nicchia, E.; Cappelli, E.; Ravera, S.; De Rocco, D.; Faleschini, M.; Corsolini, F.; Pierri, F.; Calvillo, M.; Russo, G.; et al. Hypomorphic FANCA mutations correlate with mild mitochondrial and clinical phenotype in Fanconi anemia. Haematologica 2018, 103, 417–426. [Google Scholar] [CrossRef] [Green Version]
- Pagano, G.; Talamanca, A.A.; Castello, G.; Pallardó, F.V.; Zatterale, A.; Degan, P. Oxidative stress in Fanconi anaemia: From cells and molecules towards prospects in clinical management. Biol. Chem. 2012, 393, 11–21. [Google Scholar] [CrossRef]
- Du, W.; Adam, Z.; Rani, R.; Zhang, X.; Pang, Q. Oxidative stress in Fanconi anemia hematopoiesis and disease progression. Antioxid. Redox Signal. 2008, 10, 1909–1921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El-Bassyouni, H.; Afifi, H.; Eid, M.; Kamal, R.; El-Gebali, H.; El-Saeed, G.; Thomas, M.; Abdel-Maksoud, S. Oxidative stress -a phenotypic hallmark of Fanconi anemia and Down syndrome: The effect of antioxidants. Ann. Med. Health Sci. Res. 2015, 5, 205. [Google Scholar] [CrossRef] [Green Version]
- Ravera, S.; Vaccaro, D.; Cuccarolo, P.; Columbaro, M.; Capanni, C.; Bartolucci, M.; Panfoli, I.; Morelli, A.; Dufour, C.; Cappelli, E.; et al. Mitochondrial respiratory chain Complex i defects in Fanconi anemia complementation group A. Biochimie 2013, 95, 1828–1837. [Google Scholar] [CrossRef]
- Cappelli, E.; Cuccarolo, P.; Stroppiana, G.; Miano, M.; Bottega, R.; Cossu, V.; Degan, P.; Ravera, S. Defects in mitochondrial energetic function compels Fanconi Anemia cells to glycolytic metabolism. BBA-Mol. Basis Dis. 2017, 1863, 1214–1221. [Google Scholar] [CrossRef]
- Jayabal, P.; Ma, C.; Nepal, M.; Shen, Y.; Che, R.; Turkson, J.; Fei, P. Involvement of FANCD2 in Energy Metabolism via ATP5α. Sci. Rep. 2017, 7, 4921. [Google Scholar] [CrossRef] [PubMed]
- Kumari, U.; Ya Jun, W.; Huat Bay, B.; Lyakhovich, A. Evidence of mitochondrial dysfunction and impaired ROS detoxifying machinery in Fanconi anemia cells. Oncogene 2014, 33, 165–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumpter, R.J.; Sirasanagandla, S.; Fernández, Á.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.; Clapp, D.; et al. Fanconi Anemia Proteins Function in Mitophagy and Immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richardson, C.; Yan, S.; Vestal, C.G. Oxidative stress, bone marrow failure, and genome instability in hematopoietic stem cells. Int. J. Mol. Sci. 2015, 16, 2366–2385. [Google Scholar] [CrossRef] [Green Version]
- Lyakhovich, A. Damaged mitochondria and overproduction of ROS in Fanconi anemia cells. Rare Dis. 2013, 1, e24048. [Google Scholar] [CrossRef] [Green Version]
- Capanni, C.; Bruschi, M.; Columbaro, M.; Cuccarolo, P.; Ravera, S.; Dufour, C.; Candiano, G.; Petretto, A.; Degan, P.; Cappelli, E. Changes in vimentin, lamin A/C and mitofilin induce aberrant cell organization in fibroblasts from Fanconi anemia complementation group A (FA-A) patients. Biochimie 2013, 95, 1838–1847. [Google Scholar] [CrossRef]
- Columbaro, M.; Ravera, S.; Capanni, C.; Panfoli, I.; Cuccarolo, P.; Stroppiana, G.; Degan, P.; Cappell, E. Treatment of FANCA cells with resveratrol and N-acetylcysteine: A comparative study. PLoS ONE 2014, 9, e104857. [Google Scholar] [CrossRef]
- Ravera, S.; Degan, P.; Sabatini, F.; Columbaro, M.; Dufour, C.; Cappelli, E. Altered lipid metabolism could drive the bone marrowfailure in fanconi anemia. Br. J. Haematol. 2018, 184, 693–696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 2004, 114, 1752–1761. [Google Scholar] [CrossRef]
- Giri, N.; Batista, D.L.; Alter, B.P.; Stratakis, C.A. Endocrine Abnormalities in Patients with Fanconi Anemia. J. Clin. Endocrinol. Metab. 2007, 92, 2624–2631. [Google Scholar] [CrossRef]
- Houstis, N.; Rosen, E.D.; Lander, E.S. Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 2006, 440, 944–948. [Google Scholar] [CrossRef]
- Cappelli, E.; Degan, P.; Bruno, S.; Pierri, F.; Miano, M.; Raggi, F.; Farruggia, P.; Mecucci, C.; Crescenzi, B.; Naim, V.; et al. The passage from bone marrow niche to bloodstream triggers the metabolic impairment in Fanconi Anemia mononuclear cells. Redox Biol. 2020, 36, 101618. [Google Scholar] [CrossRef]
- Pérez-De La Cruz, V.; Konigsberg, M.; Pedraza-Chaverri, J.; Herrera-Mundo, N.; Díaz-Muñoz, M.; Morán, J.; Fortoul-van der Goes, T.; Rondán-Zárate, A.; Maldonado, P.D.; Ali, S.F.; et al. Cytoplasmic calcium mediates oxidative damage in an excitotoxic/energetic deficit synergic model in rats. Eur. J. Neurosci. 2008, 27, 1075–1085. [Google Scholar] [CrossRef]
- Ramanathan, A.; Schreiber, S.L. Direct control of mitochondrial function by mTOR. Proc. Natl. Acad. Sci. USA. 2009, 106, 22229–22232. [Google Scholar] [CrossRef] [Green Version]
- Ravera, S.; Podestà, M.; Sabatini, F.; Dagnino, M.; Cilloni, D.; Fiorini, S.; Barla, A.; Frassoni, F. Discrete Changes in Glucose Metabolism Define Aging. Sci. Rep. 2019, 9, 10347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pang, Q.; Andreassen, P.R. Fanconi Anemia Proteins and Endogenous Stresses. Mutat. Res. 2009, 668, 42–53. [Google Scholar] [CrossRef] [Green Version]
- Pinti, M.V.; Fink, G.K.; Hathaway, Q.A.; Durr, A.J.; Kunovac, A.; Hollander, J.M.; Mv, P.; Gk, F.; Qa, H.; Aj, D.; et al. Mitochondria Dysfunction in Aging and Metabolic Diseases. J. Physiol. Endocrinol. Metab. 2019, 316, 268–285. [Google Scholar] [CrossRef]
- Cadenas, E.; Davies, K.J.A. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef]
- Pagano, G.; Talamanca, A.A.; Castello, G.; d’Ischia, M.; Pallardó, F.V.; Petrović, S.; Porto, B.; Tiano, L.; Zatterale, A. Bone marrow cell transcripts from Fanconi anaemia patients reveal in vivo alterations in mitochondrial, redox and DNA repair pathways. Eur. J. Haematol. 2013, 91, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Rezaei-Sadabady, R.; Eidi, A.; Zarghami, N.; Barzegar, A. Intracellular ROS protection efficiency and free radical-scavenging activity of quercetin and quercetin-encapsulated liposomes. Artif. Cells Nanomed. Biotechnol. 2016, 44, 128–134. [Google Scholar] [CrossRef]
- Houghton, M.J.; Kerimi, A.; Tumova, S.; Boyle, J.P.; Williamson, G. Quercetin preserves redox status and stimulates mitochondrial function in metabolically-stressed HepG2 cells. Free Radic. Biol. Med. 2018, 129, 296–309. [Google Scholar] [CrossRef] [Green Version]
- Qiu, L.; Luo, Y.; Chen, X. Quercetin attenuates mitochondrial dysfunction and biogenesis via upregulated AMPK/SIRT1 signaling pathway in OA rats. Biomed. Pharmacother. 2018, 103, 1585–1591. [Google Scholar] [CrossRef]
- Rayamajhi, N.; Kim, S.-K.; Go, H.; Joe, Y.; Callaway, Z.; Kang, J.-G.; Ryter, S.W.; Chung, H.T. Quercetin induces mitochondrial biogenesis through activation of HO-1 in HepG2 cells. Oxid. Med. Cell. Longev. 2013, 2013, 154279. [Google Scholar] [CrossRef] [Green Version]
- Nesci, S.; Trombetti, F.; Pagliarani, A.; Ventrella, V.; Algieri, C.; Tioli, G.; Lenaz, G. Molecular and Supramolecular Structure of the Mitochondrial Oxidative Phosphorylation System: Implications for Pathology. Life 2021, 11, 242. [Google Scholar] [CrossRef] [PubMed]
- Pisonero-Vaquero, S.; Martínez-Ferreras, Á.; García-Mediavilla, M.V.; Martínez-Flórez, S.; Fernández, A.; Benet, M.; Olcoz, J.L.; Jover, R.; González-Gallego, J.; Sánchez-Campos, S. Quercetin ameliorates dysregulation of lipid metabolism genes via the PI3K/AKT pathway in a diet-induced mouse model of nonalcoholic fatty liver disease. Mol. Nutr. Food Res. 2015, 59, 879–893. [Google Scholar] [CrossRef] [Green Version]
- Kobori, M.; Masumoto, S.; Akimoto, Y.; Oike, H. Chronic dietary intake of quercetin alleviates hepatic fat accumulation associated with consumption of a Western-style diet in C57/BL6J mice. Mol. Nutr. Food Res. 2011, 55, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Sipple, J.; Maynard, S.; Mehta, P.A.; Rose, S.R.; Davies, S.M.; Pang, Q. Fanconi Anemia Links Reactive Oxygen Species to Insulin Resistance and Obesity. Antioxid. Redox Signal. 2012, 17, 1083–1098. [Google Scholar] [CrossRef]
- Haddad, P.; Eid, H. The Antidiabetic Potential of Quercetin: Underlying Mechanisms. Curr. Med. Chem. 2017, 24, 355–364. [Google Scholar] [CrossRef]
- Udupa, A.S.; Nahar, P.S.; Shah, S.H.; Kshirsagar, M.J.; Ghongane, B.B. Study of comparative effects of antioxidants on insulin sensitivity in type 2 diabetes mellitus. J. Clin. Diagn. Res. 2012, 6, 1469–1473. [Google Scholar]
- Straub, L.G.; Efthymiou, V.; Grandl, G.; Balaz, M.; Challa, T.D.; Truscello, L.; Horvath, C.; Moser, C.; Rachamin, Y.; Arnold, M.; et al. Antioxidants protect against diabetes by improving glucose homeostasis in mouse models of inducible insulin resistance and obesity. Diabetologia 2019, 62, 2094–2105. [Google Scholar] [CrossRef] [Green Version]
- Damiot, A.; Demangel, R.; Noone, J.; Chery, I.; Zahariev, A.; Normand, S.; Brioche, T.; Crampes, F.; de Glisezinski, I.; Lefai, E.; et al. A nutrient cocktail prevents lipid metabolism alterations induced by 20 days of daily steps reduction and fructose overfeeding: Result from a randomized study. J. Appl. Physiol. 2019, 126, 88–101. [Google Scholar] [CrossRef] [Green Version]
- Davidson, M.H. Antioxidants and lipid metabolism. Implications for the present and direction for the future. Am. J. Cardiol. 1993, 71, 32B–36B. [Google Scholar] [CrossRef]
- Nolan, M.; Knudson, K.; Holz, M.K.; Chaudhury, I. Fanconi anemia and mTOR pathways functionally interact during stalled replication fork recovery. FEBS Lett. 2021, 595, 595–603. [Google Scholar] [CrossRef]
- Guo, F.; Li, J.; Du, W.; Zhang, S.; O’Connor, M.; Thomas, G.; Kozma, S.; Zingarelli, B.; Pang, Q.; Zheng, Y. mTOR regulates DNA damage response through NF-κB-mediated FANCD2 pathway in hematopoietic cells. Leukemia 2013, 27, 2040–2046. [Google Scholar] [CrossRef] [Green Version]
- Shen, C.; Oswald, D.; Phelps, D.; Cam, H.; Pelloski, C.E.; Pang, Q.; Houghton, P.J. Regulation of FANCD2 by the mTOR Pathway Contributes to the Resistance of Cancer Cells to DNA Double-Strand Breaks. Ther. Targets Chem. Biol. 2013, 73, 3393–3401. [Google Scholar] [CrossRef] [Green Version]
- de la Cruz López, K.G.; Toledo Guzmán, M.E.; Sánchez, E.O.; García Carrancá, A. mTORC1 as a Regulator of Mitochondrial Functions and a Therapeutic Target in Cancer. Front. Oncol. 2019, 9, 1373. [Google Scholar] [CrossRef] [Green Version]
- Shyamsunder, P.; Esner, M.; Barvalia, M.; Wu, Y.J.; Loja, T.; Boon, H.B.; Lleonart, M.E.; Verma, R.S.; Krejci, L.; Lyakhovich, A. Impaired mitophagy in Fanconi anemia is dependent on mitochondrial fission. Oncotarget 2016, 7, 58065–58074. [Google Scholar] [CrossRef]
- Cappelli, E.; Ravera, S.; Vaccaro, D.; Cuccarolo, P.; Bartolucci, M.; Panfoli, I.; Dufour, C.; Degan, P. Mitochondrial respiratory complex I defects in Fanconi anemia. Trends Mol. Med. 2013, 19, 513–514. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.-L.; Zhou, Y.-M.; Tang, D.-B.; Zhou, N.; Zheng, W.-W.; Tang, Z.-H.; Duan, C.-W.; Chen, J. Rapamycin ameliorates immune-mediated aplastic anemia by inhibiting the proliferation and metabolism of T cells. Biochem. Biophys. Res. Commun. 2019, 518, 212–218. [Google Scholar] [CrossRef] [PubMed]
- Niu, H.; Qi, W.; Wang, Y.; Xing, L.; Fu, R.; Shao, Z.; Wang, H. Successful sirolimus therapy of an aplastic anemia patient with chronic kidney disease. Medicine 2020, 99, e20669. [Google Scholar] [CrossRef]
- Feng, X.; Lin, Z.; Sun, W.; Hollinger, M.K.; Desierto, M.J.; Keyvanfar, K.; Malide, D.; Muranski, P.; Chen, J.; Young, N.S. Rapamycin is highly effective in murine models of immune-mediated bone marrow failure. Haematologica 2017, 102, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Calap-Quintana, P.; Soriano, S.; Llorens, J.V.; Al-Ramahi, I.; Botas, J.; Moltó, M.D.; Martínez-Sebastián, M.J. TORC1 Inhibition by Rapamycin Promotes Antioxidant Defences in a Drosophila Model of Friedreich’s Ataxia. PLoS ONE 2015, 10, e0132376. [Google Scholar] [CrossRef]
- Jiang, J.; Jiang, J.; Zuo, Y.; Gu, Z. Rapamycin protects the mitochondria against oxidative stress and apoptosis in a rat model of Parkinson’s disease. Int. J. Mol. Med. 2013, 31, 825–832. [Google Scholar] [CrossRef] [Green Version]
- Ropolo, M.; Daga, A.; Griffero, F.; Foresta, M.; Casartelli, G.; Zunino, A.; Poggi, A.; Cappelli, E.; Zona, G.; Spaziante, R.; et al. Comparative analysis of DNA repair in stem and nonstem glioma cell cultures. Mol. Cancer Res. 2009, 7, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Sakai, W.; Sugasawa, K. Importance of finding the bona fide target of the Fanconi anemia pathway. Genes Environ. Off. J. Japanese Environ. Mutagen Soc. 2019, 41, 6. [Google Scholar]
- Degan, P.; Cappelli, E.; Regis, S.; Ravera, S. New Insights and Perspectives in Fanconi Anemia Research. Trends Mol. Med. 2019, 25, 167–170. [Google Scholar] [CrossRef]
- Degan, P.; Cappelli, E.; Longobardi, M.G.; Pulliero, A.; Cuccarolo, P.; Dufour, C.; Ravera, S.; Calzia, D.; Izzotti, A. A Global MicroRNA Profile in Fanconi Anemia: A Pilot Study. Metab. Syndr. Relat. Disord. 2019, 17, 53–59. [Google Scholar] [CrossRef]
- Filocamo, M.; Mazzotti, R.; Corsolini, F.; Stroppiano, M.; Stroppiana, G.; Grossi, S.; Lualdi, S.; Tappino, B.; Lanza, F.; Galotto, S.; et al. Cell Line and DNA Biobank From Patients Affected by Genetic Diseases. Open J. Bioresour. 2014, 1, e2. [Google Scholar] [CrossRef] [Green Version]
- Hanenberg, H.; Batish, S.D.; Pollok, K.E.; Vieten, L.; Verlander, P.C.; Leurs, C.; Cooper, R.J.; Göttsche, K.; Haneline, L.; Clapp, D.W.; et al. Phenotypic correction of primary Fanconi anemia T cells with retroviral vectors as a diagnostic tool. Exp. Hematol. 2002, 30, 410–420. [Google Scholar] [CrossRef]
- Bruno, S.; Ghiotto, F.; Tenca, C.; Mazzarello, A.N.; Bono, M.; Luzzi, P.; Casciaro, S.; Recchia, A.; DeCensi, A.; Morabito, F.; et al. N-(4-hydroxyphenyl)retinamide promotes apoptosis of resting and proliferating B-cell chronic lymphocytic leukemia cells and potentiates fludarabine and ABT-737 cytotoxicity. Leukemia 2012, 26, 2260–2268. [Google Scholar] [CrossRef] [PubMed]
- Hinkle, P.C. P/O ratios of mitochondrial oxidative phosphorylation. Biochim. Biophys. Acta 2005, 1706, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Villa, F.; Bruno, S.; Costa, A.; Li, M.; Russo, M.; Cimino, J.; Altieri, P.; Ruggeri, C.; Gorgun, C.; De Biasio, P.; et al. The Human Fetal and Adult Stem Cell Secretome Can Exert Cardioprotective Paracrine Effects against Cardiotoxicity and Oxidative Stress from Cancer Treatment. Cancers 2021, 13, 3729. [Google Scholar] [CrossRef] [PubMed]
- Shimakata, T.; Fujita, Y.; Kusaka, T. Purification and Characterization of 3-Hydroxyacyl-CoA Dehydrogenase of Mycobacterium smegmatis. J. Biochem. 1979, 86, 1191–1198. [Google Scholar] [CrossRef] [PubMed]
- Ravera, S.; Bartolucci, M.; Cuccarolo, P.; Litamè, E.; Illarcio, M.; Calzia, D.; Degan, P.; Morelli, A.; Panfoli, I. Oxidative stress in myelin sheath: The other face of the extramitochondrial oxidative phosphorylation ability. Free Radic. Res. 2015, 49, 1156–1164. [Google Scholar] [CrossRef]
- Snoeck, H.-W. Mitochondrial regulation of hematopoietic stem cells. Curr. Opin. Cell Biol. 2017, 49, 91–98. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cappelli, E.; Bertola, N.; Bruno, S.; Degan, P.; Regis, S.; Corsolini, F.; Banelli, B.; Dufour, C.; Ravera, S. A Multidrug Approach to Modulate the Mitochondrial Metabolism Impairment and Relative Oxidative Stress in Fanconi Anemia Complementation Group A. Metabolites 2022, 12, 6. https://doi.org/10.3390/metabo12010006
Cappelli E, Bertola N, Bruno S, Degan P, Regis S, Corsolini F, Banelli B, Dufour C, Ravera S. A Multidrug Approach to Modulate the Mitochondrial Metabolism Impairment and Relative Oxidative Stress in Fanconi Anemia Complementation Group A. Metabolites. 2022; 12(1):6. https://doi.org/10.3390/metabo12010006
Chicago/Turabian StyleCappelli, Enrico, Nadia Bertola, Silvia Bruno, Paolo Degan, Stefano Regis, Fabio Corsolini, Barbara Banelli, Carlo Dufour, and Silvia Ravera. 2022. "A Multidrug Approach to Modulate the Mitochondrial Metabolism Impairment and Relative Oxidative Stress in Fanconi Anemia Complementation Group A" Metabolites 12, no. 1: 6. https://doi.org/10.3390/metabo12010006
APA StyleCappelli, E., Bertola, N., Bruno, S., Degan, P., Regis, S., Corsolini, F., Banelli, B., Dufour, C., & Ravera, S. (2022). A Multidrug Approach to Modulate the Mitochondrial Metabolism Impairment and Relative Oxidative Stress in Fanconi Anemia Complementation Group A. Metabolites, 12(1), 6. https://doi.org/10.3390/metabo12010006