
Modelling Metabolic Shifts during Cardiomyocyte Differentiation, Iron Deficiency and Transferrin Rescue Using Human Pluripotent Stem Cells

 and
and
Abstract
:
1. Introduction
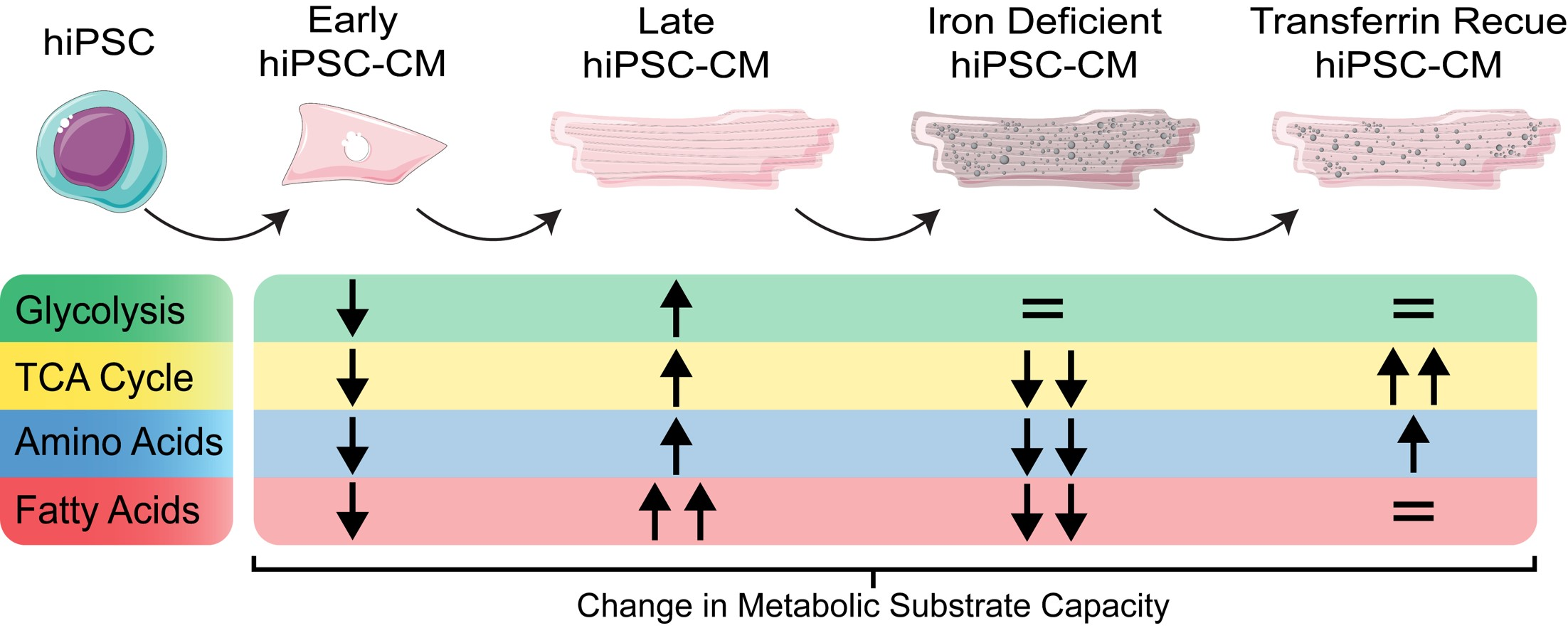
2. Results
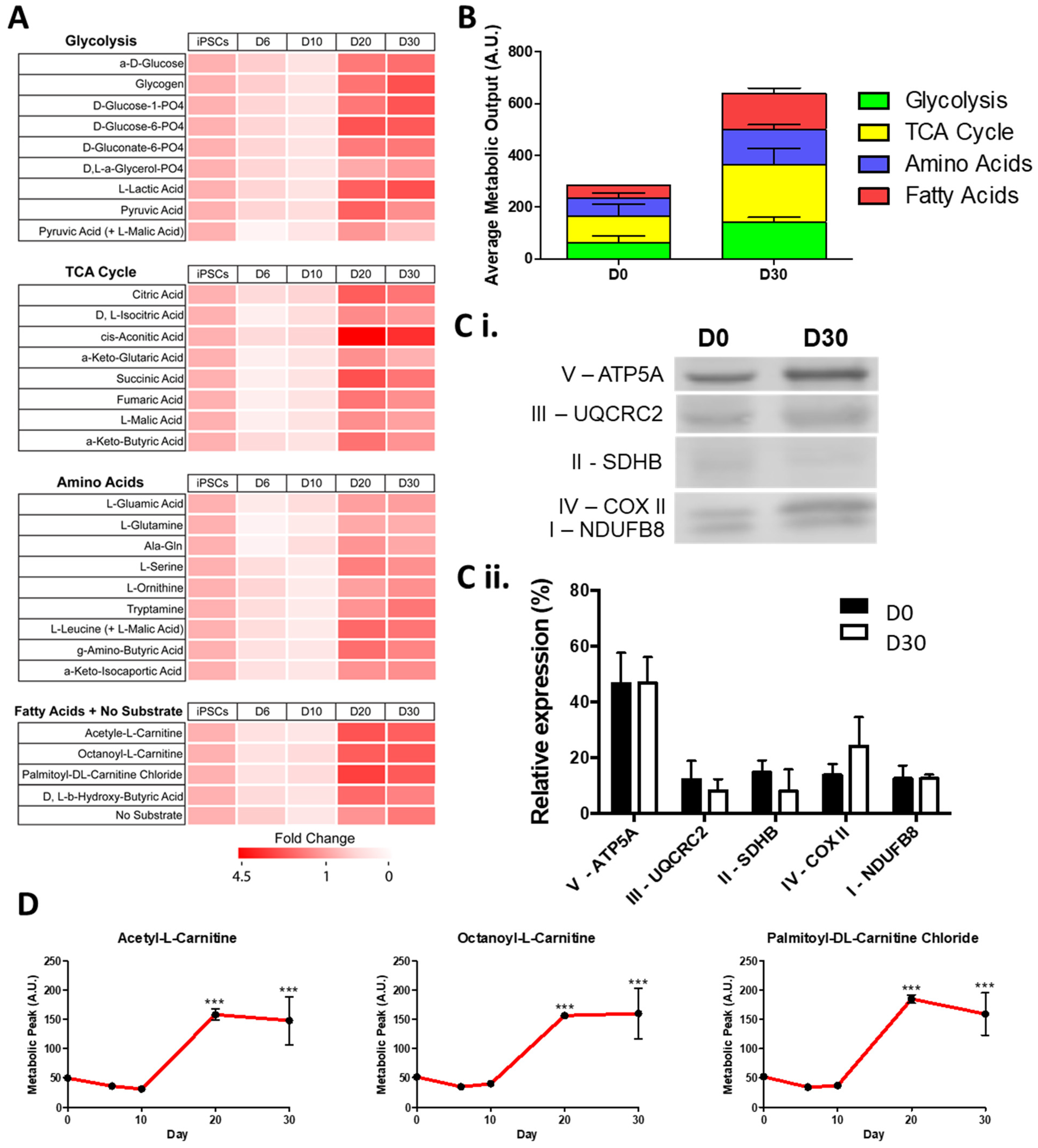
2.1. hiPSC-CMs Undergo Critical Metabolic Changes between Day 10 and Day 20 of Differentiation
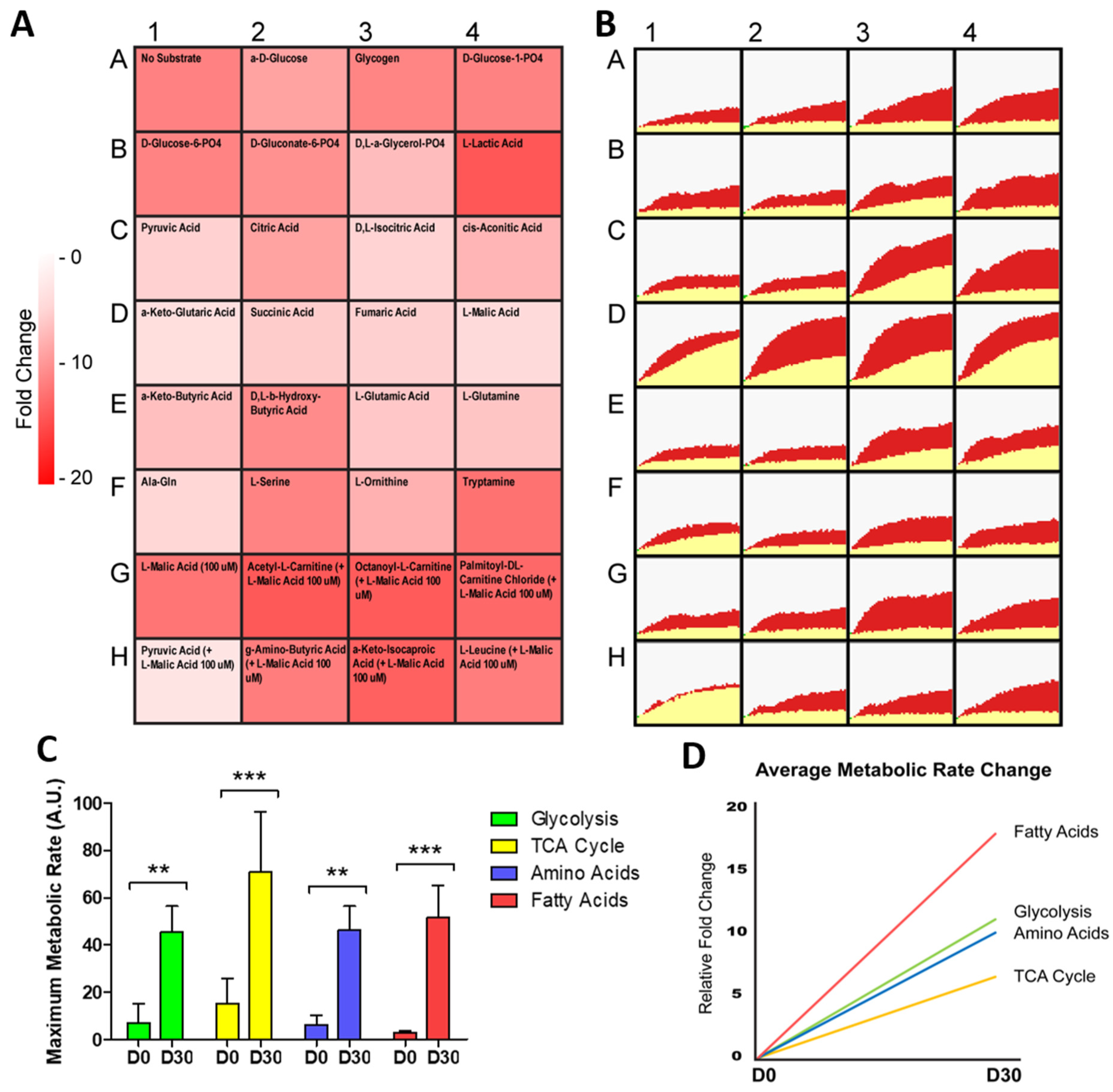
2.2. hiPSC-CMs Differentiation Leads to a Marked Increase in Fatty Acid Metabolism
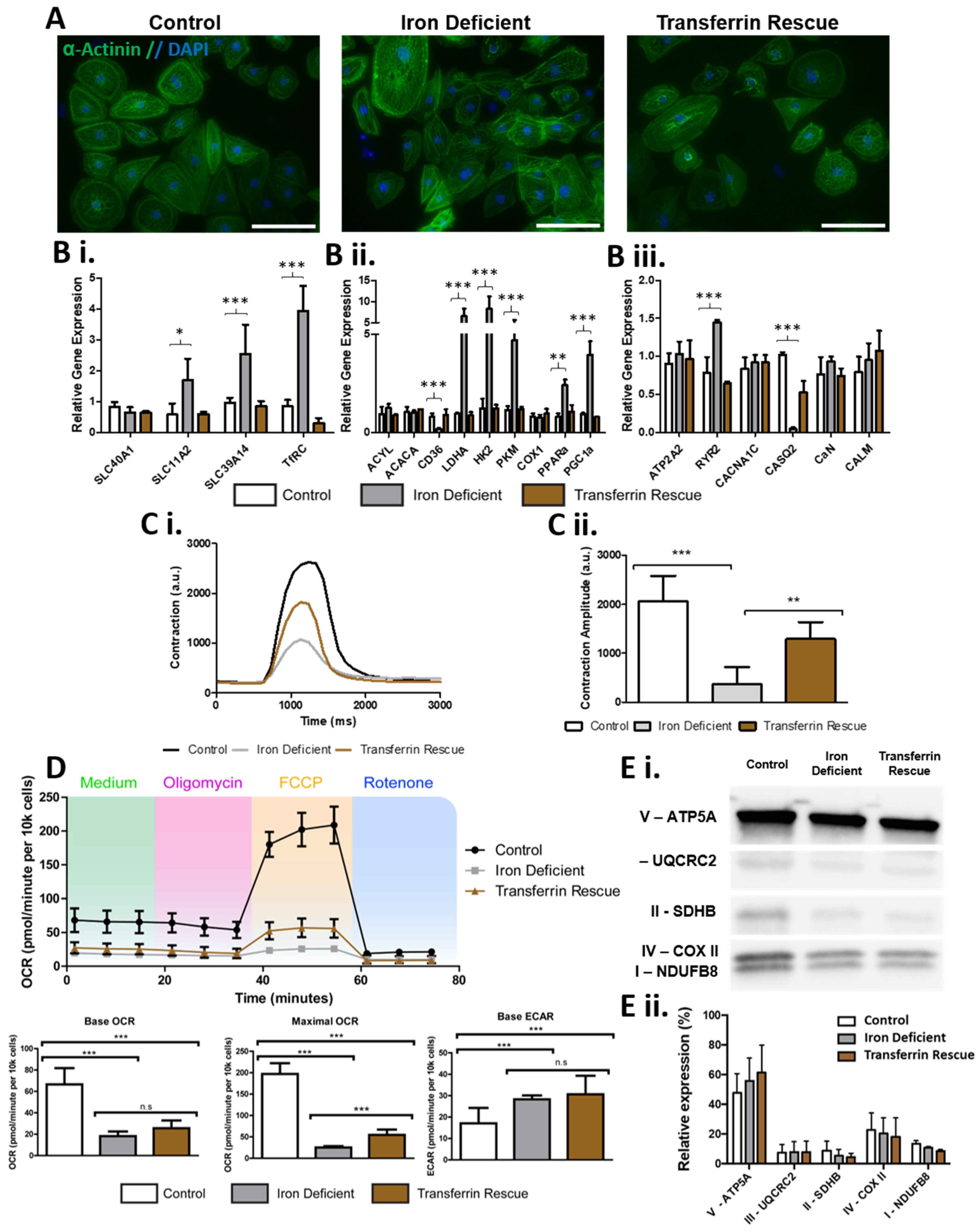
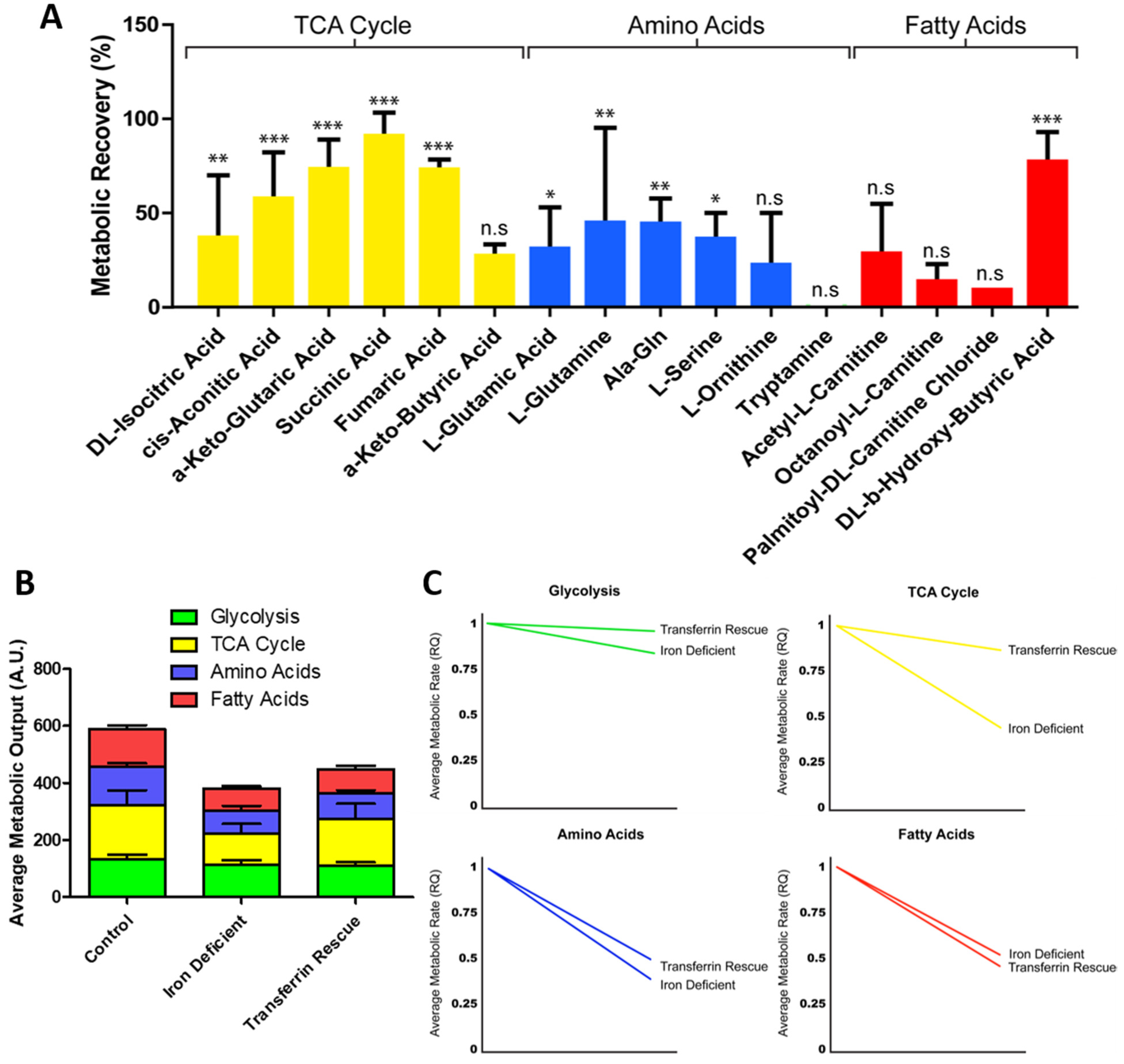
2.3. ID Leads to a Metabolic Shift Only Partially Rescued by Transferrin
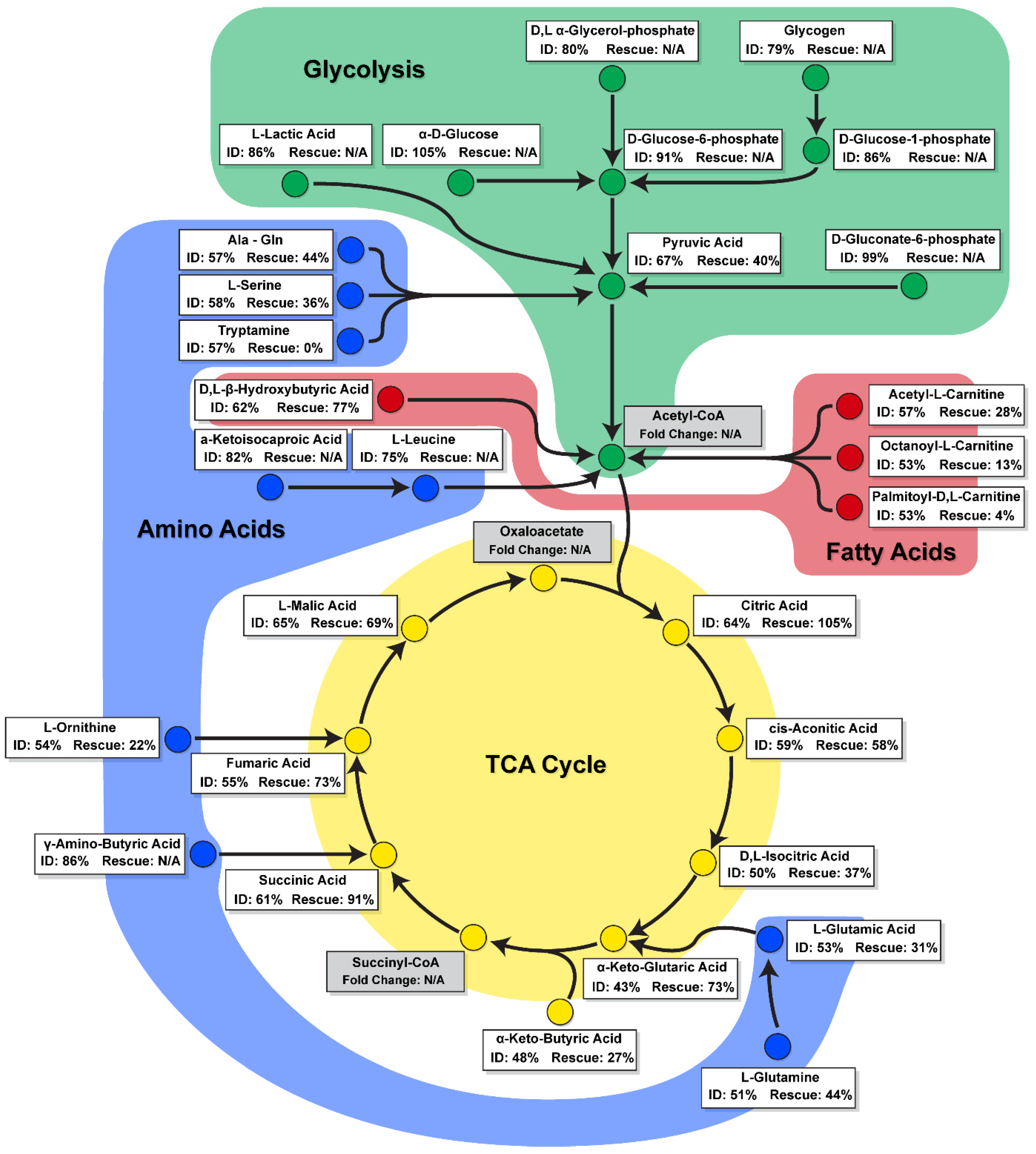
2.4. ID Leads to a Unique Set of Metabolic Substrate Alterations, including Reduced TCA, Amino Acid and Fatty Acid Substrates
2.5. Transferrin Rescue Restores TCA Cycle Substrate Metabolism, but Amino Acid and Fatty Acid Metabolism Remain Perturbed
3. Discussion
4. Methods
4.1. hiPSC Culture and Cardiomyocyte Differentiation
4.2. Immunocytochemistry and Imaging
4.3. Gene and Protein Expression Analysis
4.4. Contraction Analysis
4.5. Metabolic Analysis
4.6. Data Interpretation
4.7. Quantification and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Huss, J.M.; Kelly, D.P. Mitochondrial energy metabolism in heart failure: A question of balance. J. Clin. Investig. 2005, 115, 547–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Epiquereau, J.; Ecaffin, F.; Enovotova, M.; Lemaire, C.; Eveksler, V.; Egarnier, A.; Eventura-Clapier, R.; Ejoubert, F. Mitochondrial dynamics in the adult cardiomyocytes: Which roles for a highly specialized cell? Front. Physiol. 2013, 4, 102. [Google Scholar] [CrossRef] [Green Version]
- Dong, S.; Qian, L.; Cheng, Z.; Chen, C.; Wang, K.; Hu, S.; Zhang, X.; Wu, T. Lactate and Myocadiac Energy Metabolism. Front. Physiol. 2021, 12, 715081. [Google Scholar] [CrossRef]
- Fukushima, A.; Milner, K.; Gupta, A.; Lopaschuk, G. Myocardial Energy Substrate Metabolism in Heart Failure: From Pathways to Therapeutic Targets. Curr. Pharm. Des. 2015, 21, 3654–3664. [Google Scholar] [CrossRef] [PubMed]
- Aubert, G.; Vega, R.B.; Kelly, D.P. Perturbations in the gene regulatory pathways controlling mitochondrial energy production in the failing heart. Biochim. Biophys. Acta (BBA)-Bioenerg. 2013, 1833, 840–847. [Google Scholar] [CrossRef] [Green Version]
- Neubauer, S. The Failing Heart—An Engine Out of Fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casademont, J.; Miró, Ò. Electron Transport Chain Defects in Heart Failure. Heart Fail. Rev. 2002, 7, 131–139. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Ussher, J.R.; Folmes, C.; Jaswal, J.S.; Stanley, W.C. Myocardial Fatty Acid Metabolism in Health and Disease. Physiol. Rev. 2010, 90, 207–258. [Google Scholar] [CrossRef]
- Aubert, G.; Martin, O.J.; Horton, J.L.; Lai, L.; Vega, R.B.; Leone, T.C.; Koves, T.; Gardell, S.J.; Krüger, M.; Hoppel, C.L.; et al. The Failing Heart Relies on Ketone Bodies as a Fuel. Circulation 2016, 133, 698–705. [Google Scholar] [CrossRef] [PubMed]
- Collins-Nakai, R.L.; Noseworthy, D.; Lopaschuk, G.D. Epinephrine increases ATP production in hearts by preferentially increasing glucose metabolism. Am. J. Physiol. Circ. Physiol. 1994, 267, H1862–H1871. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef]
- Smith, J.G.; Owen, T.; Bhagwan, J.; Mosqueira, D.; Scott, E.; Mannhardt, I.; Patel, A.K.; Barriales-Villa, R.; Monserrat, L.; Hansen, A.; et al. Isogenic Pairs of hiPSC-CMs with Hypertrophic Cardiomyopathy/LVNC-Associated ACTC1 E99K Mutation Unveil Differential Functional Deficits. Stem Cell Rep. 2018, 11, 1226–1243. [Google Scholar] [CrossRef] [Green Version]
- Bhagwan, J.R.; Mosqueira, D.; Chairez-Cantu, K.; Mannhardt, I.; Bodbin, S.E.; Bakar, M.; Smith, J.; Denning, C. Isogenic models of hypertrophic cardiomyopathy unveil differential phenotypes and mechanism-driven therapeutics. J. Mol. Cell. Cardiol. 2020, 145, 43–53. [Google Scholar] [CrossRef] [PubMed]
- Mosqueira, D.; Mannhardt, I.; Bhagwan, J.; Lis-Slimak, K.; Katili, P.; Scott, E.; Hassan, M.; Prondzynski, M.; Harmer, S.; Tinker, A.; et al. CRISPR/Cas9 editing in human pluripotent stem cell-cardiomyocytes highlights arrhythmias, hypocontractility, and energy depletion as potential therapeutic targets for hypertrophic cardiomyopathy. Eur. Heart J. 2018, 39, 3879–3892. [Google Scholar] [CrossRef] [PubMed]
- Jahng, J.W.; Zhang, M.; Wu, J.C. The role of metabolism in directed differentiation versus trans-differentiation of cardiomyocytes. Semin. Cell Dev. Biol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Morita, Y.; Tohyama, S. Metabolic Regulation of Cardiac Differentiation and Maturation in Pluripotent Stem Cells: A Lesson from Heart Development. JMA J. 2020, 3, 193–200. [Google Scholar] [CrossRef]
- Correia, C.; Koshkin, A.; Duarte, P.; Hu, D.; Teixeira, A.; Domian, I.; Serra, M.; Alves, P.M. Distinct carbon sources affect structural and functional maturation of cardiomyocytes derived from human pluripotent stem cells. Sci. Rep. 2017, 7, 8590. [Google Scholar] [CrossRef]
- Ordoño, J.; Pérez-Amodio, S.; Ball, K.; Aguirre, A.; Engel, E. Lactate promotes cardiomyocyte dedifferentiation through metabolic reprogramming. BioRxiv Prepr. 2020. [Google Scholar] [CrossRef]
- Rouault, T.A.; Tong, W.-H. Iron–sulphur cluster biogenesis and mitochondrial iron homeostasis. Nat. Rev. Mol. Cell Biol. 2005, 6, 345–351. [Google Scholar] [CrossRef]
- Beard, J.L. Iron Biology in Immune Function, Muscle Metabolism and Neuronal Functioning. J. Nutr. 2001, 131, 568S–580S. [Google Scholar] [CrossRef]
- Brzóska, K.; Meczyńska, S.; Kruszewski, M. Iron-sulfur cluster proteins: Electron transfer and beyond. Acta Biochim. Pol. 2006, 53, 685–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, J.; Vinothkumar, K.R.; Hirst, J. Structure of mammalian respiratory complex I. Nat. Cell Biol. 2016, 536, 354–358. [Google Scholar] [CrossRef] [Green Version]
- Crooks, D.R.; Maio, N.; Lane, A.N.; Jarnik, M.; Higashi, R.M.; Haller, R.G.; Yang, Y.; Fan, T.W.-M.; Linehan, W.M.; Rouault, T.A. Acute loss of iron–sulfur clusters results in metabolic reprogramming and generation of lipid droplets in mammalian cells. J. Biol. Chem. 2018, 293, 8297–8311. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouchani, E.T.; Methner, C.; Buonincontri, G.; Hu, C.-H.; Logan, A.; Sawiak, S.J.; Murphy, M.P.; Krieg, T. Complex I Deficiency Due to Selective Loss of Ndufs4 in the Mouse Heart Results in Severe Hypertrophic Cardiomyopathy. PLoS ONE 2014, 9, e94157. [Google Scholar] [CrossRef] [Green Version]
- Klip, I.T.; Comin-Colet, J.; Voors, A.A.; Ponikowski, P.; Enjuanes, C.; Banasiak, W.; Lok, D.J.; Rosentryt, P.; Torrens, A.; Polonski, L.; et al. Iron deficiency in chronic heart failure: An international pooled analysis. Am. Heart J. 2013, 165, 575–582.e3. [Google Scholar] [CrossRef]
- Van Veldhuisen, D.J.; Anker, S.D.; Ponikowski, P.; MacDougall, I.C. Anemia and iron deficiency in heart failure: Mechanisms and therapeutic approaches. Nat. Rev. Cardiol. 2011, 8, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Anker, S.D.; Comin Colet, J.; Filippatos, G.; Willenheimer, R.; Dickstein, K.; Drexler, H.; Lüscher, T.F.; Bart, B.; Banasiak, W.; Niegowska, J.; et al. Ferric Carboxymaltose in Patients with Heart Failure and Iron Deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef] [Green Version]
- Ponikowski, P.; Van Veldhuisen, D.J.; Comin-Colet, J.; Ertl, G.; Komajda, M.; Mareev, V.; McDonagh, T.; Parkhomenko, A.; Tavazzi, L.; Levesque, V.; et al. Beneficial effects of long-term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur. Heart J. 2015, 36, 657–668. [Google Scholar] [CrossRef]
- Chung, Y.J.; Swietach, P.; Curtis, M.K.; Ball, V.; Robbins, P.A.; Lakhal-Littleton, S. Iron-Deficiency Anemia Results in Transcriptional and Metabolic Remodeling in the Heart Toward a Glycolytic Phenotype. Front. Cardiovasc. Med. 2021, 7, 616920. [Google Scholar] [CrossRef]
- Hoes, M.; Beverborg, N.G.; Kijlstra, J.D.; Kuipers, J.; Swinkels, D.W.; Giepmans, B.; Rodenburg, R.J.; Van Veldhuisen, D.J.; De Boer, R.A.; Van Der Meer, P. Iron deficiency impairs contractility of human cardiomyocytes through decreased mitochondrial function. Eur. J. Heart Fail. 2018, 20, 910–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radogna, F.; Gérard, D.; Dicato, M.; Diederich, M. Assessment of Mitochondrial Cell by Assays. Springer Protoc. Handb. 2021, 2276, 129–141. [Google Scholar] [CrossRef]
- Kordus, R.J.; Hossain, A.; Malter, H.E.; Lavoie, H.A. Mitochondrial metabolic substrate utilization in granulosa cells reflects body mass index and total follicle stimulating hormone dosage in in vitro fertilization patients. J. Assist. Reprod. Genet. 2020, 37, 2743–2756. [Google Scholar] [CrossRef]
- Garcia, D.; Carr, J.F.; Chan, F.; Peterson, A.L.; Ellis, K.A.; Scaffa, A.; Ghio, A.J.; Yao, H.; Dennery, P.A. Short exposure to hyperoxia causes cultured lung epithelial cell mitochondrial dysregulation and alveolar simplification in mice. Pediatr. Res. 2021, 90, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Kuzniewska, B.; Cysewski, D.; Wasilewski, M.; Sakowska, P.; Milek, J.; Kulinski, T.M.; Winiarski, M.; Kozielewicz, P.; Knapska, E.; Dadlez, M.; et al. Mitochondrial protein biogenesis in the synapse is supported by local translation. EMBO Rep. 2020, 21, e48882. [Google Scholar] [CrossRef]
- Gandhirajan, R.K.; Meyer, D.; Sagwal, S.K.; Weltmann, K.-D.; von Woedtke, T.; Bekeschus, S. The amino acid metabolism is essential for evading physical plasma-induced tumour cell death. Br. J. Cancer 2021, 124, 1854–1863. [Google Scholar] [CrossRef]
- Bhalla, K.; Jaber, S.; Reagan, K.; Hamburg, A.; Underwood, K.F.; Jhajharia, A.; Singh, M.; Bhandary, B.; Bhat, S.; Nanaji, N.M.; et al. SIRT3, a metabolic target linked to ataxia-telangiectasia mutated (ATM) gene deficiency in diffuse large B-cell lymphoma. Sci. Rep. 2020, 10, 21159. [Google Scholar] [CrossRef]
- Pantopoulos, K. Iron Metabolism and the IRE/IRP Regulatory System: An Update. Ann. N. Y. Acad. Sci. 2004, 1012, 1–13. [Google Scholar] [CrossRef]
- Yang, X.; Rodriguez, M.L.; Leonard, A.; Sun, L.; Fischer, K.A.; Wang, Y.; Ritterhoff, J.; Zhao, L.; Kolwicz, S.C., Jr.; Pabon, L.; et al. Fatty Acids Enhance the Maturation of Cardiomyocytes Derived from Human Pluripotent Stem Cells. Stem Cell Rep. 2019, 13, 657–668. [Google Scholar] [CrossRef] [Green Version]
- Poon, E.; Luo, X.-L.; Webb, S.; Yan, B.; Zhao, R.; Wu, S.C.M.; Yang, Y.; Zhang, P.; Bai, H.; Shao, J.; et al. The cell surface marker CD36 selectively identifies matured, mitochondria-rich hPSC-cardiomyocytes. Cell Res. 2020, 30, 626–629. [Google Scholar] [CrossRef] [Green Version]
- Sato, Y.; Kobayashi, H.; Higuchi, T.; Shimada, Y.; Ida, H.; Ohashi, T. Metabolomic Profiling of Pompe Disease-Induced Pluripotent Stem Cell-Derived Cardiomyocytes Reveals That Oxidative Stress Is Associated with Cardiac and Skeletal Muscle Pathology. Stem Cells Transl. Med. 2016, 6, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Sayed, N.; Liu, C.; Wu, J.C. Translation of Human-Induced Pluripotent Stem Cells: From Clinical Trial in a Dish to Precision Medicine. J. Am. Coll. Cardiol. 2016, 67, 2161–2176. [Google Scholar] [CrossRef]
- Tohyama, S.; Hattori, F.; Sano, M.; Hishiki, T.; Nagahata, Y.; Matsuura, T.; Hashimoto, H.; Suzuki, T.; Yamashita, H.; Satoh, Y.; et al. Distinct Metabolic Flow Enables Large-Scale Purification of Mouse and Human Pluripotent Stem Cell-Derived Cardiomyocytes. Cell Stem Cell 2013, 12, 127–137. [Google Scholar] [CrossRef] [Green Version]
- Beinert, H.; Kennedy, M.C. Aconitase, a two-faced protein: Enzyme and iron regulatory factor 1 2. FASEB J. 1993, 7, 1442–1449. [Google Scholar] [CrossRef] [PubMed]
- Gray, N.; Pantopoulos, K.; Dandekar, T.; Ackrell, B.A.; Hentze, M. Translational regulation of mammalian and Drosophila citric acid cycle enzymes via iron-responsive elements. Proc. Natl. Acad. Sci. USA 1996, 93, 4925–4930. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-Y.; LaVaute, T.; Iwai, K.; Klausner, R.D.; Rouault, T.A. Identification of a Conserved and Functional Iron-responsive Element in the 5′-Untranslated Region of Mammalian Mitochondrial Aconitase. J. Biol. Chem. 1996, 271, 24226–24230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schalinske, K.L.; Chen, O.S.; Eisenstein, R.S. Implications for iron regulatory proteins as regulators of mitochondrial citrate utilization. J. Biol. Chem. 1998, 273, 3740–3746. [Google Scholar] [CrossRef] [Green Version]
- Oexle, H.; Gnaiger, E.; Weiss, G. Iron-dependent changes in cellular energy metabolism: Influence on citric acid cycle and oxidative phosphorylation. Biochim. Biophys. Acta-Bioenerg. 1999, 1413, 99–107. [Google Scholar] [CrossRef] [Green Version]
- Feyen, D.A.; McKeithan, W.L.; Bruyneel, A.A.; Spiering, S.; Hörmann, L.; Ulmer, B.; Zhang, H.; Briganti, F.; Schweizer, M.; Hegyi, B.; et al. Metabolic Maturation Media Improve Physiological Function of Human iPSC-Derived Cardiomyocytes. Cell Rep. 2020, 32, 107925. [Google Scholar] [CrossRef]
- Kim, T.; Dyck, J.R. The role of CD36 in the regulation of myocardial lipid metabolism. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2016, 1861, 1450–1460. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Peng, Y.; Hang, W.; Nie, J.; Zhou, N.; Wang, D.W. The role of CD36 in cardiovascular disease. Cardiovasc. Res. 2020, cvaa319. [Google Scholar] [CrossRef]
- van der Meer, D.L.M.; Degenhardt, T.; Väisänen, S.; de Groot, P.J.; Heinäniemi, M.; de Vries, S.C.; Müller, M.; Carlberg, C.; Kersten, S. Profiling of promoter occupancy by PPARα in human hepatoma cells via ChIP-chip analysis. Nucleic Acids Res. 2010, 38, 2839–2850. [Google Scholar] [CrossRef] [Green Version]
- Pakzad, K.K.; Tan, J.J.; Anderson, S.; Board, M.; Clarke, K.; Carr, C.A. Metabolic maturation of differentiating cardiosphere-derived cells. Stem Cell Res. 2021, 54, 102422. [Google Scholar] [CrossRef]
- Chung, Y.J.; Luo, A.; Park, K.C.; Loonat, A.A.; Lakhal-Littleton, S.; Robbins, P.A.; Swietach, P. Iron-deficiency anemia reduces cardiac contraction by downregulating RyR2 channels and suppressing SERCA pump activity. JCI Insight 2019, 4, 4. [Google Scholar] [CrossRef] [PubMed]
- Handhle, A.; Ormonde, C.E.; Thomas, N.L.; Bralesford, C.; Williams, A.; Lai, T.; Zissimopoulos, S. Calsequestrin interacts directly with the cardiac ryanodine receptor luminal domain. J. Cell Sci. 2016, 129, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Reinhold, J.; Papadopoulou, C.; Baral, R.; Vassiliou, V.S. Iron deficiency for prognosis in acute coronary syndrome–A systematic review and meta-analysis. Int. J. Cardiol. 2021, 328, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Wenninger, J.; Meinitzer, A.; Holasek, S.; Schnedl, W.J.; Zelzer, S.; Mangge, H.; Herrmann, M.; Enko, D. Associations between tryptophan and iron metabolism observed in individuals with and without iron deficiency. Sci. Rep. 2019, 9, 14548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGranaghan, P.; Kirwan, J.A.; Garcia-Rivera, M.A.; Pieske, B.; Edelmann, F.; Blaschke, F.; Appunni, S.; Saxena, A.; Rubens, M.; Veledar, E.; et al. Lipid Metabolite Biomarkers in Cardiovascular Disease: Discovery and Biomechanism Translation from Human Studies. Metabolites 2021, 11, 621. [Google Scholar] [CrossRef]
- Burridge, P.W.; Matsa, E.; Shukla, P.; Lin, Z.C.; Churko, J.M.; Ebert, A.D.; Lan, F.; Diecke, S.; Huber, B.; Mordwinkin, N.M.; et al. Chemically defined generation of human cardiomyocytes. Nat. Methods 2014, 11, 855–860. [Google Scholar] [CrossRef] [Green Version]
- Sala, L.; van Meer, B.J.; Tertoolen, L.G.; Bakkers, J.; Bellin, M.; Davis, R.P.; Denning, C.N.; Dieben, M.A.; Eschenhagen, T.; Giacomelli, E.; et al. MUSCLEMOTIONA: Versatile Open Software Tool to Quantify Cardiomyocyte and Cardiac Muscle Contraction In Vitro and In Vivo. Circ. Res. 2018, 122, e5–e16. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Primer | Forward Sequence (5′-3′) | Reverse Sequence (5′-3′) |
|---|---|---|
| GAPDH | GTCTCCTCTGACTTCAACAGCG | ACCACCCTGTTGCTGTAGCCAA |
| SLC11A2 | GGACTGTGGGCATACGGTAA | ACACTGGCTCTGATGGCTAC |
| SLC39A14 | TTGCGCTAGCTGGAGGAATG | TGGAATCAAGATGCTGCCCTT |
| SLC40A1 | CTAGTGTCATGACCAGGGCG | CACATCCGATCTCCCCAAGT |
| TFRC | TGGCAGTTCAGAATGATGGA | AGGCTGAACCGGGTATATGA |
| ACACA | TTCACTCCACCTTGTCAGCGGA | GTCAGAGAAGCAGCCCATCACT |
| ACLY | GCTCTGCCTATGACAGCACCAT | GTCCGATGATGGTCACTCCCTT |
| PKM | ATGGCTGACACATTCCTGGAGC | CCTTCAACGTCTCCACTGATCG |
| HK2 | GAGTTTGACCTGGATGTGGTTGC | CCTCCATGTAGCAGGCATTGCT |
| LDHA | GGATCTCCAACATGGCAGCCTT | AGACGGCTTTCTCCCTCTTGCT |
| PGC-1a | AGGCTAGTCCTTCCTCCATGC | GTTGGCTGGTGCCAGTAAGAG |
| COX1 | TCCTTATTCGAGCCGAGCTG | GGGCTGTGACGATAACGTTG |
| RYR2 | CCTTGCCTGAGTGCAGTTG | TTGAGGTATCAACAGGTTGTGG |
| CASQ2 | AGCTTGTGGAGTTTGTGAAG | GGATTGTCAGTGTTGTCCC |
| CALM1 | TGCGGAAGTTAGGAGTGCTG | GCACAGCATAATGGAAGGCG |
| CaN | AGTAACTTTCGAGCCAGCCC | GGGGGTCTGACCACAAGATG |
| ATP2A2 | CTCGGATCCAACACTACAGGTGTTGAATGG | CGGAATTCATGCGCAGTGATAAATTGAC |
| CACNA1C | AAGGCTACCTGGATTGGATCAC | GCCACGTTTTCGGTGTTGAC |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, B.B.; Reinhold, J.; Holmes, T.L.; Moore, J.A.; Cowell, V.; Bernardo, A.S.; Rushworth, S.A.; Vassiliou, V.; Smith, J.G.W. Modelling Metabolic Shifts during Cardiomyocyte Differentiation, Iron Deficiency and Transferrin Rescue Using Human Pluripotent Stem Cells. Metabolites 2022, 12, 9. https://doi.org/10.3390/metabo12010009
Johnson BB, Reinhold J, Holmes TL, Moore JA, Cowell V, Bernardo AS, Rushworth SA, Vassiliou V, Smith JGW. Modelling Metabolic Shifts during Cardiomyocyte Differentiation, Iron Deficiency and Transferrin Rescue Using Human Pluripotent Stem Cells. Metabolites. 2022; 12(1):9. https://doi.org/10.3390/metabo12010009
Chicago/Turabian StyleJohnson, Benjamin B., Johannes Reinhold, Terri L. Holmes, Jamie A. Moore, Verity Cowell, Andreia S. Bernardo, Stuart A. Rushworth, Vassilios Vassiliou, and James G. W. Smith. 2022. "Modelling Metabolic Shifts during Cardiomyocyte Differentiation, Iron Deficiency and Transferrin Rescue Using Human Pluripotent Stem Cells" Metabolites 12, no. 1: 9. https://doi.org/10.3390/metabo12010009
APA StyleJohnson, B. B., Reinhold, J., Holmes, T. L., Moore, J. A., Cowell, V., Bernardo, A. S., Rushworth, S. A., Vassiliou, V., & Smith, J. G. W. (2022). Modelling Metabolic Shifts during Cardiomyocyte Differentiation, Iron Deficiency and Transferrin Rescue Using Human Pluripotent Stem Cells. Metabolites, 12(1), 9. https://doi.org/10.3390/metabo12010009