Altered Urinary Metabolomics in Hereditary Angioedema

Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Participants
2.2. Sample Collection
2.3. Metabolite Extraction
2.4. HPLC-MS/MS Analysis
2.5. Metabolite Identification and Quantification
2.6. Genetic Analysis
2.7. Statistical Analysis
3. Results
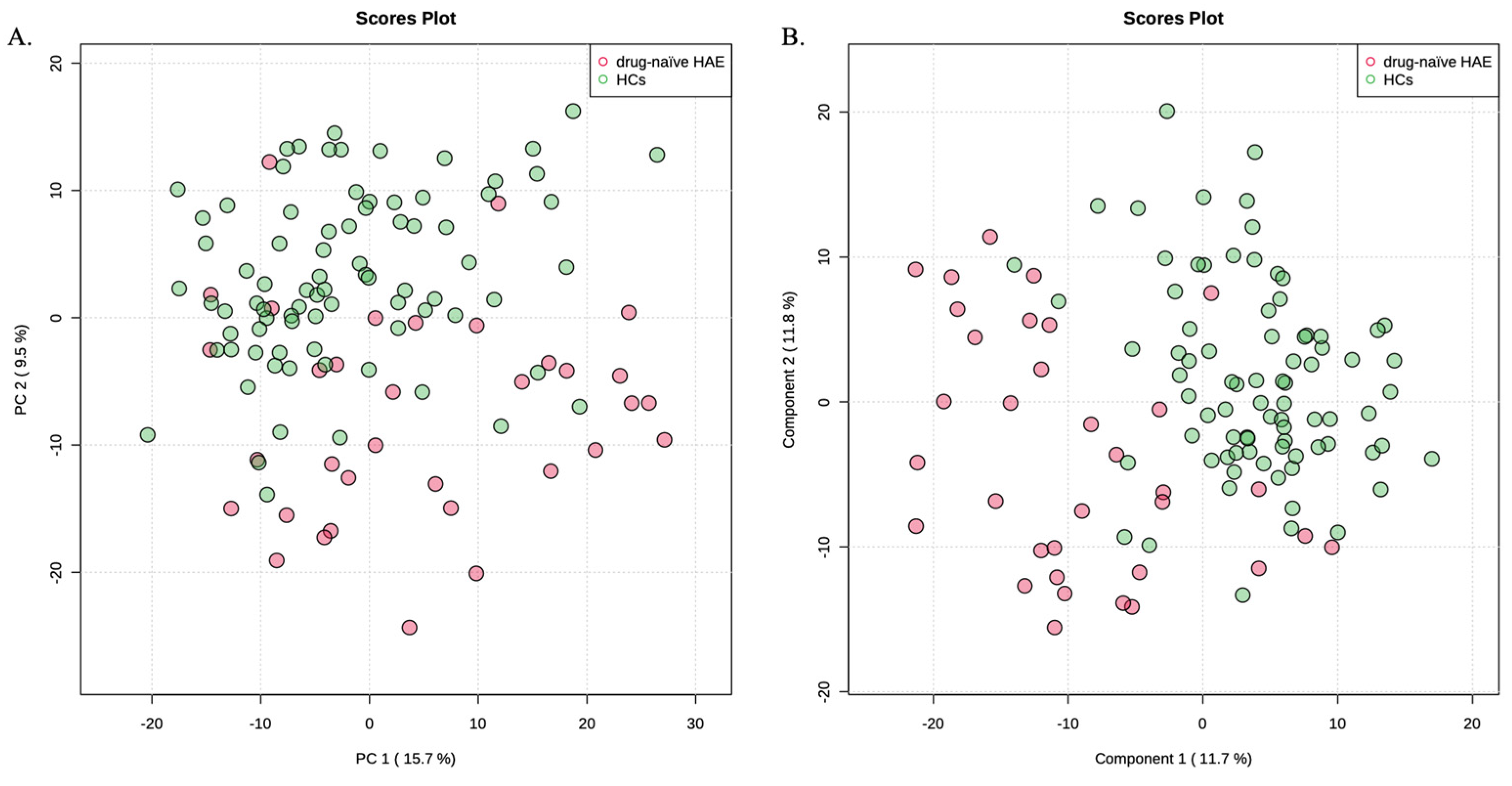
3.1. Metabolic Signatures of HAE Patients
3.2. Metabolic Signatures of HAE Caused by a Premature Stop Codon
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Liu, S.; Xu, Y.; Liu, Y.; Zhi, Y. Hereditary angioedema: A Chinese perspective. Eur. J. Dermatol. 2019, 29, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Maurer, M.; Magerl, M.; Betschel, S.; Aberer, W.; Ansotegui, I.J.; Aygoren-Pursun, E.; Banerji, A.; Bara, N.A.; Boccon-Gibod, I.; Bork, K.; et al. The international WAO/EAACI guideline for the management of hereditary angioedema-The 2021 revision and update. Allergy 2022, 77, 1961–1990. [Google Scholar] [CrossRef] [PubMed]
- Ponard, D.; Gaboriaud, C.; Charignon, D.; Ghannam, A.; Wagenaar-Bos, I.G.A.; Roem, D.; Lopez-Lera, A.; Lopez-Trascasa, M.; Tosi, M.; Drouet, C. SERPING1 mutation update: Mutation spectrum and C1 Inhibitor phenotypes. Hum. Mutat. 2020, 41, 38–57. [Google Scholar] [CrossRef] [PubMed]
- Busse, P.J.; Christiansen, S.C. Hereditary Angioedema. N. Engl. J. Med. 2020, 382, 1136–1148. [Google Scholar] [CrossRef]
- Dewald, G.; Bork, K. Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem. Biophys. Res. Commun. 2006, 343, 1286–1289. [Google Scholar] [CrossRef]
- Bafunno, V.; Firinu, D.; D’Apolito, M.; Cordisco, G.; Loffredo, S.; Leccese, A.; Bova, M.; Barca, M.P.; Santacroce, R.; Cicardi, M.; et al. Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J. Allergy Clin. Immunol. 2018, 141, 1009–1017. [Google Scholar] [CrossRef] [Green Version]
- Bork, K.; Wulff, K.; Steinmuller-Magin, L.; Braenne, I.; Staubach-Renz, P.; Witzke, G.; Hardt, J. Hereditary angioedema with a mutation in the plasminogen gene. Allergy 2018, 73, 442–450. [Google Scholar] [CrossRef]
- Bork, K.; Wulff, K.; Rossmann, H.; Steinmuller-Magin, L.; Braenne, I.; Witzke, G.; Hardt, J. Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the N-terminal cleavage site of bradykinin. Allergy 2019, 74, 2479–2481. [Google Scholar] [CrossRef]
- Ariano, A.; D’Apolito, M.; Bova, M.; Bellanti, F.; Loffredo, S.; D’Andrea, G.; Intrieri, M.; Petraroli, A.; Maffione, A.B.; Spadaro, G.; et al. A myoferlin gain-of-function variant associates with a new type of hereditary angioedema. Allergy 2020, 75, 2989–2992. [Google Scholar] [CrossRef]
- Bork, K.; Wulff, K.; Mohl, B.S.; Steinmuller-Magin, L.; Witzke, G.; Hardt, J.; Meinke, P. Novel hereditary angioedema linked with a heparan sulfate 3-O-sulfotransferase 6 gene mutation. J. Allergy Clin. Immunol. 2021, 148, 1041–1048. [Google Scholar] [CrossRef]
- Firinu, D.; Costanzo, G.; Del Giacco, S.R. Oxidative stress in hereditary angioedema caused by C1 inhibitor deficiency: An interesting finding that deserves further studies. Pol. Arch. Intern. Med. 2020, 130, 73–75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcoleo, F.; Pizzo, M.L.; Misiano, G.; Milano, S.; Romano, G.C.; Muggeo, V.; Cillari, E. The complex alteration in the network of IL-17-type cytokines in patients with hereditary angioedema. Clin. Exp. Med. 2018, 18, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Hollywood, K.; Brison, D.R.; Goodacre, R. Metabolomics: Current technologies and future trends. Proteomics 2006, 6, 4716–4723. [Google Scholar] [CrossRef] [PubMed]
- Olsson, M.; Hellman, U.; Wixner, J.; Anan, I. Metabolomics analysis for diagnosis and biomarker discovery of transthyretin amyloidosis. Amyloid 2021, 28, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Bocca, C.; Le Paih, V.; de la Barca, J.M.C.; Kouassy Nzoughet, J.; Amati-Bonneau, P.; Blanchet, O.; Vedie, B.; Geromin, D.; Simard, G.; Procaccio, V.; et al. A plasma metabolomic signature of Leber hereditary optic neuropathy showing taurine and nicotinamide deficiencies. Hum. Mol. Genet. 2021, 30, 21–29. [Google Scholar] [CrossRef]
- Wang, X.; Han, W.; Yang, J.; Westaway, D.; Li, L. Development of chemical isotope labeling LC-MS for tissue metabolomics and its application for brain and liver metabolome profiling in Alzheimer’s disease mouse model. Anal. Chim Acta 2019, 1050, 95–104. [Google Scholar] [CrossRef]
- Mamas, M.; Dunn, W.B.; Neyses, L.; Goodacre, R. The role of metabolites and metabolomics in clinically applicable biomarkers of disease. Arch. Toxicol. 2011, 85, 5–17. [Google Scholar] [CrossRef]
- Bork, K.; Anderson, J.T.; Caballero, T.; Craig, T.; Johnston, D.T.; Li, H.H.; Longhurst, H.J.; Radojicic, C.; Riedl, M.A. Assessment and management of disease burden and quality of life in patients with hereditary angioedema: A consensus report. Allergy Asthma Clin. Immunol. 2021, 17, 40. [Google Scholar] [CrossRef]
- Want, E.J.; O’Maille, G.; Smith, C.A.; Brandon, T.R.; Uritboonthai, W.; Qin, C.; Trauger, S.A.; Siuzdak, G. Solvent-dependent metabolite distribution, clustering, and protein extraction for serum profiling with mass spectrometry. Anal. Chem. 2006, 78, 743–752. [Google Scholar] [CrossRef]
- Barri, T.; Dragsted, L.O. UPLC-ESI-QTOF/MS and multivariate data analysis for blood plasma and serum metabolomics: Effect of experimental artefacts and anticoagulant. Anal. Chim. Acta 2013, 768, 118–128. [Google Scholar] [CrossRef]
- Luo, P.; Dai, W.; Yin, P.; Zeng, Z.; Kong, H.; Zhou, L.; Wang, X.; Chen, S.; Lu, X.; Xu, G. Multiple reaction monitoring-ion pair finder: A systematic approach to transform nontargeted mode to pseudotargeted mode for metabolomics study based on liquid chromatography-mass spectrometry. Anal. Chem. 2015, 87, 5050–5055. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.Y.; Zhi, Y.X.; Yin, J.; Wang, L.L.; Wen, L.P.; Gu, J.Q.; Guan, K.; Craig, T.; Zhang, H.Y. Mutational spectrum and geno-phenotype correlation in Chinese families with hereditary angioedema. Allergy 2012, 67, 1430–1436. [Google Scholar] [CrossRef] [PubMed]
- Gosswein, T.; Kocot, A.; Emmert, G.; Kreuz, W.; Martinez-Saguer, I.; Aygoren-Pursun, E.; Rusicke, E.; Bork, K.; Oldenburg, J.; Muller, C.R. Mutational spectrum of the C1INH (SERPING1) gene in patients with hereditary angioedema. Cytogenet Genome Res. 2008, 121, 181–188. [Google Scholar] [CrossRef]
- Wang, X.; Lei, S.; Xu, Y.; Liu, S.; Zhi, Y. Mutation update of SERPING1 related to hereditary angioedema in the Chinese population. Hereditas 2022, 159, 28. [Google Scholar] [CrossRef] [PubMed]
- Pappalardo, E.; Caccia, S.; Suffritti, C.; Tordai, A.; Zingale, L.C.; Cicardi, M. Mutation screening of C1 inhibitor gene in 108 unrelated families with hereditary angioedema: Functional and structural correlates. Mol. Immunol. 2008, 45, 3536–3544. [Google Scholar] [CrossRef] [PubMed]
- Guryanova, I.; Suffritti, C.; Parolin, D.; Zanichelli, A.; Ishchanka, N.; Polyakova, E.; Belevtsev, M.; Perego, F.; Cicardi, M.; Zharankova, Y.; et al. Hereditary angioedema due to C1 inhibitor deficiency in Belarus: Epidemiology, access to diagnosis and seven novel mutations in SERPING1 gene. Clin. Mol. Allergy 2021, 19, 3. [Google Scholar] [CrossRef] [PubMed]
- Hashimura, C.; Kiyohara, C.; Fukushi, J.I.; Hirose, T.; Ohsawa, I.; Tahira, T.; Horiuchi, T. Clinical and genetic features of hereditary angioedema with and without C1-inhibitor (C1-INH) deficiency in Japan. Allergy 2021, 76, 3529–3534. [Google Scholar] [CrossRef] [PubMed]
- Obtulowicz, K.; Ksi, A.T.; Bogdali, A.; Dyga, W.; Czarnobilska, E.; Juchacz, A. Genetic variants of SERPING1 gene in Polish patients with hereditary angioedema due to C1 inhibitor deficiency. Cent. Eur. J. Immunol. 2020, 45, 301–309. [Google Scholar] [CrossRef]
- Monteiro, M.S.; Carvalho, M.; Bastos, M.L.; de Pinho, P.G. Metabolomics analysis for biomarker discovery: Advances and challenges. Curr. Med. Chem. 2013, 20, 257–271. [Google Scholar] [CrossRef]
- Kim, Y.J.; Ryu, H.M.; Choi, J.Y.; Cho, J.H.; Kim, C.D.; Park, S.H.; Kim, Y.L. Hypoxanthine causes endothelial dysfunction through oxidative stress-induced apoptosis. Biochem. Biophys Res. Commun. 2017, 482, 821–827. [Google Scholar] [CrossRef]
- George, J.; Struthers, A. The role of urate and xanthine oxidase in vascular oxidative stress: Future directions. Ther. Clin. Risk Manag. 2009, 5, 799–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olszanecki, R.; Kozlovski, V.I.; Chlopicki, S.; Gryglewski, R.J. Paradoxical augmentation of bradykinin-induced vasodilatation by xanthine/xanthine oxidase-derived free radicals in isolated guinea pig heart. J. Physiol. Pharmacol. 2002, 53, 689–699. [Google Scholar]
- Suwannasom, N.; Kao, I.; Pruss, A.; Georgieva, R.; Baumler, H. Riboflavin: The Health Benefits of a Forgotten Natural Vitamin. Int. J. Mol. Sci. 2020, 21, 950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, I.M.; McGhee, C.N.; Sherwin, T. Beneficial effect of the antioxidant riboflavin on gene expression of extracellular matrix elements, antioxidants and oxidases in keratoconic stromal cells. Clin. Exp. Optom. 2014, 97, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Li, W.; Lu, X.; Zhao, X. Riboflavin alleviates cardiac failure in Type I diabetic cardiomyopathy. Heart Int. 2011, 6, e21. [Google Scholar] [CrossRef] [Green Version]
- Del Giacco, S.R.; Firinu, D.; Minciullo, P.L.; Barca, M.P.; Manconi, P.E.; Tartarisco, G.; Cristani, M.; Saija, A.; Gangemi, S. Oxidative stress markers in patients with hereditary angioedema. Arch. Med Sci. 2019, 15, 92–98. [Google Scholar] [CrossRef] [Green Version]
- Obtulowicz, K.; Goralska, J.; Bogdali, A.; Dyga, W.; Obtulowicz, A.; Myszkowska, D.; Ziemianin, M.; Gruca, A.; Solnica, B.; Czarnobilska, E. Bradykinin and oxidative stress in patients with hereditary angioedema due to C1 inhibitor deficiency. Pol. Arch. Intern. Med. 2020, 130, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Toyosawa, T.; Suzuki, M.; Kodama, K.; Araki, S. Highly purified vitamin B2 presents a promising therapeutic strategy for sepsis and septic shock. Infect. Immun. 2004, 72, 1820–1823. [Google Scholar] [CrossRef]
- Toyosawa, T.; Suzuki, M.; Kodama, K.; Araki, S. Effects of intravenous infusion of highly purified vitamin B2 on lipopolysaccharide-induced shock and bacterial infection in mice. Eur. J. Pharmacol. 2004, 492, 273–280. [Google Scholar] [CrossRef]
- Joseph, K.; Tholanikunnel, B.G.; Kaplan, A.P. Cytokine and estrogen stimulation of endothelial cells augments activation of the prekallikrein-high molecular weight kininogen complex: Implications for hereditary angioedema. J. Allergy Clin. Immunol. 2017, 140, 170–176. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, A.P.; Joseph, K. Pathogenesis of Hereditary Angioedema: The Role of the Bradykinin-Forming Cascade. Immunol. Allergy Clin. N. Am. 2017, 37, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Seekamp, A.; Hultquist, D.E.; Till, G.O. Protection by vitamin B2 against oxidant-mediated acute lung injury. Inflammation 1999, 23, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Noll, T.; Holschermann, H.; Koprek, K.; Gunduz, D.; Haberbosch, W.; Tillmanns, H.; Piper, H.M. ATP reduces macromolecule permeability of endothelial monolayers despite increasing [Ca2+]i. Am. J. Physiol. 1999, 276, H1892–H1901. [Google Scholar] [CrossRef] [PubMed]
- Brettner, F.; Chappell, D.; Nebelsiek, T.; Hauer, D.; Schelling, G.; Becker, B.F.; Rehm, M.; Weis, F. Preinterventional hydrocortisone sustains the endothelial glycocalyx in cardiac surgery. Clin. Hemorheol. Microcirc. 2019, 71, 59–70. [Google Scholar] [CrossRef] [PubMed]
- Chappell, D.; Jacob, M.; Hofmann-Kiefer, K.; Bruegger, D.; Rehm, M.; Conzen, P.; Welsch, U.; Becker, B.F. Hydrocortisone preserves the vascular barrier by protecting the endothelial glycocalyx. Anesthesiology 2007, 107, 776–784. [Google Scholar] [CrossRef] [Green Version]
- Whitehouse, M.W. Anti-inflammatory glucocorticoid drugs: Reflections after 60 years. Inflammopharmacology 2011, 19, 1–19. [Google Scholar] [CrossRef]
- Thon, V.; Harle, P.; Scholmerich, J.; Kuklinek, P.; Lokaj, J.; Straub, R.H. Lack of dehydroepiandrosterone in type I and II hereditary angioedema and role of danazol in steroid hormone conversion. Allergy 2007, 62, 1320–1325. [Google Scholar] [CrossRef]
- Amrani, N.; Sachs, M.S.; Jacobson, A. Early nonsense: mRNA decay solves a translational problem. Nat. Rev. Mol. Cell Biol. 2006, 7, 415–425. [Google Scholar] [CrossRef]
- Xu, Y.Y.; Gu, J.Q.; Zhi, Y.X. Hereditary angioedema caused by a premature stop codon mutation in the SERPING1 gene. Clin. Transl Allergy 2020, 10, 53. [Google Scholar] [CrossRef]


{kind=link}
| Drug-Naïve HAE | Healthy Controls | |
|---|---|---|
| n | 34 | 82 |
| Female | 25 (73.5) | 54 (65.9) |
| Male | 9 (26.5) | 28 (34.1) |
| Age (y) | 44.2 ± 10.7 | 40.3 ± 10.5 |
| Hypertension | 1 (2.9) | 3 (3.6) |
| Skin edema | 31 (91.2) | NA |
| Gastrointestinal edema | 20 (58.8) | NA |
| Laryngeal edema | 24 (70.6) | NA |
| Annual attack frequency | 5 [1,2,3,4,5,6,7,8,9,10,11,12,13,14] | NA |
| Disease severity | 7.3 ± 1.5 | NA |
| Metabolite | Class | HMDB ID | QC, RSD% | FC | p-Value | q-Value | VIP Value |
|---|---|---|---|---|---|---|---|
| Quinone | Ketones | HMDB0003364 | 33.37 | 2.65 | 0.0000 | 0.0008 | 2.21 |
| N-Methylhydantoin | Organoheterocyclic compounds | HMDB0003646 | 10.51 | 1.2 | 0.0002 | 0.0042 | 2.20 |
| 2-Aminooctanoic acid | Organic acid | HMDB0000991 | 9.65 | 0.4 | 0.0000 | 0.0001 | 2.12 |
| 2,6-Dimethoxybenzoic acid | Benzoic acid | HMDB0029273 | 15.03 | 1.83 | 0.0015 | 0.0178 | 2.11 |
| Thymine | Nucleotide | HMDB0000262 | 25.15 | 2.37 | 0.0000 | 0.0013 | 2.10 |
| Propionylcholine | Cholines | HMDB0013305 | 11.72 | 0.41 | 0.0000 | 0.0001 | 2.08 |
| 1-Methylxanthine | Purines and purine derivatives | HMDB0010738 | 4.28 | 0.27 | 0.0000 | 0.0000 | 2.04 |
| D-Glyceraldehyde 3-phosphate | Organic acid | HMDB0001112 | 8.98 | 1.79 | 0.0029 | 0.0298 | 2.00 |
| O-Acetyl-L-carnitine | Carnitine | HMDB0000201 | 9.58 | 0.33 | 0.0000 | 0.0008 | 2.00 |
| Hypoxanthine | Nucleotide | HMDB0000157 | 8.52 | 0.53 | 0.0000 | 0.0013 | 1.95 |
| Acetylcarnitine | Carnitine | HMDB0000201 | 7.71 | 0.36 | 0.0000 | 0.0009 | 1.94 |
| Trehalose 6-phosphate | Carbohydrates | HMDB0001124 | 10.28 | 1.95 | 0.0047 | 0.0417 | 1.90 |
| 1,4-Dihydro-1-Methyl-4-Oxo-3-Pyridinecarboxamide | Pyridine | HMDB0004194 | 3.28 | 0.55 | 0.0000 | 0.0011 | 1.87 |
| 2,4-Dihydroxybenzoic Acid | Benzoic acid | HMDB0029666 | 17.12 | 0.54 | 0.0000 | 0.0008 | 1.86 |
| L-Octanoylcarnitine | Carnitine | HMDB0000791 | 3.12 | 0.44 | 0.0000 | 0.0009 | 1.86 |
| Oxypurinol | Nucleotide | HMDB0000786 | 4.18 | 0.54 | 0.0000 | 0.0011 | 1.86 |
| DL-Citrulline | Amino acid | HMDB0000904 | 5.15 | 2 | 0.0054 | 0.0440 | 1.85 |
| Carnitine-C8 | Carnitine | NA | 3.16 | 0.44 | 0.0000 | 0.0009 | 1.84 |
| Xanthine | Nucleotide | HMDB0000292 | 3.69 | 0.56 | 0.0000 | 0.0013 | 1.81 |
| L-Alanine | Amino acid | HMDB0000161 | 5.19 | 1.38 | 0.0053 | 0.0440 | 1.78 |
| Adrenochrome | Organoheterocyclic compounds | HMDB0012884 | 2.91 | 0.44 | 0.0000 | 0.0008 | 1.77 |
| Hippuric acid | Organic acid | HMDB0000714 | 2.59 | 0.44 | 0.0000 | 0.0008 | 1.77 |
| Taurochenodeoxycholic acid | Bile acids | NA | 6.20 | 0.55 | 0.0001 | 0.0017 | 1.77 |
| D-3-Phenyllactic acid | Organic acid | HMDB0000563 | 3.43 | 1.76 | 0.0053 | 0.0440 | 1.75 |
| Glucarate O-Phosphoric Acid | Carbohydrates | NA | 23.71 | 1.48 | 0.0064 | 0.0495 | 1.70 |
| Vitamin B2 | Vitamins | HMDB0000244 | 8.20 | 0.35 | 0.0000 | 0.0013 | 1.70 |
| Hexanoylcarnitine | Carnitine | HMDB0000705 | 3.83 | 0.55 | 0.0000 | 0.0013 | 1.69 |
| L-Homocystine | Amino acid | HMDB0000676 | 25.03 | 0.64 | 0.0000 | 0.0011 | 1.69 |
| Carnitine-C6 | Carnitine | NA | 4.40 | 0.55 | 0.0001 | 0.0016 | 1.67 |
| Decanoylcarnitine | Carnitine | HMDB0000651 | 5.00 | 0.52 | 0.0001 | 0.0016 | 1.67 |
| Dodecanoylcarnitine | Carnitine | HMDB0002250 | 3.92 | 0.55 | 0.0003 | 0.0050 | 1.67 |
| Carnitine-C12 | Carnitine | NA | 4.96 | 0.55 | 0.0003 | 0.0050 | 1.65 |
| Isovalerylcarnitine | Carnitine | HMDB0000688 | 3.59 | 0.55 | 0.0001 | 0.0031 | 1.65 |
| 5-Aminosalicylate | Organic acid | HMDB0014389 | 22.26 | 0.56 | 0.0001 | 0.0017 | 1.64 |
| Carnitine-C3 | Carnitine | NA | 3.50 | 0.49 | 0.0004 | 0.0063 | 1.63 |
| Carnitine-C5 | Carnitine | NA | 3.47 | 0.55 | 0.0001 | 0.0034 | 1.63 |
| Hydrocortisone | Hormones | HMDB0000063 | 4.42 | 0.44 | 0.0001 | 0.0016 | 1.62 |
| 2-Methylbutyroylcarnitine | Fatty acyls | HMDB0000378 | 2.46 | 0.56 | 0.0002 | 0.0035 | 1.61 |
| Cholate | Bile acids | NA | 7.07 | 2.74 | 0.0053 | 0.0440 | 1.61 |
| N-Propionylglycine | Amino acid | HMDB0000783 | 10.05 | 1.35 | 0.0057 | 0.0450 | 1.57 |
| 3-Oxo-7alpha,12alpha-hydroxy-5beta-cholanoic acid | Bile acids | NA | 18.39 | 1.67 | 0.0047 | 0.0417 | 1.56 |
| Cinnamoylglycine | Amino acid | HMDB0011621 | 4.36 | 0.57 | 0.0002 | 0.0042 | 1.56 |
| O-Acetyl-L-homoserine | Amino acid | HMDB0029423 | 4.55 | 0.49 | 0.0001 | 0.0029 | 1.55 |
| L-Carnitine | Carnitine | HMDB0000062 | 5.97 | 0.52 | 0.0002 | 0.0042 | 1.52 |
| Propionyl-L-carnitine | Carnitine | HMDB0000824 | 4.58 | 0.5 | 0.0003 | 0.0050 | 1.52 |
| L-Alanyl-L-Lysine | Amino acid | HMDB0028692 | 6.47 | 0.52 | 0.0003 | 0.0056 | 1.49 |
| Cortisone | Others | HMDB0002802 | 4.79 | 0.62 | 0.0002 | 0.0042 | 1.43 |
| Methylmalonate | Organic acid | HMDB0000202 | 4.56 | 0.59 | 0.0008 | 0.0110 | 1.43 |
| 3-Hydroxy-3-Methylpentane-1,5-Dioic acid | Fatty acyls | NA | 7.25 | 0.52 | 0.0003 | 0.0051 | 1.39 |
| DL-Norepinephrine | Hormones | NA | 39.22 | 0.57 | 0.0005 | 0.0072 | 1.34 |
| Succinic acid | TCA cycle | HMDB0000254 | 2.83 | 0.61 | 0.0011 | 0.0143 | 1.34 |
| Nonanoic acid | Fatty acyls | HMDB0000847 | 21.30 | 1.24 | 0.0055 | 0.0443 | 1.33 |
| 3-OH-anthranilate | Organic acid | NA | 16.94 | 0.59 | 0.0000 | 0.0013 | 1.31 |
| Deoxyguanosine | Nucleotide | HMDB0000085 | 9.15 | 0.65 | 0.0002 | 0.0042 | 1.31 |
| Uric acid | Organic acid | HMDB0000289 | 4.89 | 0.66 | 0.0001 | 0.0016 | 1.30 |
| Pantetheine | Amino acid | HMDB0003426 | 11.55 | 0.63 | 0.0005 | 0.0072 | 1.27 |
| Guanethidine | Organic acid | NA | 44.51 | 0.49 | 0.0000 | 0.0008 | 1.25 |
| D-Glucosamine 6-phosphate | Sugar acids | HMDB0001254 | 16.26 | 0.49 | 0.0004 | 0.0057 | 1.16 |
| Indoxylsulfuric acid | Organic acid | HMDB0000682 | 4.06 | 0.69 | 0.0028 | 0.0297 | 1.16 |
| CMPF | Fatty acyls | HMDB0061112 | 5.03 | 0.57 | 0.0019 | 0.0218 | 1.14 |
| Hydroquinone | Phenols | HMDB0002434 | 4.92 | 0.6 | 0.0009 | 0.0121 | 1.12 |
| p-cresol | Phenols | HMDB0001858 | 5.38 | 0.69 | 0.0054 | 0.0440 | 1.11 |
| 3-Hydroxy-butyryl carnitine | Carnitine | NA | 6.95 | 0.3 | 0.0010 | 0.0132 | 1.10 |
| Isocitrate | Organic acid | HMDB0001874 | 11.40 | 0.67 | 0.0040 | 0.0375 | 1.09 |
| Tauroursodeoxycholic acid Dihydrate | Bile acids | NA | 7.16 | 0.48 | 0.0036 | 0.0339 | 1.09 |
| 4-Aminobenzoate | Benzoic acid | HMDB0004992 | 17.89 | 0.61 | 0.0005 | 0.0070 | 1.08 |
| Tauro-alpha-Muricholic acid | Bile acids | NA | 34.87 | 0.54 | 0.0016 | 0.0190 | 1.08 |
| Taurocholic acid | Bile acids | HMDB0000036 | 7.35 | 0.6 | 0.0008 | 0.0116 | 1.08 |
| Furfural | Aldehydes | HMDB0032914 | 27.54 | 0.64 | 0.0001 | 0.0017 | 1.06 |
| O-Anisic Acid | Benzoic acid | HMDB0032604 | 5.27 | 0.62 | 0.0020 | 0.0220 | 1.05 |
| Biotin | Vitamins | HMDB0000030 | 7.32 | 0.67 | 0.0048 | 0.0419 | 1.03 |
| L-Glutamine O-Hexside | Amino acid | NA | 11.32 | 0.57 | 0.0023 | 0.0250 | 1.03 |
| Pyridoxamine | Pyridine | HMDB0001431 | 16.36 | 0.7 | 0.0014 | 0.0173 | 1.01 |
| Group | Sex | Age (y) | DNA Change | Protein | HAE Type | References |
|---|---|---|---|---|---|---|
| A | F | 40 | c.1A>G | p.(Met1Val) | 1 | [23] |
| A | F | 45 | c.816_818del | p.(Asn272del) | 1 | [3] |
| A | F | 32 | c.1223A>G | p.(Asp408Gly) | 1 | [24] |
| A | M | 28 | c.1289T>C | p.(Leu430Pro) | 1 | [25] |
| A | M | 28 | c.1289T>C | p.(Leu430Pro) | 1 | [25] |
| A | M | 31 | c.1289T>G | p.(Leu430Arg) | 1 | [24] |
| B | F | 51 | c.172_181del | p.(Pro58Argfs * 18) | 1 | [24] |
| B | F | 47 | c.197dup | p.(Thr67Aspfs * 15) | 1 | [24] |
| B | F | 56 | c.635dup | p.(Phe213Leufs * 44) | 1 | [24] |
| B | F | 45 | c.733_736dup | p.(Ser246Lysfs * 12) | 1 | [24] |
| B | F | 46 | c.748_749del | p.(Val250Profs * 6) | 1 | VCV001329454.1 |
| B | M | 29 | c.897del | p.(Trp299 *) | 1 | This study |
| B | F | 45 | c.941_942insTC | p.(Phe315Profs * 7) | 1 | [24] |
| B | F | 40 | c.944del | p.(Phe315Serfs * 6) | 1 | [3] |
| B | M | 44 | c.1051del | p.(His351Thrfs * 3) | 1 | [24] |
| B | F | 47 | c.1051del | p.(His351Thrfs * 3) | 1 | [24] |
| B | F | 49 | c.1106del | p.(Asp369Alafs * 28) | 1 | [26] |
| B | M | 47 | c.1480C>T | p.(Arg494 *) | 1 | [27] |
| - | F | 48 | c.1396C>A | p.(Arg466Ser) | 2 | [28] |
| - | F | 49 | c.1396C>A | p.(Arg466Ser) | 2 | [28] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, X.; Zhi, Y. Altered Urinary Metabolomics in Hereditary Angioedema. Metabolites 2022, 12, 1140. https://doi.org/10.3390/metabo12111140
Wang X, Zhi Y. Altered Urinary Metabolomics in Hereditary Angioedema. Metabolites. 2022; 12(11):1140. https://doi.org/10.3390/metabo12111140
Chicago/Turabian StyleWang, Xue, and Yuxiang Zhi. 2022. "Altered Urinary Metabolomics in Hereditary Angioedema" Metabolites 12, no. 11: 1140. https://doi.org/10.3390/metabo12111140
APA StyleWang, X., & Zhi, Y. (2022). Altered Urinary Metabolomics in Hereditary Angioedema. Metabolites, 12(11), 1140. https://doi.org/10.3390/metabo12111140