
Qualitative and Quantitative Effects of Fatty Acids Involved in Heart Diseases

 ,
,
Abstract
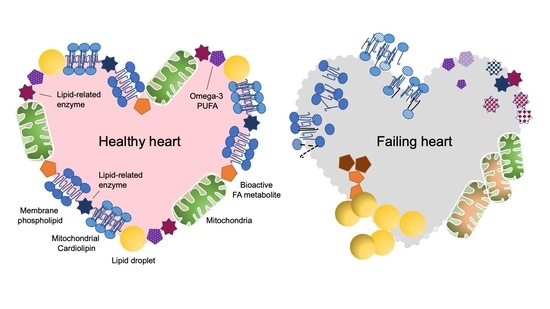
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. FAs Control the Heart as an Energy Source
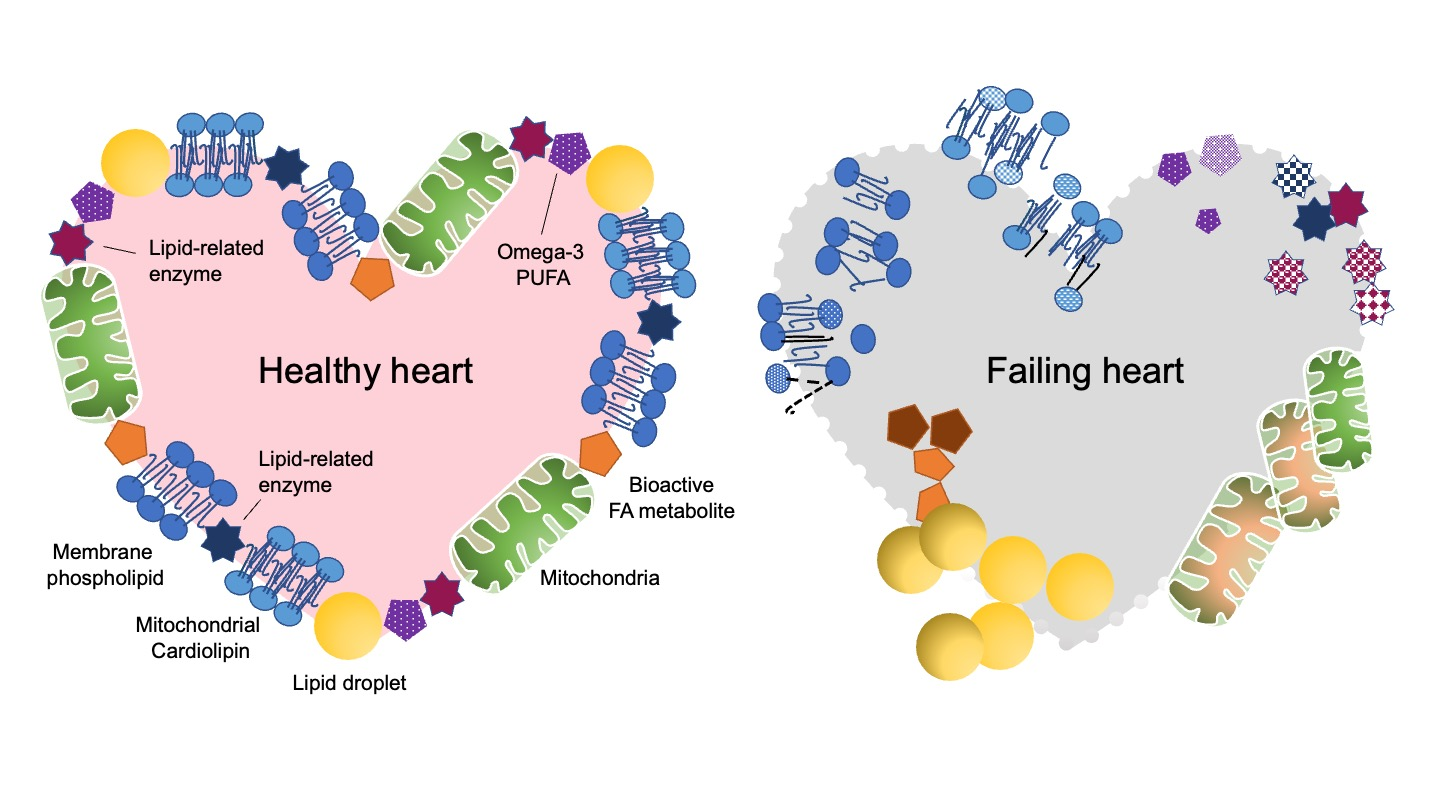
2.1. Cardiac Lipotoxicity Due to the Imbalance between Supply and Oxidation of FAs
2.2. Oversupplied FAs Are Stored in LDs
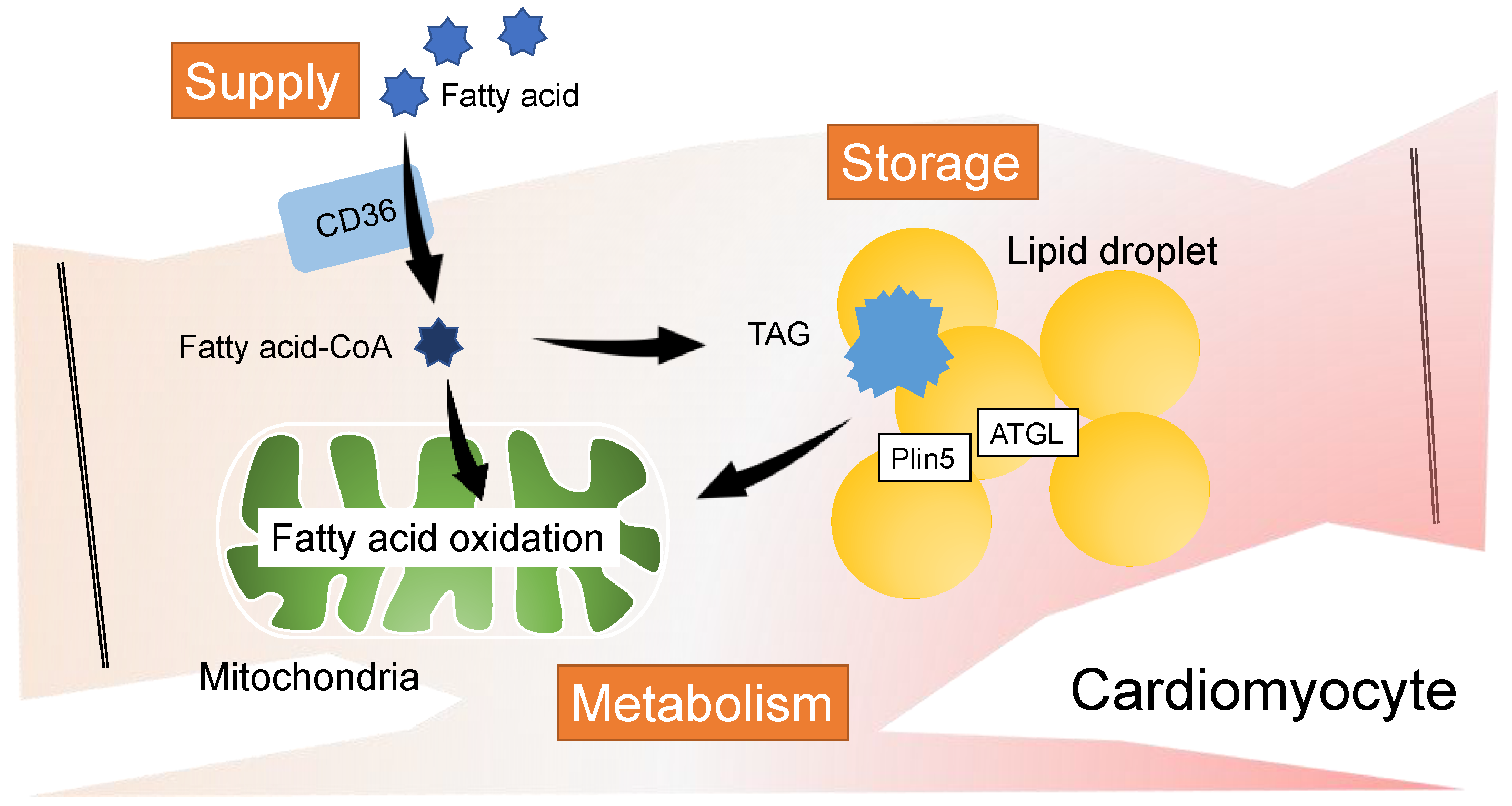
3. FAs Control the Heart as a Component of Membrane Phospholipids
3.1. Balance of FA Saturation in Membrane Phospholipids
3.2. FA Remodeling in Cardiolipin and Heart Disease
4. FAs Control the Heart as Bioactive Mediators
4.1. n-6 PUFA-Derived Mediators
4.2. n-3 PUFA-Derived Mediators
5. n-3 PUFAs Protect the Heart
5.1. n-3 PUFAs in Clinical Trials
5.2. Pleiotropic Effects of n-3 PUFAs
5.3. The Differences between EPA and DHA
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Drosatos, K.; Schulze, P.C. Cardiac lipotoxicity: Molecular pathways and therapeutic implications. Curr. Heart Fail Rep. 2013, 10, 109–121. [Google Scholar] [CrossRef] [Green Version]
- Dunlay, S.M.; Roger, V.L.; Redfield, M.M. Epidemiology of heart failure with preserved ejection fraction. Nat. Rev. Cardiol 2017, 14, 591–602. [Google Scholar] [CrossRef] [PubMed]
- Leggat, J.; Bidault, G.; Vidal-Puig, A. Lipotoxicity: A driver of heart failure with preserved ejection fraction? Clin. Sci. (Lond.) 2021, 135, 2265–2283. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Trent, C.M.; Schulze, P.C. Lipid metabolism and toxicity in the heart. Cell Metab. 2012, 15, 805–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brittain, E.L.; Talati, M.; Fessel, J.P.; Zhu, H.; Penner, N.; Calcutt, M.W.; West, J.D.; Funke, M.; Lewis, G.D.; Gerszten, R.E.; et al. Fatty Acid Metabolic Defects and Right Ventricular Lipotoxicity in Human Pulmonary Arterial Hypertension. Circulation 2016, 133, 1936–1944. [Google Scholar] [CrossRef] [PubMed]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef] [PubMed]
- Mazumder, P.K.; O’Neill, B.T.; Roberts, M.W.; Buchanan, J.; Yun, U.J.; Cooksey, R.C.; Boudina, S.; Abel, E.D. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes 2004, 53, 2366–2374. [Google Scholar] [CrossRef] [Green Version]
- Peterson, L.R.; Herrero, P.; Schechtman, K.B.; Racette, S.B.; Waggoner, A.D.; Kisrieva-Ware, Z.; Dence, C.; Klein, S.; Marsala, J.; Meyer, T.; et al. Effect of obesity and insulin resistance on myocardial substrate metabolism and efficiency in young women. Circulation 2004, 109, 2191–2196. [Google Scholar] [CrossRef] [Green Version]
- Kolwicz, S.C., Jr.; Olson, D.P.; Marney, L.C.; Garcia-Menendez, L.; Synovec, R.E.; Tian, R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ. Res. 2012, 111, 728–738. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.; Kolwicz, S.C., Jr.; Wang, P.; Roe, N.D.; Villet, O.; Nishi, K.; Hsu, Y.A.; Flint, G.V.; Caudal, A.; Wang, W.; et al. Increasing Fatty Acid Oxidation Prevents High-Fat Diet-Induced Cardiomyopathy Through Regulating Parkin-Mediated Mitophagy. Circulation 2020, 142, 983–997. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Shah, A.M.; Borlaug, B.A. Heart Failure With Preserved Ejection Fraction In Perspective. Circ. Res. 2019, 124, 1598–1617. [Google Scholar] [CrossRef] [PubMed]
- Borlaug, B.A. Evaluation and management of heart failure with preserved ejection fraction. Nat. Rev. Cardiol. 2020, 17, 559–573. [Google Scholar] [CrossRef] [PubMed]
- Szczepaniak, L.S.; Victor, R.G.; Orci, L.; Unger, R.H. Forgotten but not gone: The rediscovery of fatty heart, the most common unrecognized disease in America. Circ. Res. 2007, 101, 759–767. [Google Scholar] [CrossRef] [Green Version]
- Sharma, S.; Adrogue, J.V.; Golfman, L.; Uray, I.; Lemm, J.; Youker, K.; Noon, G.P.; Frazier, O.H.; Taegtmeyer, H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. Faseb J. 2004, 18, 1692–1700. [Google Scholar] [CrossRef]
- Schiattarella, G.G.; Altamirano, F.; Tong, D.; French, K.M.; Villalobos, E.; Kim, S.Y.; Luo, X.; Jiang, N.; May, H.I.; Wang, Z.V.; et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature 2019, 568, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Schiattarella, G.G.; Altamirano, F.; Kim, S.Y.; Tong, D.; Ferdous, A.; Piristine, H.; Dasgupta, S.; Wang, X.; French, K.M.; Villalobos, E.; et al. Xbp1s-FoxO1 axis governs lipid accumulation and contractile performance in heart failure with preserved ejection fraction. Nat. Commun. 2021, 12, 1684. [Google Scholar] [CrossRef]
- Olzmann, J.A.; Carvalho, P. Dynamics and functions of lipid droplets. Nat. Rev. Mol. Cell Biol. 2019, 20, 137–155. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Reue, K.; Abumrad, N.A.; Bickel, P.E.; Cohen, S.; Fisher, E.A.; Galis, Z.S.; Granneman, J.G.; Lewandowski, E.D.; Murphy, R.; et al. Deciphering the Role of Lipid Droplets in Cardiovascular Disease: A Report From the 2017 National Heart, Lung, and Blood Institute Workshop. Circulation 2018, 138, 305–315. [Google Scholar] [CrossRef]
- Kienesberger, P.C.; Pulinilkunnil, T.; Nagendran, J.; Dyck, J.R. Myocardial triacylglycerol metabolism. J. Mol. Cell Cardiol. 2013, 55, 101–110. [Google Scholar] [CrossRef]
- Kuramoto, K.; Okamura, T.; Yamaguchi, T.; Nakamura, T.Y.; Wakabayashi, S.; Morinaga, H.; Nomura, M.; Yanase, T.; Otsu, K.; Usuda, N.; et al. Perilipin 5, a lipid droplet-binding protein, protects heart from oxidative burden by sequestering fatty acid from excessive oxidation. J. Biol. Chem. 2012, 287, 23852–23863. [Google Scholar] [CrossRef] [Green Version]
- Haemmerle, G.; Moustafa, T.; Woelkart, G.; Büttner, S.; Schmidt, A.; van de Weijer, T.; Hesselink, M.; Jaeger, D.; Kienesberger, P.C.; Zierler, K.; et al. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-α and PGC-1. Nat. Med. 2011, 17, 1076–1085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirano, K.; Ikeda, Y.; Zaima, N.; Sakata, Y.; Matsumiya, G. Triglyceride deposit cardiomyovasculopathy. N. Engl. J. Med. 2008, 359, 2396–2398. [Google Scholar] [CrossRef] [PubMed]
- Kienesberger, P.C.; Pulinilkunnil, T.; Nagendran, J.; Young, M.E.; Bogner-Strauss, J.G.; Hackl, H.; Khadour, R.; Heydari, E.; Haemmerle, G.; Zechner, R.; et al. Early structural and metabolic cardiac remodelling in response to inducible adipose triglyceride lipase ablation. Cardiovasc Res. 2013, 99, 442–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukhorukov, V.N.; Khotina, V.A.; Chegodaev, Y.S.; Ivanova, E.; Sobenin, I.A.; Orekhov, A.N. Lipid Metabolism in Macrophages: Focus on Atherosclerosis. Biomedicines 2020, 8, 262. [Google Scholar] [CrossRef]
- Bailey, A.P.; Koster, G.; Guillermier, C.; Hirst, E.M.; MacRae, J.I.; Lechene, C.P.; Postle, A.D.; Gould, A.P. Antioxidant Role for Lipid Droplets in a Stem Cell Niche of Drosophila. Cell 2015, 163, 340–353. [Google Scholar] [CrossRef] [Green Version]
- Onal, G.; Kutlu, O.; Gozuacik, D.; Dokmeci Emre, S. Lipid Droplets in Health and Disease. Lipids Health Dis. 2017, 16, 128. [Google Scholar] [CrossRef] [Green Version]
- Khor, V.K.; Ahrends, R.; Lin, Y.; Shen, W.J.; Adams, C.M.; Roseman, A.N.; Cortez, Y.; Teruel, M.N.; Azhar, S.; Kraemer, F.B. The proteome of cholesteryl-ester-enriched versus triacylglycerol-enriched lipid droplets. PLoS ONE 2014, 9, e105047. [Google Scholar] [CrossRef] [Green Version]
- van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef]
- Kennedy, E.P.; Weiss, S.B. The function of cytidine coenzymes in the biosynthesis of phospholipides. J. Biol. Chem. 1956, 222, 193–214. [Google Scholar] [CrossRef]
- Lands, W.E. Metabolism of glycerolipides; a comparison of lecithin and triglyceride synthesis. J. Biol. Chem. 1958, 231, 883–888. [Google Scholar] [CrossRef]
- Shindou, H.; Hishikawa, D.; Harayama, T.; Eto, M.; Shimizu, T. Generation of membrane diversity by lysophospholipid acyltransferases. J. Biochem. 2013, 154, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Vries, J.E.; Vork, M.M.; Roemen, T.H.; de Jong, Y.F.; Cleutjens, J.P.; van der Vusse, G.J.; van Bilsen, M. Saturated but not mono-unsaturated fatty acids induce apoptotic cell death in neonatal rat ventricular myocytes. J. Lipid Res. 1997, 38, 1384–1394. [Google Scholar] [CrossRef]
- Yamamoto, T.; Endo, J.; Kataoka, M.; Matsuhashi, T.; Katsumata, Y.; Shirakawa, K.; Yoshida, N.; Isobe, S.; Moriyama, H.; Goto, S.; et al. Decrease in membrane phospholipids unsaturation correlates with myocardial diastolic dysfunction. PLoS ONE 2018, 13, e0208396. [Google Scholar] [CrossRef] [PubMed]
- Matsui, H.; Yokoyama, T.; Sekiguchi, K.; Iijima, D.; Sunaga, H.; Maniwa, M.; Ueno, M.; Iso, T.; Arai, M.; Kurabayashi, M. Stearoyl-CoA desaturase-1 (SCD1) augments saturated fatty acid-induced lipid accumulation and inhibits apoptosis in cardiac myocytes. PLoS ONE 2012, 7, e33283. [Google Scholar]
- Yamamoto, T.; Endo, J.; Kataoka, M.; Matsuhashi, T.; Katsumata, Y.; Shirakawa, K.; Yoshida, N.; Isobe, S.; Moriyama, H.; Goto, S.; et al. Sirt1 counteracts decrease in membrane phospholipid unsaturation and diastolic dysfunction during saturated fatty acid overload. J. Mol. Cell Cardiol. 2019, 133, 1–11. [Google Scholar] [CrossRef]
- Le, C.H.; Mulligan, C.M.; Routh, M.A.; Bouma, G.J.; Frye, M.A.; Jeckel, K.M.; Sparagna, G.C.; Lynch, J.M.; Moore, R.L.; McCune, S.A.; et al. Delta-6-desaturase links polyunsaturated fatty acid metabolism with phospholipid remodeling and disease progression in heart failure. Circ. Heart Fail. 2014, 7, 172–183. [Google Scholar] [CrossRef] [Green Version]
- Mulligan, C.M.; Le, C.H.; deMooy, A.B.; Nelson, C.B.; Chicco, A.J. Inhibition of delta-6 desaturase reverses cardiolipin remodeling and prevents contractile dysfunction in the aged mouse heart without altering mitochondrial respiratory function. J. Gerontol. A Biol. Sci. Med. Sci. 2014, 69, 799–809. [Google Scholar] [CrossRef] [Green Version]
- Fry, M.; Green, D.E. Cardiolipin requirement for electron transfer in complex I and III of the mitochondrial respiratory chain. J. Biol. Chem. 1981, 256, 1874–1880. [Google Scholar] [CrossRef]
- Sabbah, H.N. Targeting the Mitochondria in Heart Failure: A Translational Perspective. JACC Basic Transl. Sci. 2020, 5, 88–106. [Google Scholar] [CrossRef]
- Kagan, V.E.; Chu, C.T.; Tyurina, Y.Y.; Cheikhi, A.; Bayir, H. Cardiolipin asymmetry, oxidation and signaling. Chem. Phys. Lipids 2014, 179, 64–69. [Google Scholar] [CrossRef] [Green Version]
- Kagan, V.E.; Tyurin, V.A.; Jiang, J.; Tyurina, Y.Y.; Ritov, V.B.; Amoscato, A.A.; Osipov, A.N.; Belikova, N.A.; Kapralov, A.A.; Kini, V.; et al. Cytochrome c acts as a cardiolipin oxygenase required for release of proapoptotic factors. Nat. Chem. Biol. 2005, 1, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T.; Ji, J.; Dagda, R.K.; Jiang, J.F.; Tyurina, Y.Y.; Kapralov, A.A.; Tyurin, V.A.; Yanamala, N.; Shrivastava, I.H.; Mohammadyani, D.; et al. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat. Cell Biol. 2013, 15, 1197–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiebish, M.A.; Bell, R.; Yang, K.; Phan, T.; Zhao, Z.; Ames, W.; Seyfried, T.N.; Gross, R.W.; Chuang, J.H.; Han, X. Dynamic simulation of cardiolipin remodeling: Greasing the wheels for an interpretative approach to lipidomics. J. Lipid Res. 2010, 51, 2153–2170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saric, A.; Andreau, K.; Armand, A.S.; Møller, I.M.; Petit, P.X. Barth Syndrome: From Mitochondrial Dysfunctions Associated with Aberrant Production of Reactive Oxygen Species to Pluripotent Stem Cell Studies. Front. Genet. 2015, 6, 359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sparagna, G.C.; Lesnefsky, E.J. Cardiolipin remodeling in the heart. J. Cardiovasc. Pharmacol. 2009, 53, 290–301. [Google Scholar] [CrossRef]
- Sparagna, G.C.; Johnson, C.A.; McCune, S.A.; Moore, R.L.; Murphy, R.C. Quantitation of cardiolipin molecular species in spontaneously hypertensive heart failure rats using electrospray ionization mass spectrometry. J. Lipid Res. 2005, 46, 1196–1204. [Google Scholar] [CrossRef] [Green Version]
- Sparagna, G.C.; Chicco, A.J.; Murphy, R.C.; Bristow, M.R.; Johnson, C.A.; Rees, M.L.; Maxey, M.L.; McCune, S.A.; Moore, R.L. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J. Lipid Res. 2007, 48, 1559–1570. [Google Scholar] [CrossRef] [Green Version]
- Koop, A.M.C.; Hagdorn, Q.A.J.; Bossers, G.P.L.; van Leusden, T.; Gerding, A.; van Weeghel, M.; Vaz, F.M.; Koonen, D.P.Y.; Sillje, H.H.W.; Berger, R.M.F.; et al. Right ventricular pressure overload alters cardiac lipid composition. Int. J. Cardiol. 2019, 287, 96–105. [Google Scholar] [CrossRef] [PubMed]
- Ji, J.; Kline, A.E.; Amoscato, A.; Samhan-Arias, A.K.; Sparvero, L.J.; Tyurin, V.A.; Tyurina, Y.Y.; Fink, B.; Manole, M.D.; Puccio, A.M.; et al. Lipidomics identifies cardiolipin oxidation as a mitochondrial target for redox therapy of brain injury. Nat. Neurosci. 2012, 15, 1407–1413. [Google Scholar] [CrossRef] [Green Version]
- Chan, R.B.; Di Paolo, G. Knockout punch: Cardiolipin oxidation in trauma. Nat. Neurosci. 2012, 15, 1325–1327. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Mitochondrial bioenergetics and cardiolipin alterations in myocardial ischemia-reperfusion injury: Implications for pharmacological cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H1341–H1352. [Google Scholar] [CrossRef] [PubMed]
- Koleini, N.; Nickel, B.E.; Edel, A.L.; Fandrich, R.R.; Ravandi, A.; Kardami, E. Oxidized phospholipids in Doxorubicin-induced cardiotoxicity. Chem. Biol. Interact. 2019, 303, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Spencer, C.T.; Bryant, R.M.; Day, J.; Gonzalez, I.L.; Colan, S.D.; Thompson, W.R.; Berthy, J.; Redfearn, S.P.; Byrne, B.J. Cardiac and clinical phenotype in Barth syndrome. Pediatrics 2006, 118, e337–e346. [Google Scholar] [CrossRef] [PubMed]
- Schlame, M.; Ren, M.; Xu, Y.; Greenberg, M.L.; Haller, I. Molecular symmetry in mitochondrial cardiolipins. Chem. Phys. Lipids 2005, 138, 38–49. [Google Scholar] [CrossRef] [PubMed]
- Phoon, C.K.; Acehan, D.; Schlame, M.; Stokes, D.L.; Edelman-Novemsky, I.; Yu, D.; Xu, Y.; Viswanathan, N.; Ren, M. Tafazzin knockdown in mice leads to a developmental cardiomyopathy with early diastolic dysfunction preceding myocardial noncompaction. J. Am. Heart Assoc. 2012, 1, e000455. [Google Scholar] [CrossRef] [Green Version]
- Cole, L.K.; Mejia, E.M.; Sparagna, G.C.; Vandel, M.; Xiang, B.; Han, X.; Dedousis, N.; Kaufman, B.A.; Dolinsky, V.W.; Hatch, G.M. Cardiolipin deficiency elevates susceptibility to a lipotoxic hypertrophic cardiomyopathy. J. Mol. Cell Cardiol. 2020, 144, 24–34. [Google Scholar] [CrossRef]
- Liu, X.; Wang, S.; Guo, X.; Li, Y.; Ogurlu, R.; Lu, F.; Prondzynski, M.; de la Serna Buzon, S.; Ma, Q.; Zhang, D.; et al. Increased Reactive Oxygen Species-Mediated Ca(2+)/Calmodulin-Dependent Protein Kinase II Activation Contributes to Calcium Handling Abnormalities and Impaired Contraction in Barth Syndrome. Circulation 2021, 143, 1894–1911. [Google Scholar] [CrossRef]
- Chowdhury, A.; Aich, A.; Jain, G.; Wozny, K.; Lüchtenborg, C.; Hartmann, M.; Bernhard, O.; Balleiniger, M.; Alfar, E.A.; Zieseniss, A.; et al. Defective Mitochondrial Cardiolipin Remodeling Dampens HIF-1α Expression in Hypoxia. Cell Rep. 2018, 25, 561–570.e6. [Google Scholar] [CrossRef] [Green Version]
- Ojala, T.; Polinati, P.; Manninen, T.; Hiippala, A.; Rajantie, J.; Karikoski, R.; Suomalainen, A.; Tyni, T. New mutation of mitochondrial DNAJC19 causing dilated and noncompaction cardiomyopathy, anemia, ataxia, and male genital anomalies. Pediatr. Res. 2012, 72, 432–437. [Google Scholar] [CrossRef] [Green Version]
- Richter-Dennerlein, R.; Korwitz, A.; Haag, M.; Tatsuta, T.; Dargazanli, S.; Baker, M.; Decker, T.; Lamkemeyer, T.; Rugarli, E.I.; Langer, T. DNAJC19, a mitochondrial cochaperone associated with cardiomyopathy, forms a complex with prohibitins to regulate cardiolipin remodeling. Cell Metab. 2014, 20, 158–171. [Google Scholar] [CrossRef] [Green Version]
- Cao, J.; Liu, Y.; Lockwood, J.; Burn, P.; Shi, Y. A novel cardiolipin-remodeling pathway revealed by a gene encoding an endoplasmic reticulum-associated acyl-CoA:lysocardiolipin acyltransferase (ALCAT1) in mouse. J. Biol. Chem. 2004, 279, 31727–31734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Romestaing, C.; Han, X.; Li, Y.; Hao, X.; Wu, Y.; Sun, C.; Liu, X.; Jefferson, L.S.; Xiong, J.; et al. Cardiolipin remodeling by ALCAT1 links oxidative stress and mitochondrial dysfunction to obesity. Cell Metab. 2010, 12, 154–165. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liu, X.; Wang, H.; Zhang, W.; Chan, D.C.; Shi, Y. Lysocardiolipin acyltransferase 1 (ALCAT1) controls mitochondrial DNA fidelity and biogenesis through modulation of MFN2 expression. Proc. Natl. Acad. Sci. USA 2012, 109, 6975–6980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Ye, B.; Miller, S.; Yuan, H.; Zhang, H.; Tian, L.; Nie, J.; Imae, R.; Arai, H.; Li, Y.; et al. Ablation of ALCAT1 mitigates hypertrophic cardiomyopathy through effects on oxidative stress and mitophagy. Mol. Cell Biol. 2012, 32, 4493–4504. [Google Scholar] [CrossRef] [Green Version]
- Song, C.; Zhang, J.; Qi, S.; Liu, Z.; Zhang, X.; Zheng, Y.; Andersen, J.P.; Zhang, W.; Strong, R.; Martinez, P.A.; et al. Cardiolipin remodeling by ALCAT1 links mitochondrial dysfunction to Parkinson’s diseases. Aging Cell 2019, 18, e12941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Liu, X.; Nie, J.; Zhang, J.; Kimball, S.R.; Zhang, H.; Zhang, W.J.; Jefferson, L.S.; Cheng, Z.; Ji, Q.; et al. ALCAT1 controls mitochondrial etiology of fatty liver diseases, linking defective mitophagy to steatosis. Hepatology 2015, 61, 486–496. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.S.; Mathew, B.; Li, H.; Zhao, Y.; Ma, S.F.; Noth, I.; Reddy, S.P.; Harijith, A.; Usatyuk, P.V.; Berdyshev, E.V.; et al. The mitochondrial cardiolipin remodeling enzyme lysocardiolipin acyltransferase is a novel target in pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 189, 1402–1415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imae, R.; Inoue, T.; Nakasaki, Y.; Uchida, Y.; Ohba, Y.; Kono, N.; Nakanishi, H.; Sasaki, T.; Mitani, S.; Arai, H. LYCAT, a homologue of C. elegans acl-8, acl-9, and acl-10, determines the fatty acid composition of phosphatidylinositol in mice. J. Lipid Res. 2012, 53, 335–347. [Google Scholar] [CrossRef] [Green Version]
- Grevengoed, T.J.; Martin, S.A.; Katunga, L.; Cooper, D.E.; Anderson, E.J.; Murphy, R.C.; Coleman, R.A. Acyl-CoA synthetase 1 deficiency alters cardiolipin species and impairs mitochondrial function. J. Lipid Res. 2015, 56, 1572–1582. [Google Scholar] [CrossRef] [Green Version]
- Goldenberg, J.R.; Carley, A.N.; Ji, R.; Zhang, X.; Fasano, M.; Schulze, P.C.; Lewandowski, E.D. Preservation of Acyl Coenzyme A Attenuates Pathological and Metabolic Cardiac Remodeling Through Selective Lipid Trafficking. Circulation 2019, 139, 2765–2777. [Google Scholar] [CrossRef]
- O’Connell, T.D.; Mason, R.P.; Budoff, M.J.; Navar, A.M.; Shearer, G.C. Mechanistic insights into cardiovascular protection for omega-3 fatty acids and their bioactive lipid metabolites. Eur. Heart J. Suppl. 2020, 22 (Suppl. J), J3–J20. [Google Scholar] [CrossRef] [PubMed]
- Takayama, K.; Yuhki, K.; Ono, K.; Fujino, T.; Hara, A.; Yamada, T.; Kuriyama, S.; Karibe, H.; Okada, Y.; Takahata, O.; et al. Thromboxane A2 and prostaglandin F2alpha mediate inflammatory tachycardia. Nat. Med. 2005, 11, 562–566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kayama, Y.; Minamino, T.; Toko, H.; Sakamoto, M.; Shimizu, I.; Takahashi, H.; Okada, S.; Tateno, K.; Moriya, J.; Yokoyama, M.; et al. Cardiac 12/15 lipoxygenase-induced inflammation is involved in heart failure. J. Exp. Med. 2009, 206, 1565–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, H.; Kayama, Y.; Sakamoto, M.; Iuchi, H.; Shimizu, I.; Yoshino, T.; Katoh, D.; Nagoshi, T.; Tojo, K.; Minamino, T.; et al. Arachidonate 12/15-lipoxygenase-induced inflammation and oxidative stress are involved in the development of diabetic cardiomyopathy. Diabetes 2015, 64, 618–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hara, A.; Yuhki, K.; Fujino, T.; Yamada, T.; Takayama, K.; Kuriyama, S.; Takahata, O.; Karibe, H.; Okada, Y.; Xiao, C.Y.; et al. Augmented cardiac hypertrophy in response to pressure overload in mice lacking the prostaglandin I2 receptor. Circulation 2005, 112, 84–92. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.Y.; Hara, A.; Yuhki, K.; Fujino, T.; Ma, H.; Okada, Y.; Takahata, O.; Yamada, T.; Murata, T.; Narumiya, S.; et al. Roles of prostaglandin I(2) and thromboxane A(2) in cardiac ischemia-reperfusion injury: A study using mice lacking their respective receptors. Circulation 2001, 104, 2210–2215. [Google Scholar] [CrossRef] [Green Version]
- Xiao, C.Y.; Yuhki, K.; Hara, A.; Fujino, T.; Kuriyama, S.; Yamada, T.; Takayama, K.; Takahata, O.; Karibe, H.; Taniguchi, T.; et al. Prostaglandin E2 protects the heart from ischemia-reperfusion injury via its receptor subtype EP4. Circulation 2004, 109, 2462–2468. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Meyer-Kirchrath, J.; Kaber, G.; Jacoby, C.; Flögel, U.; Schrader, J.; Rüther, U.; Schrör, K.; Hohlfeld, T. Cardiospecific overexpression of the prostaglandin EP3 receptor attenuates ischemia-induced myocardial injury. Circulation 2005, 112, 400–406. [Google Scholar] [CrossRef] [Green Version]
- Tokudome, S.; Sano, M.; Shinmura, K.; Matsuhashi, T.; Morizane, S.; Moriyama, H.; Tamaki, K.; Hayashida, K.; Nakanishi, H.; Yoshikawa, N.; et al. Glucocorticoid protects rodent hearts from ischemia/reperfusion injury by activating lipocalin-type prostaglandin D synthase-derived PGD2 biosynthesis. J. Clin. Investig. 2009, 119, 1477–1488. [Google Scholar] [CrossRef]
- Katsumata, Y.; Shinmura, K.; Sugiura, Y.; Tohyama, S.; Matsuhashi, T.; Ito, H.; Yan, X.; Ito, K.; Yuasa, S.; Ieda, M.; et al. Endogenous prostaglandin D2 and its metabolites protect the heart against ischemia-reperfusion injury by activating Nrf2. Hypertension 2014, 63, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.; Chen, C. The Role of Epoxyeicosatrienoic Acids in Cardiac Remodeling. Front. Physiol. 2021, 12, 642470. [Google Scholar] [CrossRef]
- Dhanasekaran, A.; Gruenloh, S.K.; Buonaccorsi, J.N.; Zhang, R.; Gross, G.J.; Falck, J.R.; Patel, P.K.; Jacobs, E.R.; Medhora, M. Multiple antiapoptotic targets of the PI3K/Akt survival pathway are activated by epoxyeicosatrienoic acids to protect cardiomyocytes from hypoxia/anoxia. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H724–H735. [Google Scholar] [CrossRef] [PubMed]
- Lynes, M.D.; Leiria, L.O.; Lundh, M.; Bartelt, A.; Shamsi, F.; Huang, T.L.; Takahashi, H.; Hirshman, M.F.; Schlein, C.; Lee, A.; et al. The cold-induced lipokine 12,13-diHOME promotes fatty acid transport into brown adipose tissue. Nat. Med. 2017, 23, 631–637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanford, K.I.; Lynes, M.D.; Takahashi, H.; Baer, L.A.; Arts, P.J.; May, F.J.; Lehnig, A.C.; Middelbeek, R.J.W.; Richard, J.J.; So, K.; et al. 12,13-diHOME: An Exercise-Induced Lipokine that Increases Skeletal Muscle Fatty Acid Uptake. Cell Metab. 2018, 27, 1111–1120.e3. [Google Scholar] [CrossRef] [Green Version]
- Pinckard, K.M.; Shettigar, V.K.; Wright, K.R.; Abay, E.; Baer, L.A.; Vidal, P.; Dewal, R.S.; Das, D.; Duarte-Sanmiguel, S.; Hernández-Saavedra, D.; et al. A Novel Endocrine Role for the BAT-Released Lipokine 12,13-diHOME to Mediate Cardiac Function. Circulation 2021, 143, 145–159. [Google Scholar] [CrossRef] [PubMed]
- Endo, J.; Arita, M. Cardioprotective mechanism of omega-3 polyunsaturated fatty acids. J. Cardiol. 2016, 67, 22–27. [Google Scholar] [CrossRef] [Green Version]
- Endo, J.; Sano, M.; Isobe, Y.; Fukuda, K.; Kang, J.X.; Arai, H.; Arita, M. 18-HEPE, an n-3 fatty acid metabolite released by macrophages, prevents pressure overload-induced maladaptive cardiac remodeling. J. Exp. Med. 2014, 211, 1673–1687. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.X.; Wang, J.; Wu, L.; Kang, Z.B. Transgenic mice: Fat-1 mice convert n-6 to n-3 fatty acids. Nature 2004, 427, 504. [Google Scholar] [CrossRef]
- Kang, J.X. A transgenic mouse model for gene-nutrient interactions. J. Nutr. Nutr. 2008, 1, 172–177. [Google Scholar] [CrossRef]
- Schwab, J.M.; Chiang, N.; Arita, M.; Serhan, C.N. Resolvin E1 and protectin D1 activate inflammation-resolution programmes. Nature 2007, 447, 869–874. [Google Scholar] [CrossRef] [Green Version]
- Arita, M.; Bianchini, F.; Aliberti, J.; Sher, A.; Chiang, N.; Hong, S.; Yang, R.; Petasis, N.A.; Serhan, C.N. Stereochemical assignment, antiinflammatory properties, and receptor for the omega-3 lipid mediator resolvin E1. J. Exp. Med. 2005, 201, 713–722. [Google Scholar] [CrossRef] [PubMed]
- Basil, M.C.; Levy, B.D. Specialized pro-resolving mediators: Endogenous regulators of infection and inflammation. Nat. Rev. Immunol. 2016, 16, 51–67. [Google Scholar] [CrossRef] [PubMed]
- Connor, K.M.; SanGiovanni, J.P.; Lofqvist, C.; Aderman, C.M.; Chen, J.; Higuchi, A.; Hong, S.; Pravda, E.A.; Majchrzak, S.; Carper, D.; et al. Increased dietary intake of omega-3-polyunsaturated fatty acids reduces pathological retinal angiogenesis. Nat. Med. 2007, 13, 868–873. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcheselli, V.L.; Hong, S.; Lukiw, W.J.; Tian, X.H.; Gronert, K.; Musto, A.; Hardy, M.; Gimenez, J.M.; Chiang, N.; Serhan, C.N.; et al. Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J. Biol. Chem. 2003, 278, 43807–43817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duffield, J.S.; Hong, S.; Vaidya, V.S.; Lu, Y.; Fredman, G.; Serhan, C.N.; Bonventre, J.V. Resolvin D series and protectin D1 mitigate acute kidney injury. J. Immunol. 2006, 177, 5902–5911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keyes, K.T.; Ye, Y.; Lin, Y.; Zhang, C.; Perez-Polo, J.R.; Gjorstrup, P.; Birnbaum, Y. Resolvin E1 protects the rat heart against reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 2010, 299, H153–H164. [Google Scholar] [CrossRef] [Green Version]
- Kain, V.; Ingle, K.A.; Colas, R.A.; Dalli, J.; Prabhu, S.D.; Serhan, C.N.; Joshi, M.; Halade, G.V. Resolvin D1 activates the inflammation resolving response at splenic and ventricular site following myocardial infarction leading to improved ventricular function. J. Mol. Cell Cardiol. 2015, 84, 24–35. [Google Scholar] [CrossRef] [Green Version]
- Simopoulos, A.P. The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp. Biol. Med. (Maywood) 2008, 233, 674–688. [Google Scholar] [CrossRef]
- Dyerberg, J.; Bang, H.O. Haemostatic function and platelet polyunsaturated fatty acids in Eskimos. Lancet 1979, 2, 433–435. [Google Scholar] [CrossRef]
- Dietary supplementation with n-3 polyunsaturated fatty acids and vitamin E after myocardial infarction: Results of the GISSI-Prevenzione trial. Gruppo Italiano per lo Studio della Sopravvivenza nell’Infarto miocardico. Lancet 1999, 354, 447–455.
- Yokoyama, M.; Origasa, H.; Matsuzaki, M.; Matsuzawa, Y.; Saito, Y.; Ishikawa, Y.; Oikawa, S.; Sasaki, J.; Hishida, H.; Itakura, H.; et al. Effects of eicosapentaenoic acid on major coronary events in hypercholesterolaemic patients (JELIS): A randomised open-label, blinded endpoint analysis. Lancet 2007, 369, 1090–1098. [Google Scholar] [CrossRef]
- Bhatt, D.L.; Steg, P.G.; Miller, M.; Brinton, E.A.; Jacobson, T.A.; Ketchum, S.B.; Doyle, R.T., Jr.; Juliano, R.A.; Jiao, L.; Granowitz, C.; et al. Cardiovascular Risk Reduction with Icosapent Ethyl for Hypertriglyceridemia. N. Engl. J. Med. 2019, 380, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Tavazzi, L.; Maggioni, A.P.; Marchioli, R.; Barlera, S.; Franzosi, M.G.; Latini, R.; Lucci, D.; Nicolosi, G.L.; Porcu, M.; Tognoni, G. Effect of n-3 polyunsaturated fatty acids in patients with chronic heart failure (the GISSI-HF trial): A randomised, double-blind, placebo-controlled trial. Lancet 2008, 372, 1223–1230. [Google Scholar] [PubMed]
- Nodari, S.; Triggiani, M.; Campia, U.; Manerba, A.; Milesi, G.; Cesana, B.M.; Gheorghiade, M.; Dei Cas, L. Effects of n-3 polyunsaturated fatty acids on left ventricular function and functional capacity in patients with dilated cardiomyopathy. J. Am. Coll Cardiol. 2011, 57, 870–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, R.C.; Liu, L.; Herrington, D.M.; Huang, S.; Tsai, M.Y.; O’Connell, T.D.; Shearer, G.C. Predicting Risk for Incident Heart Failure With Omega-3 Fatty Acids: From MESA. JACC Heart Fail 2019, 7, 651–661. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Shearer, G.C.; Chen, Q.; Healy, C.L.; Beyer, A.J.; Nareddy, V.B.; Gerdes, A.M.; Harris, W.S.; O’Connell, T.D.; Wang, D. Omega-3 fatty acids prevent pressure overload-induced cardiac fibrosis through activation of cyclic GMP/protein kinase G signaling in cardiac fibroblasts. Circulation 2011, 123, 584–593. [Google Scholar] [CrossRef] [Green Version]
- Toko, H.; Morita, H.; Katakura, M.; Hashimoto, M.; Ko, T.; Bujo, S.; Adachi, Y.; Ueda, K.; Murakami, H.; Ishizuka, M.; et al. Omega-3 fatty acid prevents the development of heart failure by changing fatty acid composition in the heart. Sci. Rep. 2020, 10, 15553. [Google Scholar] [CrossRef]
- Mason, R.P.; Libby, P.; Bhatt, D.L. Emerging Mechanisms of Cardiovascular Protection for the Omega-3 Fatty Acid Eicosapentaenoic Acid. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 1135–1147. [Google Scholar] [CrossRef]
- Briscoe, C.P.; Tadayyon, M.; Andrews, J.L.; Benson, W.G.; Chambers, J.K.; Eilert, M.M.; Ellis, C.; Elshourbagy, N.A.; Goetz, A.S.; Minnick, D.T.; et al. The orphan G protein-coupled receptor GPR40 is activated by medium and long chain fatty acids. J. Biol. Chem. 2003, 278, 11303–11311. [Google Scholar] [CrossRef] [Green Version]
- Itoh, Y.; Kawamata, Y.; Harada, M.; Kobayashi, M.; Fujii, R.; Fukusumi, S.; Ogi, K.; Hosoya, M.; Tanaka, Y.; Uejima, H.; et al. Free fatty acids regulate insulin secretion from pancreatic beta cells through GPR40. Nature 2003, 422, 173–176. [Google Scholar] [CrossRef]
- O’Connell, T.D.; Block, R.C.; Huang, S.P.; Shearer, G.C. Omega3-Polyunsaturated fatty acids for heart failure: Effects of dose on efficacy and novel signaling through free fatty acid receptor 4. J. Mol. Cell Cardiol. 2017, 103, 74–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eclov, J.A.; Qian, Q.; Redetzke, R.; Chen, Q.; Wu, S.C.; Healy, C.L.; Ortmeier, S.B.; Harmon, E.; Shearer, G.C.; O’Connell, T.D. EPA, not DHA, prevents fibrosis in pressure overload-induced heart failure: Potential role of free fatty acid receptor 4. J. Lipid Res. 2015, 56, 2297–2308. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.E.; Lambert, M.H.; Montana, V.G.; Parks, D.J.; Blanchard, S.G.; Brown, P.J.; Sternbach, D.D.; Lehmann, J.M.; Wisely, G.B.; Willson, T.M.; et al. Molecular recognition of fatty acids by peroxisome proliferator-activated receptors. Mol. Cell 1999, 3, 397–403. [Google Scholar] [CrossRef]
- Forman, B.M.; Chen, J.; Evans, R.M. Hypolipidemic drugs, polyunsaturated fatty acids, and eicosanoids are ligands for peroxisome proliferator-activated receptors alpha and delta. Proc. Natl. Acad. Sci. USA 1997, 94, 4312–4317. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.D.; Goldstein, J.L.; Brown, M.S. SREBPs: Activators of the complete program of cholesterol and fatty acid synthesis in the liver. J. Clin. Investig. 2002, 109, 1125–1131. [Google Scholar] [CrossRef] [PubMed]
- Park, Y.; Harris, W.S. Omega-3 fatty acid supplementation accelerates chylomicron triglyceride clearance. J. Lipid Res. 2003, 44, 455–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raposo, H.F.; Patrício, P.R.; Simões, M.C.; Oliveira, H.C. Fibrates and fish oil, but not corn oil, up-regulate the expression of the cholesteryl ester transfer protein (CETP) gene. J. Nutr. Biochem. 2014, 25, 669–674. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, M.; Berneis, K. Who needs to care about small, dense low-density lipoproteins? Int. J. Clin. Pract. 2007, 61, 1949–1956. [Google Scholar] [CrossRef]
- Rizvi, A.A.; Stoian, A.P.; Janez, A.; Rizzo, M. Lipoproteins and Cardiovascular Disease: An Update on the Clinical Significance of Atherogenic Small, Dense LDL and New Therapeutical Options. Biomedicines 2021, 9, 1579. [Google Scholar] [CrossRef]
- Doublet, A.; Robert, V.; Vedie, B.; Rousseau-Ralliard, D.; Reboulleau, A.; Grynberg, A.; Paul, J.L.; Fournier, N. Contrasting effects of arachidonic acid and docosahexaenoic acid membrane incorporation into cardiomyocytes on free cholesterol turnover. Biochim. Biophys. Acta 2014, 1842, 1413–1421. [Google Scholar] [CrossRef]
- Hallaq, H.; Smith, T.W.; Leaf, A. Modulation of dihydropyridine-sensitive calcium channels in heart cells by fish oil fatty acids. Proc. Natl. Acad. Sci. USA 1992, 89, 1760–1764. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, Y.F.; Kang, J.X.; Morgan, J.P.; Leaf, A. Blocking effects of polyunsaturated fatty acids on Na+ channels of neonatal rat ventricular myocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 11000–11004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manson, J.E.; Cook, N.R.; Lee, I.M.; Christen, W.; Bassuk, S.S.; Mora, S.; Gibson, H.; Gordon, D.; Copeland, T.; D’Agostino, D.; et al. Vitamin D Supplements and Prevention of Cancer and Cardiovascular Disease. N. Engl. J. Med. 2019, 380, 33–44. [Google Scholar] [CrossRef] [PubMed]
- Bowman, L.; Mafham, M.; Wallendszus, K.; Stevens, W.; Buck, G.; Barton, J.; Murphy, K.; Aung, T.; Haynes, R.; Cox, J.; et al. Effects of n-3 Fatty Acid Supplements in Diabetes Mellitus. N. Engl. J. Med. 2018, 379, 1540–1550. [Google Scholar] [PubMed]
- Heydari, B.; Abdullah, S.; Pottala, J.V.; Shah, R.; Abbasi, S.; Mandry, D.; Francis, S.A.; Lumish, H.; Ghoshhajra, B.B.; Hoffmann, U.; et al. Effect of Omega-3 Acid Ethyl Esters on Left Ventricular Remodeling After Acute Myocardial Infarction: The OMEGA-REMODEL Randomized Clinical Trial. Circulation 2016, 134, 378–391. [Google Scholar] [CrossRef]
- Nguyen, L.N.; Ma, D.; Shui, G.; Wong, P.; Cazenave-Gassiot, A.; Zhang, X.; Wenk, M.R.; Goh, E.L.; Silver, D.L. Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature 2014, 509, 503–506. [Google Scholar] [CrossRef]
- Wong, B.H.; Chan, J.P.; Cazenave-Gassiot, A.; Poh, R.W.; Foo, J.C.; Galam, D.L.; Ghosh, S.; Nguyen, L.N.; Barathi, V.A.; Yeo, S.W.; et al. Mfsd2a Is a Transporter for the Essential ω-3 Fatty Acid Docosahexaenoic Acid (DHA) in Eye and Is Important for Photoreceptor Cell Development. J. Biol. Chem. 2016, 291, 10501–10514. [Google Scholar] [CrossRef] [Green Version]
- Rice, D.S.; Calandria, J.M.; Gordon, W.C.; Jun, B.; Zhou, Y.; Gelfman, C.M.; Li, S.; Jin, M.; Knott, E.J.; Chang, B.; et al. Adiponectin receptor 1 conserves docosahexaenoic acid and promotes photoreceptor cell survival. Nat. Commun. 2015, 6, 6228. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moriyama, H.; Endo, J.; Ikura, H.; Kitakata, H.; Momoi, M.; Shinya, Y.; Ko, S.; Ichihara, G.; Hiraide, T.; Shirakawa, K.; et al. Qualitative and Quantitative Effects of Fatty Acids Involved in Heart Diseases. Metabolites 2022, 12, 210. https://doi.org/10.3390/metabo12030210
Moriyama H, Endo J, Ikura H, Kitakata H, Momoi M, Shinya Y, Ko S, Ichihara G, Hiraide T, Shirakawa K, et al. Qualitative and Quantitative Effects of Fatty Acids Involved in Heart Diseases. Metabolites. 2022; 12(3):210. https://doi.org/10.3390/metabo12030210
Chicago/Turabian StyleMoriyama, Hidenori, Jin Endo, Hidehiko Ikura, Hiroki Kitakata, Mizuki Momoi, Yoshiki Shinya, Seien Ko, Genki Ichihara, Takahiro Hiraide, Kohsuke Shirakawa, and et al. 2022. "Qualitative and Quantitative Effects of Fatty Acids Involved in Heart Diseases" Metabolites 12, no. 3: 210. https://doi.org/10.3390/metabo12030210
APA StyleMoriyama, H., Endo, J., Ikura, H., Kitakata, H., Momoi, M., Shinya, Y., Ko, S., Ichihara, G., Hiraide, T., Shirakawa, K., Anzai, A., Katsumata, Y., & Sano, M. (2022). Qualitative and Quantitative Effects of Fatty Acids Involved in Heart Diseases. Metabolites, 12(3), 210. https://doi.org/10.3390/metabo12030210