
Association between Visceral Adipose Tissue Metabolism and Alzheimer’s Disease Pathology

Abstract
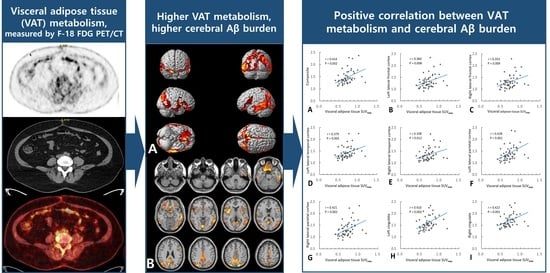
:
1. Introduction
2. Results
2.1. Population Characteristics
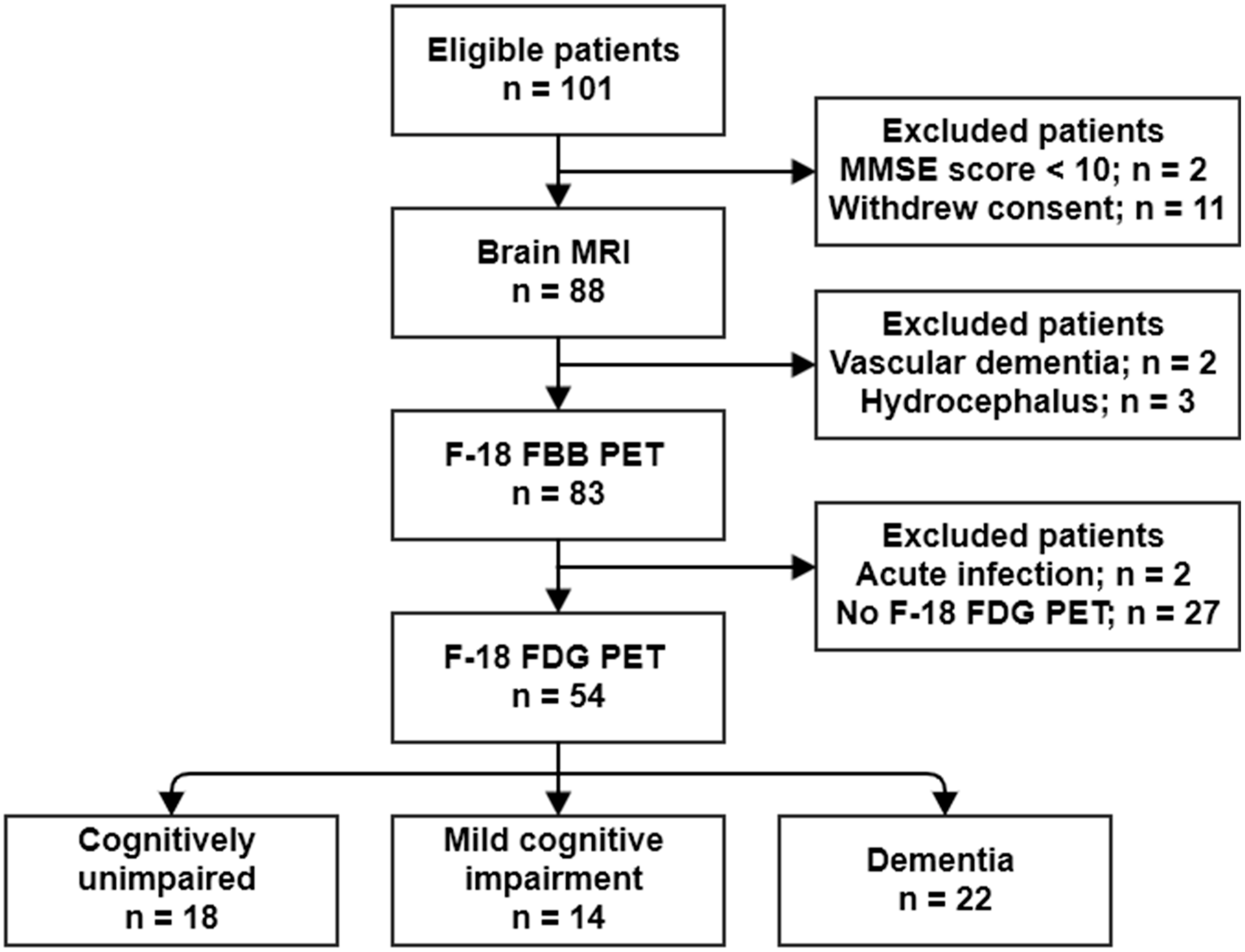
2.2. Association of VAT Metabolism with AD Pathology
3. Discussion
4. Materials and Methods
4.1. Study Population
4.2. Brain MRI
4.3. 18F-FDG PET
4.4. 18F-FBB PET
4.5. Voxel-Based Analysis
4.6. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–185. [Google Scholar] [CrossRef] [PubMed]
- Kiliaan, A.J.; Arnoldussen, I.A.; Gustafson, D.R. Adipokines: A link between obesity and dementia? Lancet Neurol. 2014, 13, 913–923. [Google Scholar] [CrossRef] [Green Version]
- Misiak, B.; Leszek, J.; Kiejna, A. Metabolic syndrome, mild cognitive impairment and Alzheimer’s disease—The emerging role of systemic low-grade inflammation and adiposity. Brain Res. Bull. 2012, 89, 144–149. [Google Scholar] [CrossRef] [PubMed]
- Pichiah, P.B.T.; Sankarganesh, D.; Arunachalam, S.; Achiraman, S. Adipose-Derived Molecules-Untouched Horizons in Alzheimer’s Disease Biology. Front. Aging Neurosci. 2020, 12, 17. [Google Scholar] [CrossRef]
- Guo, D.H.; Yamamoto, M.; Hernandez, C.M.; Khodadadi, H.; Baban, B.; Stranahan, A.M. Visceral adipose NLRP3 impairs cognition in obesity via IL-1R1 on CX3CR1+ cells. J. Clin. Investig. 2020, 130, 1961–1976. [Google Scholar] [CrossRef] [Green Version]
- Nazeri, A.; Crandall, J.P.; Fraum, T.J.; Wahl, R.L. Repeatability of Radiomic Features of Brown Adipose Tissue. J. Nucl. Med. 2021, 62, 700–706. [Google Scholar] [CrossRef]
- Reijrink, M.; de Boer, S.A.; Antunes, I.F.; Spoor, D.S.; Heerspink, H.J.; Lodewijk, M.E.; Mastik, M.F.; Boellaard, R.; Greuter, M.J.; Benjamens, S. [18 F] FDG Uptake in Adipose Tissue Is Not Related to Inflammation in Type 2 Diabetes Mellitus. Mol. Imaging Biol. 2021, 23, 117–126. [Google Scholar] [CrossRef]
- Pahk, K.; Kim, E.J.; Lee, Y.J.; Kim, S.; Seo, H.S. Characterization of glucose uptake metabolism in visceral fat by 18 F-FDG PET/CT reflects inflammatory status in metabolic syndrome. PLoS ONE 2020, 15, e0228602. [Google Scholar] [CrossRef] [Green Version]
- Bucerius, J.; Mani, V.; Wong, S.; Moncrieff, C.; Izquierdo-Garcia, D.; Machac, J.; Fuster, V.; Farkouh, M.E.; Rudd, J.H.; Fayad, Z.A. Arterial and fat tissue inflammation are highly correlated: A prospective 18F-FDG PET/CT study. Eur. J. Nucl. Med. Mol. Imaging 2014, 41, 934–945. [Google Scholar] [CrossRef] [Green Version]
- Trayhurn, P.; Beattie, J.H. Physiological role of adipose tissue: White adipose tissue as an endocrine and secretory organ. Proc. Nutr. Soc. 2001, 60, 329–339. [Google Scholar] [CrossRef] [Green Version]
- Ishii, M.; Iadecola, C. Adipocyte-derived factors in age-related dementia and their contribution to vascular and Alzheimer pathology. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2016, 1862, 966–974. [Google Scholar] [CrossRef]
- Kahn, C.R.; Wang, G.; Lee, K.Y. Altered adipose tissue and adipocyte function in the pathogenesis of metabolic syndrome. J. Clin. Investig. 2019, 129, 3990–4000. [Google Scholar] [CrossRef]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2019, 10, 1607. [Google Scholar] [CrossRef]
- Naderali, E.K.; Ratcliffe, S.H.; Dale, M.C. Obesity and Alzheimer’s disease: A link between body weight and cognitive function in old age. Am. J. Alzheimers Dis. Other Dement. 2009, 24, 445–449. [Google Scholar] [CrossRef]
- Tziomalos, K.; Dimitroula, H.V.; Katsiki, N.; Savopoulos, C.; Hatzitolios, A.I. Effects of lifestyle measures, antiobesity agents, and bariatric surgery on serological markers of inflammation in obese patients. Mediat. Inflamm. 2010, 2010, 364957. [Google Scholar] [CrossRef] [Green Version]
- Sun, Z.; Wang, Z.-T.; Sun, F.-R.; Shen, X.-N.; Xu, W.; Ma, Y.-H.; Dong, Q.; Tan, L.; Yu, J.-T.; Alzheimer’s Disease Neuroimaging Initiative. Late-life obesity is a protective factor for prodromal Alzheimer’s disease: A longitudinal study. Aging 2020, 12, 2005. [Google Scholar] [CrossRef]
- Yang, F.; Wang, G.; Wang, Z.; Sun, M.; Cao, M.; Zhu, Z.; Fu, Q.; Mao, J.; Shi, Y.; Yang, T. Visceral adiposity index may be a surrogate marker for the assessment of the effects of obesity on arterial stiffness. PLoS ONE 2014, 9, e104365. [Google Scholar] [CrossRef] [Green Version]
- Diehl-Wiesenecker, E.; von Armin, C.A.; Dupuis, L.; Muller, H.P.; Ludolph, A.C.; Kassubek, J. Adipose Tissue Distribution in Patients with Alzheimer’s Disease: A Whole Body MRI Case-Control Study. J. Alzheimers Dis. 2015, 48, 825–832. [Google Scholar] [CrossRef]
- Letra, L.; Matafome, P.; Rodrigues, T.; Duro, D.; Lemos, R.; Baldeiras, I.; Patrício, M.; Castelo-Branco, M.; Caetano, G.; Seiça, R. Association between adipokines and biomarkers of Alzheimer’s disease: A cross-sectional study. J. Alzheimers Dis. 2019, 67, 725–735. [Google Scholar] [CrossRef] [Green Version]
- Puig, K.L.; Floden, A.M.; Adhikari, R.; Golovko, M.Y.; Combs, C.K. Amyloid precursor protein and proinflammatory changes are regulated in brain and adipose tissue in a murine model of high fat diet-induced obesity. PLoS ONE 2012, 7, e30378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- An, Y.A.; Crewe, C.; Asterholm, I.W.; Sun, K.; Chen, S.; Zhang, F.; Shao, M.; Funcke, J.B.; Zhang, Z.; Straub, L.; et al. Dysregulation of Amyloid Precursor Protein Impairs Adipose Tissue Mitochondrial Function and Promotes Obesity. Nat. Metab. 2019, 1, 1243–1257. [Google Scholar] [CrossRef] [PubMed]
- Guo, Z.; Jiang, H.; Xu, X.; Duan, W.; Mattson, M.P. Leptin-mediated cell survival signaling in hippocampal neurons mediated by JAK STAT3 and mitochondrial stabilization. J. Biol. Chem. 2008, 283, 1754–1763. [Google Scholar] [CrossRef] [Green Version]
- Fewlass, D.C.; Noboa, K.; Pi-Sunyer, F.X.; Johnston, J.M.; Yan, S.D.; Tezapsidis, N. Obesity-related leptin regulates Alzheimer’s Abeta. FASEB J. 2004, 18, 1870–1878. [Google Scholar] [CrossRef] [PubMed]
- Paz-Filho, G.; Wong, M.L.; Licinio, J. The procognitive effects of leptin in the brain and their clinical implications. Int. J. Clin. Pract. 2010, 64, 1808–1812. [Google Scholar] [CrossRef] [Green Version]
- Mangge, H.; Almer, G.; Haj-Yahya, S.; Grandits, N.; Gasser, R.; Pilz, S.; Moller, R.; Horejsi, R. Nuchal thickness of subcutaneous adipose tissue is tightly associated with an increased LMW/total adiponectin ratio in obese juveniles. Atherosclerosis 2009, 203, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Mangge, H.; Almer, G.; Truschnig-Wilders, M.; Schmidt, A.; Gasser, R.; Fuchs, D. Inflammation, adiponectin, obesity and cardiovascular risk. Curr. Med. Chem. 2010, 17, 4511–4520. [Google Scholar] [CrossRef]
- Haase, J.; Weyer, U.; Immig, K.; Klöting, N.; Blüher, M.; Eilers, J.; Bechmann, I.; Gericke, M. Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia 2014, 57, 562–571. [Google Scholar] [CrossRef]
- Monteiro, R.; Azevedo, I. Chronic inflammation in obesity and the metabolic syndrome. Mediat. Inflamm. 2010, 2010. [Google Scholar] [CrossRef]
- Banks, W.A. Blood-brain barrier transport of cytokines: A mechanism for neuropathology. Curr. Pharm. Des. 2005, 11, 973–984. [Google Scholar] [CrossRef]
- Kitazawa, M.; Cheng, D.; Tsukamoto, M.R.; Koike, M.A.; Wes, P.D.; Vasilevko, V.; Cribbs, D.H.; LaFerla, F.M. Blocking IL-1 signaling rescues cognition, attenuates tau pathology, and restores neuronal β-catenin pathway function in an Alzheimer’s disease model. J. Immunol. 2011, 187, 6539–6549. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakandakari, S.C.B.R.; Munoz, V.R.; Kuga, G.K.; Gaspar, R.C.; Sant’Ana, M.R.; Pavan, I.C.B.; da Silva, L.G.S.; Morelli, A.P.; Simabuco, F.M.; da Silva, A.S.R. Short-term high-fat diet modulates several inflammatory, ER stress, and apoptosis markers in the hippocampus of young mice. Brain Behav. Immun. 2019, 79, 284–293. [Google Scholar] [CrossRef] [PubMed]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef] [PubMed]
- Van Leijsen, E.M.C.; Bergkamp, M.I.; van Uden, I.W.M.; Ghafoorian, M.; van der Holst, H.M.; Norris, D.G.; Platel, B.; Tuladhar, A.M.; de Leeuw, F.E. Progression of White Matter Hyperintensities Preceded by Heterogeneous Decline of Microstructural Integrity. Stroke 2018, 49, 1386–1393. [Google Scholar] [CrossRef] [PubMed]
- No, H.J.; Yi, H.A.; Won, K.S.; Chang, H.W.; Kim, H.W. Association between white matter lesions and the cerebral glucose metabolism in patients with cognitive impairment. Rev. Esp. Med. Nucl. Imagen Mol. 2019, 38, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Tzourio-Mazoyer, N.; Landeau, B.; Papathanassiou, D.; Crivello, F.; Etard, O.; Delcroix, N.; Mazoyer, B.; Joliot, M. Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single-subject brain. Neuroimage 2002, 15, 273–289. [Google Scholar] [CrossRef] [PubMed]
- Bullich, S.; Seibyl, J.; Catafau, A.M.; Jovalekic, A.; Koglin, N.; Barthel, H.; Sabri, O.; De Santi, S. Optimized classification of (18)F-Florbetaben PET scans as positive and negative using an SUVR quantitative approach and comparison to visual assessment. Neuroimage Clin. 2017, 15, 325–332. [Google Scholar] [CrossRef]
- Barthel, H.; Gertz, H.J.; Dresel, S.; Peters, O.; Bartenstein, P.; Buerger, K.; Hiemeyer, F.; Wittemer-Rump, S.M.; Seibyl, J.; Reininger, C.; et al. Cerebral amyloid-beta PET with florbetaben (18F) in patients with Alzheimer’s disease and healthy controls: A multicentre phase 2 diagnostic study. Lancet Neurol. 2011, 10, 424–435. [Google Scholar] [CrossRef]
- Talairach, J.; Tournoux, P. Co-Planar Stereotaxic Atlas of the Human Brain: Three-Dimensional Proportional System; Thieme Medical: New York, NY, USA, 1988. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variables | Total (n = 54) | CU (n = 18) | MCI (n = 14) | Dementia (n = 22) | p |
|---|---|---|---|---|---|
| Age, years (SD) | 66.4 (8.4) | 62.7 (5.6) | 63.9 (8.9) | 71.1 (8.2) | 0.002 1 |
| Sex, female, n (%) | 34 (63.0) | 11 (61.1) | 7 (50.0) | 16 (72.7) | 0.380 |
| Body mass index (SD) | 23.3 (3.4) | 24.3 (4.4) | 22.6 (1.0) | 22.9 (3.5) | 0.277 |
| Education, years (SD) | 11.5 (6.1) | 14.3 (3.5) | 13.8 (6.4) | 7.6 (5.6) | <0.001 2 |
| Diabetes, n (%) | 8 (14.8) | 1 (5.6%) | 2 (14.3) | 5 (22.7) | 0.314 |
| Hypertension, n (%) | 16 (29.6) | 1 (7.1) | 4 (28.6) | 11 (50.0) | 0.0261 |
| Cardiovascular disease, n (%) | 6 (11.1) | 1 (7.1) | 2 (15.4) | 3 (14.3) | 0.768 |
| Hyperlipidemia, n (%) | 9 (16.7) | 1 (8.3) | 3 (21.4) | 5 (22.7) | 0.563 |
| WMH volume (SD) | 3.4 (5.1) | 1.1 (2.2) | 1.6 (2.0) | 6.4 (6.5) | 0.001 1 |
| MMSE (SD) | 24.6 (5.3) | 28.9 (1.2) | 25.9 (2.9) | 20.3 (5.3) | <0.001 2 |
| K-BNT (SD) | 42.4 (13.5) | 52.0 (4.0) | 47.8 (10.1) | 31.1 (12.2) | <0.001 2 |
| Aβ positivity, n (%) | 26 (48.1) | 5 (27.8) | 7 (50.0) | 14 (63.6) | 0.077 |
| Composite SUVRFBB | 1.47 (0.28) | 1.33 (0.11) | 1.48 (0.22) | 1.57 (0.36) | 0.017 1 |
| VAT SUVmax (SD) | 0.71 (0.16) | 0.69 (0.17) | 0.67 (0.11) | 0.76 (0.17) | 0.200 |
| VAT SUVmean (SD) | 0.44 (0.11) | 0.41 (0.11) | 0.41 (0.08) | 0.48 (0.12) | 0.067 |
| Variables | Total (n = 54) | Low VAT Metabolism Group (n = 31) | High VAT Metabolism Group (n = 23) | p |
|---|---|---|---|---|
| Age, years (SD) | 66.4 (8.4) | 65.3 (8.3) | 67.9 (8.5) | 0.269 |
| Sex, female, n (%) | 34 (63.0) | 18 (58.1) | 16 (69.6) | 0.412 |
| Body mass index (SD) | 23.3 (3.4) | 23.8 (3.0) | 22.5 (3.9) | 0.133 |
| Education, years (SD) | 11.5 (6.1) | 12.1 (5.9) | 10.7 (6.3) | 0.403 |
| Diabetes, n (%) | 8 (14.8) | 4 (12.9) | 4 (17.4) | 0.711 |
| Hypertension, n (%) | 16 (29.6) | 10 (37.0) | 6 (26.1) | 0.546 |
| Cardiovascular disease, n (%) | 6 (11.1) | 4 (14.8) | 2 (9.5) | 0.683 |
| Hyperlipidemia, n (%) | 9 (16.7) | 6 (23.1) | 3 (13.6) | 0.478 |
| WMH volume (SD) | 3.4 (5.08) | 2.7 (4.0) | 4.3 (6.2) | 0.245 |
| Cognitive stage | ||||
| CU, n (%) | 18 (33.3) | 13 (41.9) | 5 (21.7) | 0.234 |
| MCI, n (%) | 14 (26.0) | 8 (25.8) | 6 (26.1) | |
| Dementia, n (%) | 22 (40.7) | 10 (32.3) | 12 (52.2) | |
| MMSE (SD) | 24.6 (5.3) | 25.8 (3.8) | 23.0 (6.6) | 0.245 |
| K-BNT (SD) | 42.4 (13.5) | 45.0 (10.8) | 38.9 (15.9) | 0.107 |
| Aβ positivity, n (%) | 26 (48.1%) | 8 (25.8) | 18 (78.3) | <0.001 |
| VAT SUVmax (SD) | 0.71 (0.16) | 0.61 (0.09) | 0.85 (0.12) | <0.001 |
| VAT SUVmean (SD) | 0.44 (0.11) | 0.37 (0.06) | 0.54 (0.83) | <0.001 |
| Regions | Brodmann Area | Size | MNI Coordinates | T Value | p | ||
|---|---|---|---|---|---|---|---|
| X | Y | Z | |||||
| Right occipital lobe, lingual gyrus | BA 18 | 5896 | 2 | −84 | −8 | 3.56 | <0.001 |
| Right parietal lobe, precuneus | BA 19 | 26 | −80 | 42 | 3.52 | <0.001 | |
| Right parietal lobe, precuneus | BA 31 | 8 | −68 | 24 | 3.23 | 0.001 | |
| Right frontal lobe, precentral gyrus | BA 44 | 1315 | 62 | 8 | 4 | 3.55 | <0.001 |
| Right temporal lobe, middle temporal gyrus | BA 21 | 64 | 0 | −8 | 3.35 | <0.001 | |
| Right insula | BA 13 | 36 | 10 | 4 | 3.31 | <0.001 | |
| Left parietal lobe, precuneus | BA 7 | 4022 | −18 | −78 | 48 | 3.54 | <0.001 |
| Left temporal lobe, inferior temporal gyrus | BA 20 | −62 | −28 | −16 | 3.44 | <0.001 | |
| Left occipital lobe, inferior occipital gyrus | BA 18 | −34 | −90 | −14 | 3.2 | 0.001 | |
| Left frontal lobe, rectal gyrus | BA 11 | 2258 | −8 | 10 | −24 | 3.37 | <0.001 |
| Right frontal lobe, inferior frontal gyrus | BA 47 | 26 | 12 | −22 | 3.28 | <0.001 | |
| Left frontal lobe, medial frontal gyrus | BA 25 | −6 | 6 | −16 | 3.26 | <0.001 | |
| Left frontal lobe, superior frontal gyrus | BA 6 | 348 | −2 | 4 | 54 | 3.3 | <0.001 |
| Left frontal lobe, medial frontal gyrus | BA 6 | −4 | −8 | 58 | 3.02 | 0.002 | |
| Left frontal lobe, inferior frontal gyrus | BA 44 | 450 | −60 | 8 | 18 | 3.17 | 0.001 |
| Left cerebrum, frontal lobe, precentral gyrus | BA 6 | −60 | 6 | 30 | 3.17 | 0.001 | |
| Left frontal lobe, inferior frontal gyrus | BA 45 | −56 | 20 | 12 | 3.07 | 0.002 | |
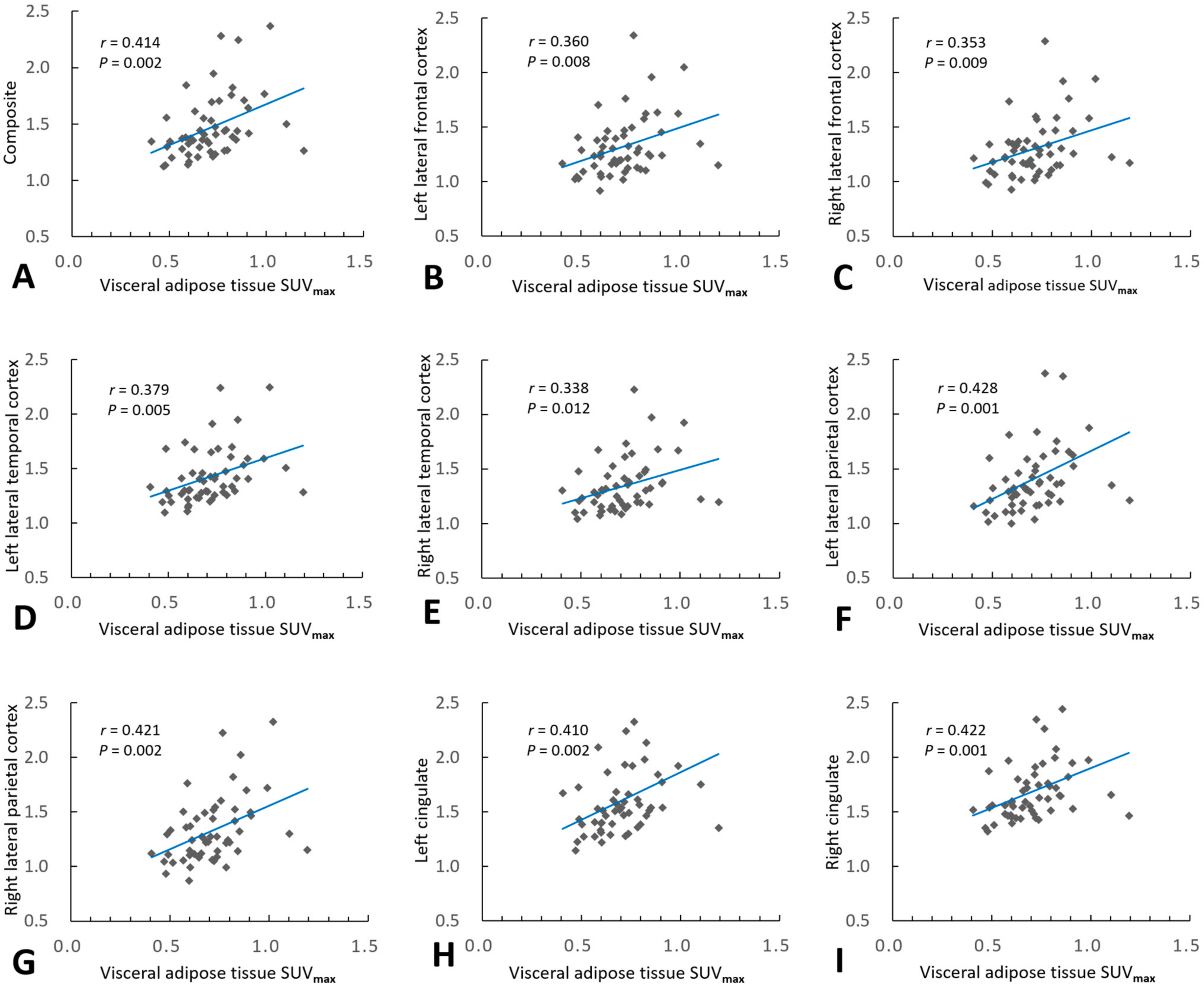
| Regions | Univariable Model | Multivariable Model | |||
|---|---|---|---|---|---|
| r | p | Adjusted R2 | Standardized β 2 | p | |
| Composite 1 | 0.414 | 0.002 | 0.195 | 0.359 | 0.007 |
| Left lateral frontal cortex | 0.360 | 0.008 | 0.113 | 0.360 | 0.008 |
| Right lateral frontal cortex | 0.353 | 0.009 | 0.140 | 0.302 | 0.025 |
| Left lateral temporal cortex | 0.379 | 0.005 | 0.127 | 0.379 | 0.005 |
| Right lateral temporal cortex | 0.338 | 0.012 | 0.147 | 0.278 | 0.038 |
| Left lateral parietal cortex | 0.428 | 0.001 | 0.218 | 0.369 | 0.005 |
| Right lateral parietal cortex | 0.421 | 0.002 | 0.201 | 0.366 | 0.005 |
| Left cingulate | 0.410 | 0.002 | 0.199 | 0.352 | 0.008 |
| Right cingulate | 0.422 | 0.001 | 0.162 | 0.422 | 0.001 |
| Regions | Univariable Model | Multivariable Model | |||
|---|---|---|---|---|---|
| r | p | Adjusted R2 | Standardized β 2 | p | |
| Composite 1 | 0.367 | 0.006 | 0.150 | 0.295 | 0.032 |
| Left lateral frontal cortex | 0.319 | 0.019 | 0.085 | 0.319 | 0.019 |
| Right lateral frontal cortex | 0.318 | 0.019 | 0.084 | 0.318 | 0.019 |
| Left lateral temporal cortex | 0.340 | 0.012 | 0.098 | 0.340 | 0.012 |
| Right lateral temporal cortex | 0.304 | 0.025 | 0.118 | 0.224 | 0.106 |
| Left lateral parietal cortex | 0.380 | 0.005 | 0.169 | 0.302 | 0.026 |
| Right lateral parietal cortex | 0.379 | 0.005 | 0.158 | 0.308 | 0.024 |
| Left cingulate | 0.360 | 0.008 | 0.152 | 0.282 | 0.039 |
| Right cingulate | 0.369 | 0.006 | 0.120 | 0369 | 0.006 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, S.; Yi, H.-A.; Won, K.S.; Lee, J.S.; Kim, H.W. Association between Visceral Adipose Tissue Metabolism and Alzheimer’s Disease Pathology. Metabolites 2022, 12, 258. https://doi.org/10.3390/metabo12030258
Kim S, Yi H-A, Won KS, Lee JS, Kim HW. Association between Visceral Adipose Tissue Metabolism and Alzheimer’s Disease Pathology. Metabolites. 2022; 12(3):258. https://doi.org/10.3390/metabo12030258
Chicago/Turabian StyleKim, Shin, Hyon-Ah Yi, Kyoung Sook Won, Ji Soo Lee, and Hae Won Kim. 2022. "Association between Visceral Adipose Tissue Metabolism and Alzheimer’s Disease Pathology" Metabolites 12, no. 3: 258. https://doi.org/10.3390/metabo12030258
APA StyleKim, S., Yi, H. -A., Won, K. S., Lee, J. S., & Kim, H. W. (2022). Association between Visceral Adipose Tissue Metabolism and Alzheimer’s Disease Pathology. Metabolites, 12(3), 258. https://doi.org/10.3390/metabo12030258