
Synthesis of Human Phase I and Phase II Metabolites of Hop (Humulus lupulus) Prenylated Flavonoids

 , , and
, , and
Abstract
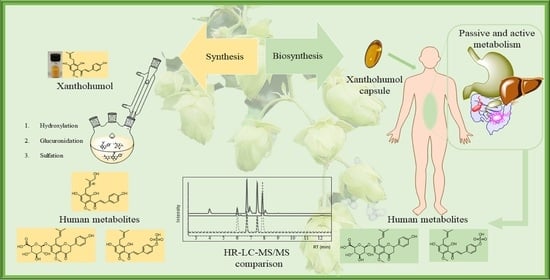
:
1. Introduction
2. Results and Discussion
2.1. Hydroxylation of the Prenyl Group
2.2. Synthesis of Sulfates
2.3. Biosynthetic Glucuronidation
XN-C-Glucuronide and IXN-C-Glucuronide
2.4. Synthesis of Glucuronides
2.5. LC-MS/MS of Synthesised Analytes
2.6. LC-MS/MS of Analytes in Human Blood after Xanthohumol Consumption
3. Materials and Methods
3.1. General
3.2. Reference Compounds
3.2.1. General Reaction of Hydroxylated Prenylated Flavonoids
3.2.2. Synthesis of Sulphate Conjugates
3.2.3. Synthesis of Synthetic Glucuronic Acid Conjugates
3.3. Biosynthesis of Glucuronic Acid Conjugates
3.3.1. Purification of Pig-Liver Microsomes
3.3.2. Glucuronidation of Flavonoids Using Pig-Liver Microsomes
3.3.3. Protein Content of Liver Microsomes
3.3.4. Enzymatic Digestion of Glucuronides
3.4. Analysis of Samples
3.4.1. HPLC
3.4.2. Mass Spectrometry
3.4.3. Nuclear Magnetic Resonance Spectroscopy
3.4.4. LC-MS/MS
3.5. Preparation of the Blood Sample
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Yang, X.; Jiang, Y.; Yang, J.; He, J.; Sun, J.; Chen, F.; Zhang, M.; Yang, B. Prenylated flavonoids, promising nutraceuticals with impressive biological activities. Trends Food Sci. Technol. 2015, 44, 93–104. [Google Scholar] [CrossRef]
- Awouafack, M.D.; Wong, C.P.; Tane, P.; Morita, H. Prenylated Flavonoids in Food. In Handbook of Dietary Phytochemicals; Xiao, J., Sarker, S.D., Asakawa, Y., Eds.; Springer: Singapore, 2020; pp. 1–23. [Google Scholar]
- Butt, M.S.; Nazir, A.; Sultan, M.T.; Schroën, K. Morus alba L. nature’s functional tonic. Trends Food Sci. Technol. 2008, 19, 505–512. [Google Scholar] [CrossRef]
- Chen, X.; Mukwaya, E.; Wong, M.-S.; Zhang, Y. A systematic review on biological activities of prenylated flavonoids. Pharm. Biol. 2014, 52, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.K.; Jiang, Y.; Yang, B. An update of prenylated phenolics: Food sources, chemistry and health benefits. Trends Food Sci. Technol. 2021, 108, 197–213. [Google Scholar] [CrossRef]
- Wen, L.; Zhou, T.; Jiang, Y.; Chang, S.K.; Yang, B. Prenylated flavonoids in foods and their applications on cancer prevention. Crit. Rev. Food Sci. Nutr. 2021, 1–14. [Google Scholar] [CrossRef]
- Stevens, J.F.; Page, J.E. Xanthohumol and related prenylflavonoids from hops and beer: To your good health! Phytochemistry 2004, 65, 1317–1330. [Google Scholar] [CrossRef]
- Mukai, R. Prenylation enhances the biological activity of dietary flavonoids by altering their bioavailability. Biosci. Biotechnol. Biochem. 2018, 82, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Neumann, H.F.; Frank, J.; Venturelli, S.; Egert, S. Bioavailability and Cardiometabolic Effects of Xanthohumol: Evidence from Animal and Human Studies. Mol. Nutr. Food Res. 2021, 66, e2100831. [Google Scholar] [CrossRef]
- Pinto, C.; Cestero, J.J.; Rodríguez-Galdón, B.; Macías, P. Xanthohumol, a prenylated flavonoid from hops (Humulus lupulus L.), protects rat tissues against oxidative damage after acute ethanol administration. Toxicol. Rep. 2014, 1, 726–733. [Google Scholar] [CrossRef] [Green Version]
- Miranda, C.L.; Stevens, J.F.; Helmrich, A.; Henderson, M.C.; Rodriguez, R.J.; Yang, Y.H.; Deinzer, M.L.; Barnes, D.W.; Buhler, D.R. Antiproliferative and cytotoxic effects of prenylated flavonoids from hops (Humulus lupulus) in human cancer cell lines. Food Chem. Toxicol. 1999, 37, 271–285. [Google Scholar] [CrossRef]
- Urmann, C.; Bieler, L.; Priglinger, E.; Aigner, L.; Couillard-Despres, S.; Riepl, H.M. Neuroregenerative Potential of Prenyl- and Pyranochalcones: A Structure–Activity Study. J. Nat. Prod. 2021, 84, 2675–2682. [Google Scholar] [CrossRef] [PubMed]
- Duan, X.; Tang, X.; Nair, M.S.; Zhang, T.; Qiu, Y.; Zhang, W.; Wang, P.; Huang, Y.; Xiang, J.; Wang, H.; et al. An Airway Organoid-Based Screen Identifies a Role for the HIF1α-Glycolysis Axis in SARS-CoV-2 Infection. Cell Rep. 2021, 37, 109920. [Google Scholar] [CrossRef] [PubMed]
- Lucas, K.; Frohlich-Nowoisky, J.; Oppitz, N.; Ackermann, M. Cinnamon and Hop Extracts as Potential Immunomodulators for Severe COVID-19 Cases. Front. Plant Sci. 2021, 12, 7. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Yan, B.; Cao, J.; Ye, Z.-W.; Liang, R.; Tang, K.; Luo, C.; Cai, J.; Chu, H.; Chung, T.W.-H.; et al. SARS-CoV-2 exploits host DGAT and ADRP for efficient replication. Cell Discov. 2021, 7, 100. [Google Scholar] [CrossRef] [PubMed]
- Langley, B.O.; Ryan, J.J.; Hanes, D.; Phipps, J.; Stack, E.; Metz, T.O.; Stevens, J.F.; Bradley, R. Xanthohumol Microbiome and Signature in Healthy Adults (the XMaS Trial): Safety and Tolerability Results of a Phase I Triple-Masked, Placebo-Controlled Clinical Trial. Mol. Nutr. Food Res. 2021, 65, 2001170. [Google Scholar] [CrossRef]
- Aghamiri, V.; Mirghafourvand, M.; Mohammad-Alizadeh-Charandabi, S.; Nazemiyeh, H. The effect of Hop (Humulus lupulus L.) on early menopausal symptoms and hot flashes: A randomized placebo-controlled trial. Complement. Ther. Clin. Pract. 2016, 23, 130–135. [Google Scholar] [CrossRef]
- Jongkees, S.A.K.; Withers, S.G. Glycoside Cleavage by a New Mechanism in Unsaturated Glucuronyl Hydrolases. J. Am. Chem. Soc. 2011, 133, 19334–19337. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Nakada, M. Allylic Oxidations in Natural Product Synthesis. Synthesis 2013, 45, 1421–1451. [Google Scholar] [CrossRef]
- Legette, L.; Karnpracha, C.; Reed, R.L.; Choi, J.; Bobe, G.; Christensen, J.M.; Rodriguez-Proteau, R.; Purnell, J.Q.; Stevens, J.F. Human pharmacokinetics of xanthohumol, an antihyperglycemic flavonoid from hops. Mol. Nutr. Food Res. 2014, 58, 248–255. [Google Scholar] [CrossRef]
- Grudniewska, A.; Popłoński, J. Simple and green method for the extraction of xanthohumol from spent hops using deep eutectic solvents. Sep. Purif. 2020, 250, 117196. [Google Scholar] [CrossRef]
- Zhang, J.L.; Yan, L.; Wei, P.; Zhou, R.Y.; Hua, C.J.; Xiao, M.; Tu, Y.P.; Gu, Z.J.; Wei, T.T. PEG-GO@XN nanocomposite suppresses breast cancer metastasis via inhibition of mitochondrial oxidative phosphorylation and blockade of epithelial-to-mesenchymal transition. Eur. J. Pharmacol. 2021, 895, 9. [Google Scholar] [CrossRef] [PubMed]
- Stompor, M.; Dancewicz, K.; Gabryś, B.; Anioł, M. Insect Antifeedant Potential of Xanthohumol, Isoxanthohumol, and Their Derivatives. J. Agric. Food Chem. 2015, 63, 6749–6756. [Google Scholar] [CrossRef] [PubMed]
- Ogunnupebi, T.A.; Oluyori, A.P.; Dada, A.O.; Oladeji, O.S.; Inyinbor, A.A.; Egharevba, G.O. Promising Natural Products in Crop Protection and Food Preservation: Basis, Advances, and Future Prospects. Int. J. Agron. 2020, 2020, 8840046. [Google Scholar] [CrossRef]
- Khatib, N.; Varidi, M.J.; Mohebbi, M.; Varidi, M.; Hosseini, S.M.H. Replacement of nitrite with lupulon–xanthohumol loaded nanoliposome in cooked beef-sausage: Experimental and model based study. J. Food Sci. Technol. 2020, 57, 2629–2639. [Google Scholar] [CrossRef]
- Nikolic, D.; Li, Y.; Chadwick, L.R.; Pauli, G.F.; van Breemen, R.B. Metabolism of xanthohumol and isoxanthohumol, prenylated flavonoids from hops (Humulus lupulus L.), by human liver microsomes. J. Mass Spectrom. 2005, 40, 289–299. [Google Scholar] [CrossRef]
- van Breemen, R.B.; Chen, L.; Tonsing-Carter, A.; Banuvar, S.; Barengolts, E.; Viana, M.; Chen, S.-N.; Pauli, G.F.; Bolton, J.L. Pharmacokinetic Interactions of a Hop Dietary Supplement with Drug Metabolism in Perimenopausal and Postmenopausal Women. J. Agric. Food Chem. 2020, 68, 5212–5220. [Google Scholar] [CrossRef]
- Nookandeh, A.; Frank, N.; Steiner, F.; Ellinger, R.; Schneider, B.; Gerhäuser, C.; Becker, H. Xanthohumol metabolites in faeces of rats. Phytochemistry 2004, 65, 561–570. [Google Scholar] [CrossRef]
- Chadwick, L.R.; Nikolic, D.; Burdette, J.E.; Overk, C.R.; Bolton, J.L.; van Breemen, R.B.; Fröhlich, R.; Fong, H.H.S.; Farnsworth, N.R.; Pauli, G.F. Estrogens and Congeners from Spent Hops (Humulus lupulus). J. Nat. Prod. 2004, 67, 2024–2032. [Google Scholar] [CrossRef]
- Herath, W.; Ferreira, D.; Khan, S.; Khan, I. Identification and Biological Activity of Microbial Metabolites of Xanthohumol. Chem. Pharm. Bull. 2004, 51, 1237–1240. [Google Scholar] [CrossRef] [Green Version]
- Ruefer, C.E.; Gerhäuser, C.; Frank, N.; Becker, H.; Kulling, S.E. In vitro phase II metabolism of xanthohumol by human UDP-glucuronosyltransferases and sulfotransferases. Mol. Nutr. Food Res. 2005, 49, 851–856. [Google Scholar] [CrossRef]
- Budziak, I.; Arczewska, M.; Kamiński, D.M. Formation of Prenylated Chalcone Xanthohumol Cocrystals: Single Crystal X-ray Diffraction, Vibrational Spectroscopic Study Coupled with Multivariate Analysis. Molecules 2019, 24, 4245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yilmazer, M.; Stevens, J.F.; Buhler, D.R. In vitro glucuronidation of xanthohumol, a flavonoid in hop and beer, by rat and human liver microsomes. FEBS Lett. 2001, 491, 252–256. [Google Scholar] [CrossRef] [Green Version]
- Schymanski, E.L.; Jeon, J.; Gulde, R.; Fenner, K.; Ruff, M.; Singer, H.P.; Hollender, J. Identifying small molecules via high resolution mass spectrometry: Communicating confidence. Environ. Sci. Technol. 2014, 48, 2097–2098. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bobe, G.; Miranda, C.L.; Lowry, M.B.; Hsu, V.L.; Lohr, C.V.; Wong, C.P.; Jump, D.B.; Robinson, M.M.; Sharpton, T.J.; et al. Tetrahydroxanthohumol, a xanthohumol derivative, attenuates high-fat diet-induced hepatic steatosis by antagonizing PPARγ. eLife 2021, 10, e66398. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zeng, L.; Xie, J.; Yue, Z.; Deng, H.; Ma, X.; Zheng, C.; Wu, X.; Luo, J.; Liu, M. Inhibition of Osteoclastogenesis and Bone Resorption in vitro and in vivo by a prenylflavonoid xanthohumol from hops. Sci. Rep. 2015, 5, 17605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Logan, I.E.; Shulzhenko, N.; Sharpton, T.J.; Bobe, G.; Liu, K.; Nuss, S.; Jones, M.L.; Miranda, C.L.; Vasquez-Perez, S.; Pennington, J.M.; et al. Xanthohumol Requires the Intestinal Microbiota to Improve Glucose Metabolism in Diet-Induced Obese Mice. Mol. Nutr. Food Res. 2021, 65, 2100389. [Google Scholar] [CrossRef]
- Pohjanvirta, R.; Nasri, A. The Potent Phytoestrogen 8-Prenylnaringenin: A Friend or a Foe? Int. J. Mol. Sci. 2022, 23, 3168. [Google Scholar] [CrossRef]
- Ferk, F.; Mišík, M.; Nersesyan, A.; Pichler, C.; Jäger, W.; Szekeres, T.; Marculescu, R.; Poulsen, H.E.; Henriksen, T.; Bono, R.; et al. Impact of xanthohumol (a prenylated flavonoid from hops) on DNA stability and other health-related biochemical parameters: Results of human intervention trials. Mol. Nutr. Food Res. 2016, 60, 773–786. [Google Scholar] [CrossRef]
- Pichler, C.; Ferk, F.; Al-Serori, H.; Huber, W.; Jäger, W.; Waldherr, M.; Mišík, M.; Kundi, M.; Nersesyan, A.; Herbacek, I.; et al. Xanthohumol Prevents DNA Damage by Dietary Carcinogens: Results of a Human Intervention Trial. Cancer Prev. Res. 2017, 10, 153–160. [Google Scholar] [CrossRef] [Green Version]
- van Breemen, R.B.; Li, Y. Caco-2 cell permeability assays to measure drug absorption. Expert Opin. Drug Metab. Toxicol. 2005, 1, 175–185. [Google Scholar] [CrossRef]
- Qi, J.; Mulabagal, V.; Liu, L.; Wilson, C.; Hayworth, J.S. A rapid UHPLC-MS/MS method for quantitation of phytoestrogens and the distribution of enterolactone in an Alabama estuary. Chemosphere 2019, 237, 124472. [Google Scholar] [CrossRef] [PubMed]
- Buckett, L.; Schinko, S.; Urmann, C.; Riepl, H.; Rychlik, M. Stable Isotope Dilution Analysis of the Major Prenylated Flavonoids Found in Beer, Hop Tea, and Hops. Front. Nutr. 2020, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Woodward, R.B.; Cava, M.P.; Ollis, W.D.; Hunger, A.; Daeniker, H.U.; Schenker, K. The Total Synthesis of Strychnine. J. Am. Chem. Soc. 1954, 76, 4749–4751. [Google Scholar] [CrossRef]
- Kamath, S.A.; Kummerow, F.A.; Narayan, K.A. A simple procedure for the isolation of rat liver microsomes. FEBS Lett. 1971, 17, 90–92. [Google Scholar] [CrossRef] [Green Version]
- Ladd, M.A.; Fitzsimmons, P.N.; Nichols, J.W. Optimization of a UDP-glucuronosyltransferase assay for trout liver S9 fractions: Activity enhancement by alamethicin, a pore-forming peptide. Xenobiotica 2016, 46, 1066–1075. [Google Scholar] [CrossRef]
- Zor, T.; Selinger, Z. Linearization of the Bradford Protein Assay Increases Its Sensitivity: Theoretical and Experimental Studies. Anal. Biochem. 1996, 236, 302–308. [Google Scholar] [CrossRef] [Green Version]
- Grebenstein, N.; Schlienz, J.; Calvo-Castro, L.A.; Burkard, M.; Venturelli, S.; Busch, C.; Frank, J. Validation of a rapid and sensitive reversed-phase liquid chromatographic method for the quantification of prenylated chalcones and flavanones in plasma and urine. NFS J. 2017, 10, 1–9. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Level 1: Confirmed Structure (Reference Standard) | Level 2: Probable Structure (Ms2) | Level 3: Tentative Structure (MS2) | Level 4: Molecular Formula | Level 5: Exact Mass of Interest (MS) |
|---|---|---|---|---|
| XN-7-O-GLc | - | IXN-4′O-Glc | - | - |
| IXN-7-O-Glc | - | 8-PN-4′-O-Glc | - | - |
| 6PN-7-O-Glc | - | 6-PN-4′-O-Glc | - | - |
| 8-PN-7-O-Glc | - | 8-PN-7-O-sulfate | - | - |
| XN-4′O-Sulfate | - | 6-PN-7-O-sulfate | - | - |
| 6-PN-7-O-sulfate | - | 6-PN-4′-O-sulfate | - | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buckett, L.; Schönberger, S.; Spindler, V.; Sus, N.; Schoergenhofer, C.; Frank, J.; Frank, O.; Rychlik, M. Synthesis of Human Phase I and Phase II Metabolites of Hop (Humulus lupulus) Prenylated Flavonoids. Metabolites 2022, 12, 345. https://doi.org/10.3390/metabo12040345
Buckett L, Schönberger S, Spindler V, Sus N, Schoergenhofer C, Frank J, Frank O, Rychlik M. Synthesis of Human Phase I and Phase II Metabolites of Hop (Humulus lupulus) Prenylated Flavonoids. Metabolites. 2022; 12(4):345. https://doi.org/10.3390/metabo12040345
Chicago/Turabian StyleBuckett, Lance, Sabrina Schönberger, Veronika Spindler, Nadine Sus, Christian Schoergenhofer, Jan Frank, Oliver Frank, and Michael Rychlik. 2022. "Synthesis of Human Phase I and Phase II Metabolites of Hop (Humulus lupulus) Prenylated Flavonoids" Metabolites 12, no. 4: 345. https://doi.org/10.3390/metabo12040345
APA StyleBuckett, L., Schönberger, S., Spindler, V., Sus, N., Schoergenhofer, C., Frank, J., Frank, O., & Rychlik, M. (2022). Synthesis of Human Phase I and Phase II Metabolites of Hop (Humulus lupulus) Prenylated Flavonoids. Metabolites, 12(4), 345. https://doi.org/10.3390/metabo12040345