
Untargeted Metabolomics of Slc13a5 Deficiency Reveal Critical Liver–Brain Axis for Lipid Homeostasis

 , , , , and
, , , , and 
Abstract
:
1. Introduction

2. Results
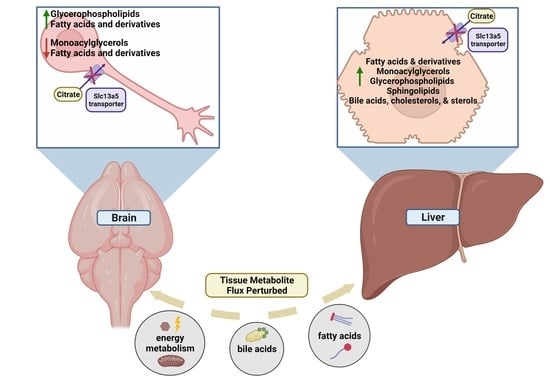
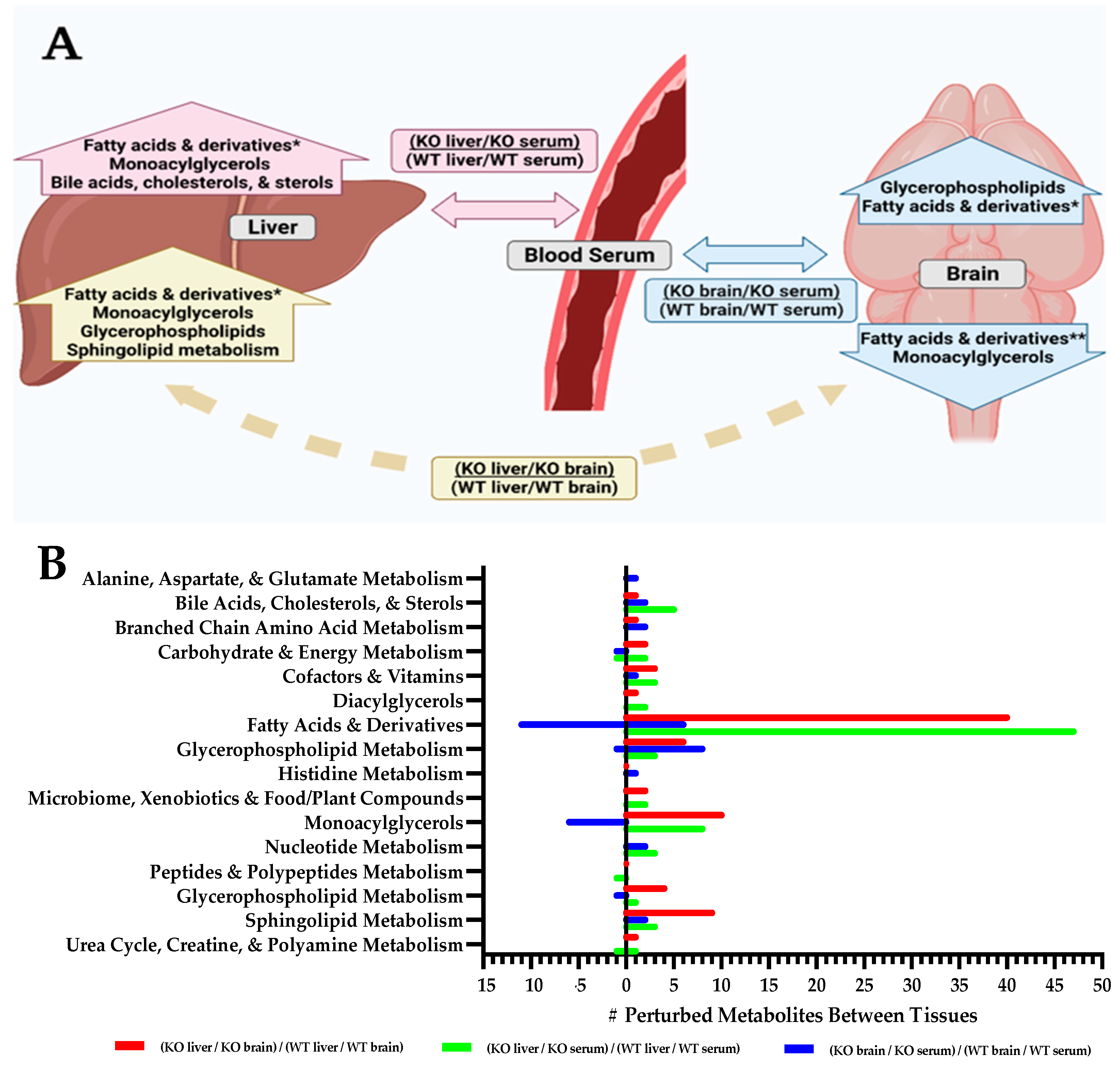
2.1. Tissue-Specific Perturbations in Slc13a5 Deficient Mice
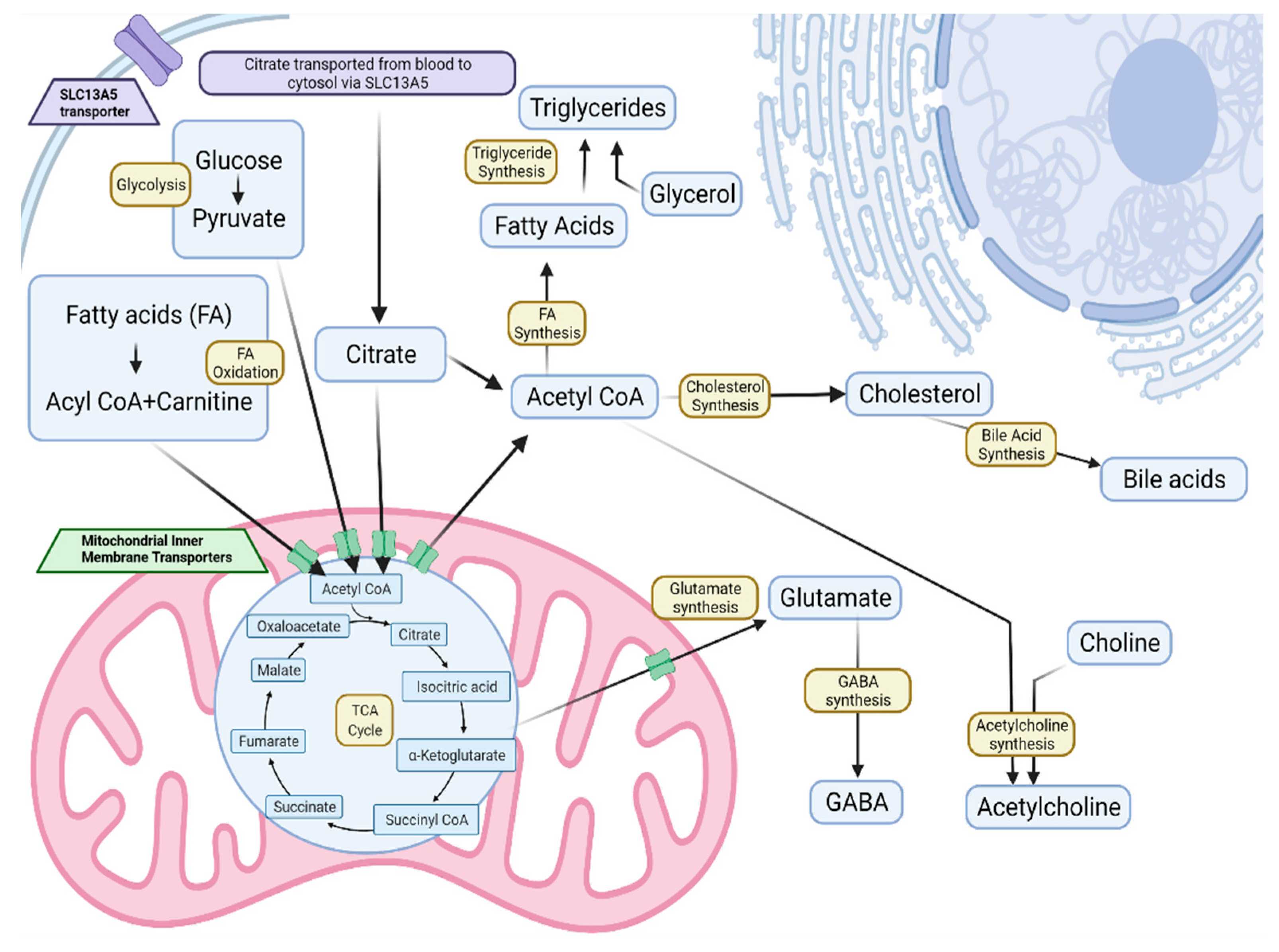
2.2. Tissue Metabolite Perturbations Suggest Abnormal Metabolite Flux in Slc13a5 Deficient Mice
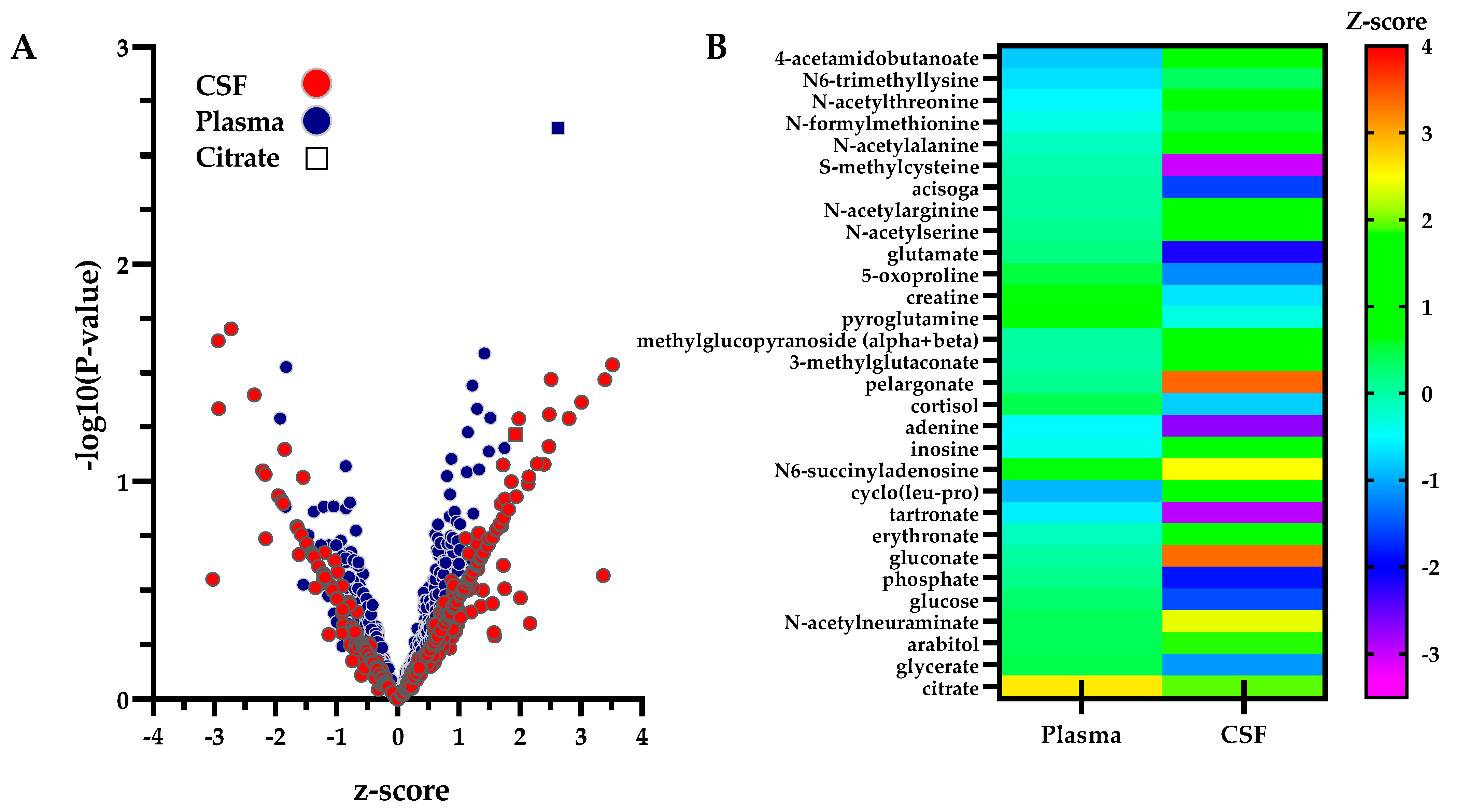
2.3. Biofluid-Specific Perturbations in Patients with Biallelic SLC13A5 Variants
2.4. Tissue Metabolite Perturbations Suggest Abnormal Metabolite Flux in SLC13A5 Deficient Patients
3. Discussion
4. Methods
4.1. Generation of Slc13a5-Deficient Mouse Model, Mouse Husbandry, and Colony Management
4.2. Mouse Tissue Sample Preparation
4.3. Human Plasma and CSF Sample Collection and Analysis
4.4. Untargeted Metabolomics
4.5. Metabolomics Data Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Higuchi, K.; Kopel, J.J.; Sivaprakasam, S.; Jaramillo-Martinez, V.; Sutton, R.B.; Urbatsch, I.L.; Ganapathy, V. Functional analysis of a species-specific inhibitor selective for human Na+-coupled citrate transporter (NaCT/SLC13A5/mINDY). Biochem. J. 2020, 477, 4149–4165. [Google Scholar] [CrossRef] [PubMed]
- Inoue, K.; Zhuang, L.; Maddox, D.M.; Smith, S.B.; Ganapathy, V. Structure, function, and expression pattern of a novel sodium-coupled citrate transporter (NaCT) cloned from mammalian brain. J. Biol. Chem. 2002, 277, 39469–39476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kopel, J.J.; Bhutia, Y.D.; Sivaprakasam, S.; Ganapathy, V. Consequences of NaCT/SLC13A5/mINDY deficiency: Good versus evil, separated only by the blood-brain barrier. Biochem. J. 2021, 478, 463–486. [Google Scholar] [CrossRef] [PubMed]
- Rogina, B.; Reenan, R.A.; Nilsen, S.P.; Helfand, S.L. Extended life-span conferred by cotransporter gene mutations in Drosophila. Science 2000, 290, 2137–2140. [Google Scholar] [CrossRef] [PubMed]
- Marden, J.H.; Rogina, B.; Montooth, K.L.; Helfand, S.L. Conditional tradeoffs between aging and organismal performance of Indy long-lived mutant flies. Proc. Natl. Acad. Sci. USA 2003, 100, 3369–3373. [Google Scholar] [CrossRef] [Green Version]
- Fei, Y.J.; Liu, J.C.; Inoue, K.; Zhuang, L.; Miyake, K.; Miyauchi, S.; Ganapathy, V. Relevance of NAC-2, an Na+-coupled citrate transporter, to life span, body size and fat content in Caenorhabditis elegans. Biochem. J. 2004, 379, 191–198. [Google Scholar] [CrossRef]
- Thevenon, J.; Milh, M.; Feillet, F.; St-Onge, J.; Duffourd, Y.; Juge, C.; Roubertie, A.; Heron, D.; Mignot, C.; Raffo, E.; et al. Mutations in SLC13A5 cause autosomal-recessive epileptic encephalopathy with seizure onset in the first days of life. Am. J. Hum. Genet. 2014, 95, 113–120. [Google Scholar] [CrossRef]
- Hardies, K.; de Kovel, C.G.; Weckhuysen, S.; Asselbergh, B.; Geuens, T.; Deconinck, T.; Azmi, A.; May, P.; Brilstra, E.; Becker, F.; et al. Recessive mutations in SLC13A5 result in a loss of citrate transport and cause neonatal epilepsy, developmental delay and teeth hypoplasia. Brain 2015, 138, 3238–3250. [Google Scholar] [CrossRef] [Green Version]
- Bhutia, Y.D.; Kopel, J.J.; Lawrence, J.J.; Neugebauer, V.; Ganapathy, V. Plasma Membrane Na⁺-Coupled Citrate Transporter (SLC13A5) and Neonatal Epileptic Encephalopathy. Molecules 2017, 22, 378. [Google Scholar] [CrossRef]
- Henke, C.; Töllner, K.; van Dijk, R.M.; Miljanovic, N.; Cordes, T.; Twele, F.; Bröer, S.; Ziesak, V.; Rohde, M.; Hauck, S.M.; et al. Disruption of the sodium-dependent citrate transporter SLC13A5 in mice causes alterations in brain citrate levels and neuronal network excitability in the hippocampus. Neurobiol. Dis. 2020, 143, 105018. [Google Scholar] [CrossRef]
- Matricardi, S.; De Liso, P.; Freri, E.; Costa, P.; Castellotti, B.; Magri, S.; Gellera, C.; Granata, T.; Musante, L.; Lesca, G.; et al. Neonatal developmental and epileptic encephalopathy due to autosomal recessive variants in SLC13A5 gene. Epilepsia 2020, 61, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.Z.; Spelbrink, E.M.; Nye, K.L.; Hsu, E.R.; Porter, B.E. Epilepsy and EEG Phenotype of SLC13A5 Citrate Transporter Disorder. Child Neurol. Open 2020, 7, 2329048x20931361. [Google Scholar] [CrossRef] [PubMed]
- Kopel, J.; Grooms, A.; Ganapathy, V.; Clothier, J. Metformin, valproic acid, and starvation induce seizures in a patient with partial SLC13A5 deficiency: A case of pharmaco-synergistic heterozygosity. Psychiatr. Genet. 2021, 31, 32–35. [Google Scholar] [CrossRef]
- Zwart, R.; Peeva, P.M.; Rong, J.X.; Sher, E. Electrophysiological characterization of human and mouse sodium-dependent citrate transporters (NaCT/SLC13A5) reveal species differences with respect to substrate sensitivity and cation dependence. J. Pharmacol. Exp. Ther. 2015, 355, 247–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bainbridge, M.N.; Cooney, E.; Miller, M.; Kennedy, A.D.; Wulff, J.E.; Donti, T.; Jhangiani, S.N.; Gibbs, R.A.; Elsea, S.H.; Porter, B.E.; et al. Analyses of SLC13A5-epilepsy patients reveal perturbations of TCA cycle. Mol. Genet. Metab. 2017, 121, 314–319. [Google Scholar] [CrossRef]
- Selch, S.; Chafai, A.; Sticht, H.; Birkenfeld, A.L.; Fromm, M.F.; König, J. Analysis of naturally occurring mutations in the human uptake transporter NaCT important for bone and brain development and energy metabolism. Sci. Rep. 2018, 8, 11330. [Google Scholar] [CrossRef]
- Kumar, A.; Cordes, T.; Thalacker-Mercer, A.E.; Pajor, A.M.; Murphy, A.N.; Metallo, C.M. NaCT/SLC13A5 facilitates citrate import and metabolism under nutrient-limited conditions. Cell Rep. 2021, 36, 109701. [Google Scholar] [CrossRef]
- Sauer, D.B.; Song, J.; Wang, B.; Hilton, J.K.; Karpowich, N.K.; Mindell, J.A.; Rice, W.J.; Wang, D.N. Structure and inhibition mechanism of the human citrate transporter NaCT. Nature 2021, 591, 157–161. [Google Scholar] [CrossRef]
- Brachs, S.; Winkel, A.F.; Tang, H.; Birkenfeld, A.L.; Brunner, B.; Jahn-Hofmann, K.; Margerie, D.; Ruetten, H.; Schmoll, D.; Spranger, J. Inhibition of citrate cotransporter Slc13a5/mINDY by RNAi improves hepatic insulin sensitivity and prevents diet-induced non-alcoholic fatty liver disease in mice. Mol. Metab. 2016, 5, 1072–1082. [Google Scholar] [CrossRef]
- Li, L.; Li, H.; Garzel, B.; Yang, H.; Sueyoshi, T.; Li, Q.; Shu, Y.; Zhang, J.; Hu, B.; Heyward, S.; et al. SLC13A5 is a novel transcriptional target of the pregnane X receptor and sensitizes drug-induced steatosis in human liver. Mol. Pharmacol. 2015, 87, 674–682. [Google Scholar] [CrossRef] [Green Version]
- Uhlen, M.; Fagerberg, L.; Hallstrom, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, A.; Kampf, C.; Sjostedt, E.; Asplund, A.; et al. Proteomics. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Akesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Bjork, L.; Breckels, L.M.; et al. A subcellular map of the human proteome. Science 2017, 356, aal3321. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo-Martinez, V.; Ganapathy, V.; Urbatsch, I.L. A home run for human NaCT/SLC13A5/INDY: Cryo-EM structure and homology model to predict transport mechanisms, inhibitor interactions and mutational defects. Biochem. J. 2021, 478, 2051–2057. [Google Scholar] [CrossRef] [PubMed]
- Sjostedt, E.; Zhong, W.; Fagerberg, L.; Karlsson, M.; Mitsios, N.; Adori, C.; Oksvold, P.; Edfors, F.; Limiszewska, A.; Hikmet, F.; et al. An atlas of the protein-coding genes in the human, pig, and mouse brain. Science 2020, 367. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Zhang, Y.; Sloan, S.A.; Clarke, L.E.; Caneda, C.; Plaza, C.A.; Blumenthal, P.D.; Vogel, H.; Steinberg, G.K.; Edwards, M.S.; Li, G.; et al. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron 2016, 89, 37–53. [Google Scholar] [CrossRef] [Green Version]
- BioRender.com, C.a. Available online: https://biorender.com/ (accessed on 1 March 2022).
- Wang, H.; Fei, Y.J.; Kekuda, R.; Yang-Feng, T.L.; Devoe, L.D.; Leibach, F.H.; Prasad, P.D.; Ganapathy, V. Structure, function, and genomic organization of human Na(+)-dependent high-affinity dicarboxylate transporter. Am. J. Physiol. Cell Physiol. 2000, 278, C1019–C1030. [Google Scholar] [CrossRef]
- Inoue, K.; Fei, Y.J.; Zhuang, L.; Gopal, E.; Miyauchi, S.; Ganapathy, V. Functional features and genomic organization of mouse NaCT, a sodium-coupled transporter for tricarboxylic acid cycle intermediates. Biochem. J. 2004, 378, 949–957. [Google Scholar] [CrossRef] [Green Version]
- Gopal, E.; Miyauchi, S.; Martin, P.M.; Ananth, S.; Srinivas, S.R.; Smith, S.B.; Prasad, P.D.; Ganapathy, V. Expression and functional features of NaCT, a sodium-coupled citrate transporter, in human and rat livers and cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 292, G402–G408. [Google Scholar] [CrossRef]
- Khamaysi, A.; Aharon, S.; Eini-Rider, H.; Ohana, E. A dynamic anchor domain in slc13 transporters controls metabolite transport. J. Biol. Chem. 2020, 295, 8155–8163. [Google Scholar] [CrossRef] [Green Version]
- Smith, C.L.; Eppig, J.T. The mammalian phenotype ontology: Enabling robust annotation and comparative analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2009, 1, 390–399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.Z.; Sung, C.W.; Tsai, Y.H.; Yeh, S.R.; Lin, W.S.; Wang, P.Y. Nervous System Deletion of Mammalian INDY in Mice Mimics Dietary Restriction-Induced Memory Enhancement. J. Gerontol. A Biol. Sci. Med. Sci. 2021, 76, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Irizarry, A.R.; Yan, G.; Zeng, Q.; Lucchesi, J.; Hamang, M.J.; Ma, Y.L.; Rong, J.X. Defective enamel and bone development in sodium-dependent citrate transporter (NaCT) Slc13a5 deficient mice. PLoS ONE 2017, 12, e0175465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkenfeld, A.L.; Lee, H.Y.; Guebre-Egziabher, F.; Alves, T.C.; Jurczak, M.J.; Jornayvaz, F.R.; Zhang, D.; Hsiao, J.J.; Martin-Montalvo, A.; Fischer-Rosinsky, A.; et al. Deletion of the mammalian INDY homolog mimics aspects of dietary restriction and protects against adiposity and insulin resistance in mice. Cell Metab. 2011, 14, 184–195. [Google Scholar] [CrossRef] [Green Version]
- Thistlethwaite, L.R.; Petrosyan, V.; Li, X.; Miller, M.J.; Elsea, S.H.; Milosavljevic, A. CTD: An information-theoretic algorithm to interpret sets of metabolomic and transcriptomic perturbations in the context of graphical models. PLoS Comput. Biol. 2021, 17, e1008550. [Google Scholar] [CrossRef]
- Lynes, M.D.; Leiria, L.O.; Lundh, M.; Bartelt, A.; Shamsi, F.; Huang, T.L.; Takahashi, H.; Hirshman, M.F.; Schlein, C.; Lee, A.; et al. The cold-induced lipokine 12,13-diHOME promotes fatty acid transport into brown adipose tissue. Nat. Med. 2017, 23, 631–637. [Google Scholar] [CrossRef] [Green Version]
- Alaimo, J.T.; Glinton, K.E.; Liu, N.; Xiao, J.; Yang, Y.; Reid Sutton, V.; Elsea, S.H. Integrated analysis of metabolomic profiling and exome data supplements sequence variant interpretation, classification, and diagnosis. Genet. Med. 2020, 22, 1560–1566. [Google Scholar] [CrossRef] [PubMed]
- Pesta, D.H.; Perry, R.J.; Guebre-Egziabher, F.; Zhang, D.; Jurczak, M.; Fischer-Rosinsky, A.; Daniels, M.A.; Willmes, D.M.; Bhanot, S.; Bornstein, S.R.; et al. Prevention of diet-induced hepatic steatosis and hepatic insulin resistance by second generation antisense oligonucleotides targeted to the longevity gene mIndy (Slc13a5). Aging (Albany NY) 2015, 7, 1086–1093. [Google Scholar] [CrossRef] [Green Version]
- Kannan, K.; Rogina, B. The Role of Citrate Transporter INDY in Metabolism and Stem Cell Homeostasis. Metabolites 2021, 11, 705. [Google Scholar] [CrossRef]
- Tracey, T.J.; Steyn, F.J.; Wolvetang, E.J.; Ngo, S.T. Neuronal Lipid Metabolism: Multiple Pathways Driving Functional Outcomes in Health and Disease. Front. Mol. Neurosci. 2018, 11, 10. [Google Scholar] [CrossRef] [Green Version]
- Liu, N.; Xiao, J.; Gijavanekar, C.; Pappan, K.L.; Glinton, K.E.; Shayota, B.J.; Kennedy, A.D.; Sun, Q.; Sutton, V.R.; Elsea, S.H. Comparison of Untargeted Metabolomic Profiling vs Traditional Metabolic Screening to Identify Inborn Errors of Metabolism. JAMA Netw. Open 2021, 4, e2114155. [Google Scholar] [CrossRef] [PubMed]
- McMillin, M.; DeMorrow, S. Effects of bile acids on neurological function and disease. FASEB J. 2016, 30, 3658–3668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, K.L.; Kalsbeek, A.; Soeters, M.R.; Eggink, H.M. Bile Acid Signaling Pathways from the Enterohepatic Circulation to the Central Nervous System. Front. Neurosci. 2017, 11, 617. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiriyama, Y.; Nochi, H. The Biosynthesis, Signaling, and Neurological Functions of Bile Acids. Biomolecules 2019, 9, 232. [Google Scholar] [CrossRef] [Green Version]
- Olsen, A.S.B.; Faergeman, N.J. Sphingolipids: Membrane microdomains in brain development, function and neurological diseases. Open Biol. 2017, 7, 170069. [Google Scholar] [CrossRef] [Green Version]
- Fasullo, M.; Endres, L. Nucleotide salvage deficiencies, DNA damage and neurodegeneration. Int. J. Mol. Sci. 2015, 16, 9431–9449. [Google Scholar] [CrossRef] [Green Version]
- Sebastián-Serrano, Á.; de Diego-García, L.; di Lauro, C.; Bianchi, C.; Díaz-Hernández, M. Nucleotides regulate the common molecular mechanisms that underlie neurodegenerative diseases; Therapeutic implications. Brain Res. Bull. 2019, 151, 84–91. [Google Scholar] [CrossRef]
- Patra, C.; Foster, K.; Corley, J.E.; Dimri, M.; Bradym, M.F. Stat Pearls: Biochemistry, cAMP. In StatPearls, 2022th ed.; StatPearls Publishing: Treasure Island, FL, USA, 2022. [Google Scholar]
- Klotz, J.; Porter, B.E.; Colas, C.; Schlessinger, A.; Pajor, A.M. Mutations in the Na(+)/citrate cotransporter NaCT (SLC13A5) in pediatric patients with epilepsy and developmental delay. Mol. Med. 2016, 22. [Google Scholar] [CrossRef]
- Dewulf, J.P.; Wiame, E.; Dorboz, I.; Elmaleh-Berges, M.; Imbard, A.; Dumitriu, D.; Rak, M.; Bourillon, A.; Helaers, R.; Malla, A.; et al. SLC13A3 variants cause acute reversible leukoencephalopathy and alpha-ketoglutarate accumulation. Ann. Neurol. 2019, 85, 385–395. [Google Scholar] [CrossRef]
- Birling, M.C.; Yoshiki, A.; Adams, D.J.; Ayabe, S.; Beaudet, A.L.; Bottomley, J.; Bradley, A.; Brown, S.D.M.; Bürger, A.; Bushell, W.; et al. A resource of targeted mutant mouse lines for 5,061 genes. Nat. Genet. 2021, 53, 416–419. [Google Scholar] [CrossRef]
- Koscielny, G.; Yaikhom, G.; Iyer, V.; Meehan, T.F.; Morgan, H.; Atienza-Herrero, J.; Blake, A.; Chen, C.K.; Easty, R.; Di Fenza, A.; et al. The International Mouse Phenotyping Consortium Web Portal, a unified point of access for knockout mice and related phenotyping data. Nucleic Acids Res. 2014, 42, D802–D809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Evans, A.M.; DeHaven, C.D.; Barrett, T.; Mitchell, M.; Milgram, E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal. Chem. 2009, 81, 6656–6667. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Kennedy, A.D.; Eckhart, A.D.; Burrage, L.C.; Wulff, J.E.; Miller, L.A.; Milburn, M.V.; Ryals, J.A.; Beaudet, A.L.; Sun, Q.; et al. Untargeted metabolomic analysis for the clinical screening of inborn errors of metabolism. J. Inherit. Metab. Dis. 2015, 38, 1029–1039. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ford, L.; Kennedy, A.D.; Goodman, K.D.; Pappan, K.L.; Evans, A.M.; Miller, L.A.D.; Wulff, J.E.; Wiggs, B.R.; Lennon, J.J.; Elsea, S.; et al. Precision of a Clinical Metabolomics Profiling Platform for Use in the Identification of Inborn Errors of Metabolism. J. Appl. Lab. Med. 2020, 5, 342–356. [Google Scholar] [CrossRef]
- Li, X.; Milosavljevic, A.; Elsea, S.H.; Wang, C.C.; Scaglia, F.; Syngelaki, A.; Nicolaides, K.H.; Poon, L.C. Effective Aspirin Treatment of Women at Risk for Preeclampsia Delays the Metabolic Clock of Gestation. Hypertension 2021, 78, 1398–1410. [Google Scholar] [CrossRef]
- Burrage, L.C.; Thistlethwaite, L.; Stroup, B.M.; Sun, Q.; Miller, M.J.; Nagamani, S.C.S.; Craigen, W.; Scaglia, F.; Sutton, V.R.; Graham, B.; et al. Untargeted metabolomic profiling reveals multiple pathway perturbations and new clinical biomarkers in urea cycle disorders. Genet. Med. 2019, 21, 1977–1986. [Google Scholar] [CrossRef]
- Skarnes, W.C.; Rosen, B.; West, A.P.; Koutsourakis, M.; Bushell, W.; Iyer, V.; Mujica, A.O.; Thomas, M.; Harrow, J.; Cox, T.; et al. A conditional knockout resource for the genome-wide study of mouse gene function. , 474, 337–342. Nature 2011, 474, 337–342. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CSF | Biochemical | Sub-Pathway | Mean z-Score * ±S.D. |
|---|---|---|---|
| High | pelargonate (9:0) | Fatty Acids & Derivatives | 3.4 ± 0.4 |
| gluconate | Carbohydrate & Energy Metabolism | 3.4 ± 4.4 | |
| N-acetylglutamine | Alanine, Aspartate, & Glutamate Metabolism | 2.5 ± 0.7 | |
| N6-succinyladenosine | Nucleotide Metabolism | 2.5 ± 1.0 | |
| N-acetylneuraminate | Carbohydrate & Energy Metabolism | 2.4 ± 1.4 | |
| citrate | Carbohydrate & Energy Metabolism | 1.9 ± 0.4 | |
| Low | glutamate | Alanine, Aspartate, & Glutamate Metabolism | −2.2 ± 2.0 |
| 4-guanidinobutanoate | Urea Cycle, Creatine, & Polyamine Metabolism | −2.4 ± 0.6 | |
| adenine | Nucleotide Metabolism | −2.7 ± 0.4 | |
| tartronate (hydroxymalonate) | Microbiome, Xenobiotics, & Food/Plant Compounds | −2.9 ± 0.7 | |
| S-methylcysteine | Sulfur Containing Amino Acid Metabolism | −3.0 ± 4.0 | |
| Plasma | Biochemical | Sub-Pathway | Mean z−Score * ± S.D. |
| High | citrate | Carbohydrate & Energy Metabolism | 2.6 ± 0.8 |
| malonate | Fatty Acids & Derivatives | 1.7 ± 1.1 | |
| 1-arachidonoylglyercophosphate | Glycerophospholipid Metabolism | 1.5 ± 0.9 | |
| pentadecanoate (15:0) | Fatty Acids & Derivatives | 1.5 ± 0.9 | |
| docosapentaenoate (n6 DPA; 22:5n6) | Fatty Acids & Derivatives | 1.4 ± 0.6 | |
| glycocholate sulfate * | Bile Acids, Cholesterols, & Sterols | −1.3 ± 1.4 | |
| Low | 5alpha-pregnan-3beta,20alpha-diol disulfate | Bile Acids, Cholesterols, & Sterols | −1.5 ± 1.5 |
| 5alpha-androstan-3beta,17beta-diol disulfate | Bile Acids, Cholesterols, & Sterols | −1.5 ± 2.3 | |
| 1-palmitoylglycerol (16:0) | Monoacylglycerols | −1.8 ± 1.0 | |
| suberate (octanedioate) | Fatty Acids & Derivatives | −1.9 ± 0.2 |
| Biochemical | Plasma | |
|---|---|---|
| Super Pathway | Sub-Pathway | |
| androsterone sulfate | Lipids | Bile Acids, Cholesterols, & Sterols |
| epiandrosterone sulfate | Lipids | Bile Acids, Cholesterols, & Sterols |
| 5alpha-pregnan-3beta,20alpha-diol-disulfate | Lipids | Bile Acids, Cholesterols, & Sterols |
| hyocholate | Lipids | Bile Acids, Cholesterols, & Sterols |
| glycohyocholate | Lipids | Bile Acids, Cholesterols, & Sterols |
| citrate | Carbohydrate & Energy Metabolism | Carbohydrate & Energy Metabolism |
| adipate | Lipids | Fatty Acids & Derivatives |
| myristoleate (14:1n5) | Lipids | Fatty Acids & Derivatives |
| methylpalmitate (15 or 2) | Lipids | Fatty Acids & Derivatives |
| mead acid (20:3n9) | Lipids | Fatty Acids & Derivatives |
| docosapentaenoate (n6 DPA; 22:5n6) | Lipids | Fatty Acids & Derivatives |
| 17-methylstearate | Lipids | Fatty Acids & Derivatives |
| dodecanedioate | Lipids | Fatty Acids & Derivatives |
| 2-oleoyl-GPE (18:1) | Lipids | Glycerophospholipid Metabolism |
| 3-methylxanthine | Xenobiotics | Microbiome, Xenobiotics & Food/Plant Compounds |
| Biochemical | CSF | |
| Super Pathway | Sub-Pathway | |
| N-acetylglutamine | Amino Acids | Alanine, Aspartate, & Glutamate Metabolism |
| N-acetylalanine | Amino Acids | Alanine, Aspartate, & Glutamate Metabolism |
| N-acetylneuraminate | Carbohydrate & Energy Metabolism | Carbohydrate & Energy Metabolism |
| citrate | Carbohydrate & Energy Metabolism | Carbohydrate & Energy Metabolism |
| 2-methylcitrate | Carbohydrate & Energy Metabolism | Carbohydrate & Energy Metabolism |
| erythronate | Carbohydrate & Energy Metabolism | Carbohydrate & Energy Metabolism |
| gluconate | Carbohydrate & Energy Metabolism | Carbohydrate & Energy Metabolism |
| ribitol | Carbohydrate & Energy Metabolism | Carbohydrate & Energy Metabolism |
| ribonate | Carbohydrate & Energy Metabolism | Carbohydrate & Energy Metabolism |
| pyridoxate | Carbohydrate & Energy Metabolism | Cofactors & Vitamins |
| pyridoxal | Carbohydrate & Energy Metabolism | Cofactors & Vitamins |
| malonylcarnitine | Lipids | Fatty Acids & Derivatives |
| tartronate (hydroxymalonate) | Xenobiotics | Microbiome, Xenobiotics, & Food/Plant Compounds |
| N6-succinyladenosine | Nucleotide Metabolism | Nucleotide Metabolism |
| adenine | Nucleotide Metabolism | Nucleotide Metabolism |
| gamma-glutamylthreonine | Peptides | Peptides & Polypeptides Metabolism |
| S-methylcysteine | Amino Acids | Sulfur Containing Amino Acid Metabolism |
| acisoga | Amino Acids | Urea Cycle, Creatine, & Polyamine Metabolism |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Milosavljevic, S.; Glinton, K.E.; Li, X.; Medeiros, C.; Gillespie, P.; Seavitt, J.R.; Graham, B.H.; Elsea, S.H. Untargeted Metabolomics of Slc13a5 Deficiency Reveal Critical Liver–Brain Axis for Lipid Homeostasis. Metabolites 2022, 12, 351. https://doi.org/10.3390/metabo12040351
Milosavljevic S, Glinton KE, Li X, Medeiros C, Gillespie P, Seavitt JR, Graham BH, Elsea SH. Untargeted Metabolomics of Slc13a5 Deficiency Reveal Critical Liver–Brain Axis for Lipid Homeostasis. Metabolites. 2022; 12(4):351. https://doi.org/10.3390/metabo12040351
Chicago/Turabian StyleMilosavljevic, Sofia, Kevin E. Glinton, Xiqi Li, Cláudia Medeiros, Patrick Gillespie, John R. Seavitt, Brett H. Graham, and Sarah H. Elsea. 2022. "Untargeted Metabolomics of Slc13a5 Deficiency Reveal Critical Liver–Brain Axis for Lipid Homeostasis" Metabolites 12, no. 4: 351. https://doi.org/10.3390/metabo12040351
APA StyleMilosavljevic, S., Glinton, K. E., Li, X., Medeiros, C., Gillespie, P., Seavitt, J. R., Graham, B. H., & Elsea, S. H. (2022). Untargeted Metabolomics of Slc13a5 Deficiency Reveal Critical Liver–Brain Axis for Lipid Homeostasis. Metabolites, 12(4), 351. https://doi.org/10.3390/metabo12040351