
Lactate Neuroprotection against Transient Ischemic Brain Injury in Mice Appears Independent of HCAR1 Activation

 , , and
, , and 
{kind=link}
{kind=link}
Abstract
:1. Introduction
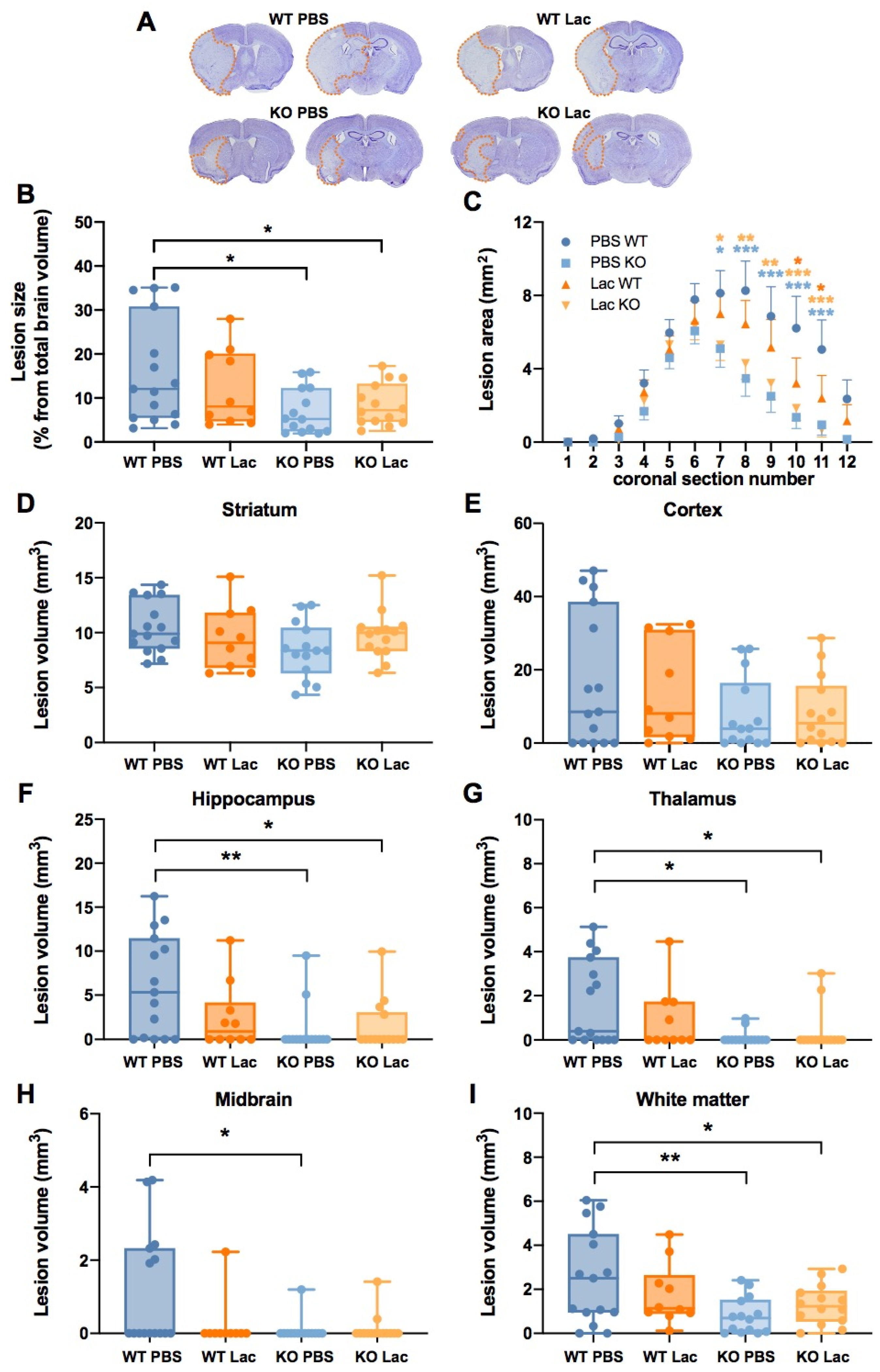
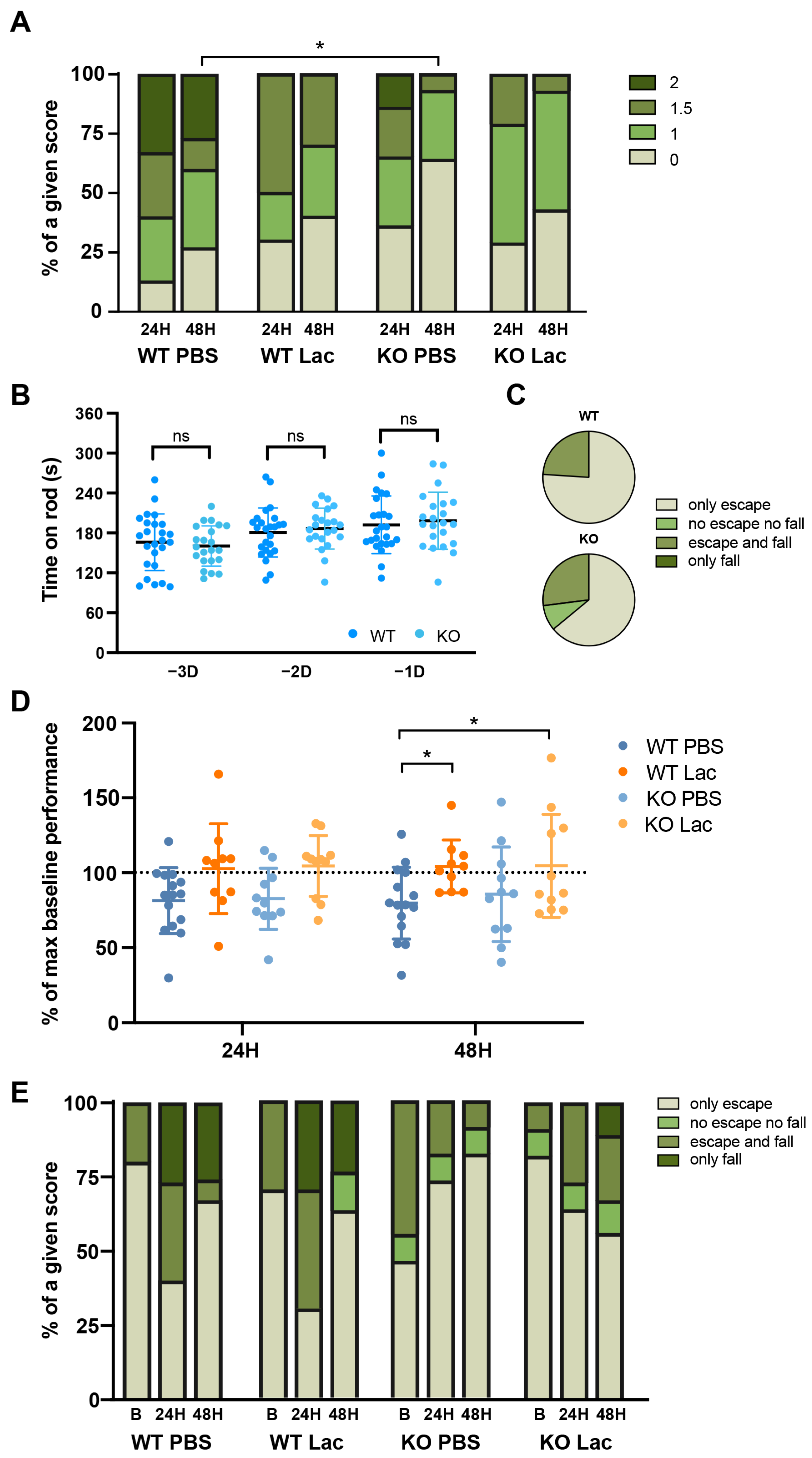
2. Results
3. Discussion
4. Material and Methods
4.1. Transient Middle Cerebral Artery Occlusion (MCAO) Model
4.2. Ischemic Lesion Volume Determination
4.3. Functional Outcome Assessment
4.4. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Berthet, C.; Castillo, X.; Magistretti, P.J.; Hirt, L. New evidence of neuroprotection by lactate after transient focal cerebral ischaemia: Extended benefit after intracerebroventricular injection and efficacy of intravenous administration. Cerebrovasc. Dis. 2012, 34, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Berthet, C.; Lei, H.; Thevenet, J.; Gruetter, R.; Magistretti, P.J.; Hirt, L. Neuroprotective role of lactate after cerebral ischemia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2009, 29, 1780–1789. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, L.; Blochet, C.; Price, M.; Magistretti, P.J.; Lei, H.; Hirt, L. Extended preclinical investigation of lactate for neuroprotection after ischemic stroke. Clin. Transl. Neurosci. 2020, 4, 2514183X20904571. [Google Scholar] [CrossRef] [Green Version]
- Castillo, X.; Rosafio, K.; Wyss, M.T.; Drandarov, K.; Buck, A.; Pellerin, L.; Weber, B.; Hirt, L. A probable dual mode of action for both L- and D-lactate neuroprotection in cerebral ischemia. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2015, 35, 1561–1569. [Google Scholar] [CrossRef]
- Rice, A.C.; Zsoldos, R.; Chen, T.; Wilson, M.S.; Alessandri, B.; Hamm, R.J.; Bullock, M.R. Lactate administration attenuates cognitive deficits following traumatic brain injury. Brain Res. 2002, 928, 156–159. [Google Scholar] [CrossRef]
- Roumes, H.; Dumont, U.; Sanchez, S.; Mazuel, L.; Blanc, J.; Raffard, G.; Chateil, J.-F.; Pellerin, L.; Bouzier-Sore, A.-K. Neuroprotective role of lactate in rat neonatal hypoxia-ischemia. J. Cereb. Blood Flow Metab. 2020, 41, 342–358. [Google Scholar] [CrossRef] [Green Version]
- Tassinari, I.D.; Andrade, M.K.G.; da Rosa, L.A.; Hoff, M.L.M.; Nunes, R.R.; Vogt, E.L.; Fabres, R.B.; Sanches, E.F.; Netto, C.A.; Paz, A.H.; et al. Lactate Administration Reduces Brain Injury and Ameliorates Behavioral Outcomes Following Neonatal Hypoxia-Ischemia. PLoS ONE 2020, 448, 191–205. [Google Scholar] [CrossRef]
- Bouzat, P.; Sala, N.; Suys, T.; Zerlauth, J.B.; Marques-Vidal, P.; Feihl, F.; Bloch, J.; Messerer, M.; Levivier, M.; Meuli, R.; et al. Cerebral metabolic effects of exogenous lactate supplementation on the injured human brain. Intensive Care Med. 2014, 40, 412–421. [Google Scholar] [CrossRef]
- Carteron, L.; Solari, D.; Patet, C.; Quintard, H.; Miroz, J.P.; Bloch, J.; Daniel, R.T.; Hirt, L.; Eckert, P.; Magistretti, P.J.; et al. Hypertonic Lactate to Improve Cerebral Perfusion and Glucose Availability After Acute Brain Injury. Crit. Care Med. 2018, 46, 1649–1655. [Google Scholar] [CrossRef]
- Ichai, C.; Payen, J.F.; Orban, J.C.; Quintard, H.; Roth, H.; Legrand, R.; Francony, G.; Leverve, X.M. Half-molar sodium lactate infusion to prevent intracranial hypertensive episodes in severe traumatic brain injured patients: A randomized controlled trial. Intensive Care Med. 2013, 39, 1413–1422. [Google Scholar] [CrossRef]
- Ahmed, K.; Tunaru, S.; Offermanns, S. GPR109A, GPR109B and GPR81, a family of hydroxy-carboxylic acid receptors. Trends Pharmacol. Sci. 2009, 30, 557–562. [Google Scholar] [CrossRef] [PubMed]
- Cai, T.Q.; Ren, N.; Jin, L.; Cheng, K.; Kash, S.; Chen, R.; Wright, S.D.; Taggart, A.K.; Waters, M.G. Role of GPR81 in lactate-mediated reduction of adipose lipolysis. Biochem. Biophys. Res. Commun. 2008, 377, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Bozzo, L.; Puyal, J.; Chatton, J.Y. Lactate modulates the activity of primary cortical neurons through a receptor-mediated pathway. PLoS ONE 2013, 8, e71721. [Google Scholar] [CrossRef] [PubMed]
- Briquet, M.; Rocher, A.-B.; Alessandri, M.; Rosenberg, N.; de Castro Abrantes, H.; Wellbourne-Wood, J.; Schmuziger, C.; Ginet, V.; Puyal, J.; Pralong, E.; et al. Activation of lactate receptor HCAR1 down-modulates neuronal activity in rodent and human brain tissue. J. Cereb. Blood Flow Metab. 2022, 0271678X221080324. [Google Scholar] [CrossRef]
- de Castro Abrantes, H.; Briquet, M.; Schmuziger, C.; Restivo, L.; Puyal, J.; Rosenberg, N.; Rocher, A.-B.; Offermanns, S.; Chatton, J.-Y. The Lactate Receptor HCAR1 Modulates Neuronal Network Activity through the Activation of Gα and Gβγ Subunits. J. Neurosci. 2019, 39, 4422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauritzen, K.H.; Morland, C.; Puchades, M.; Holm-Hansen, S.; Hagelin, E.M.; Lauritzen, F.; Attramadal, H.; Storm-Mathisen, J.; Gjedde, A.; Bergersen, L.H. Lactate receptor sites link neurotransmission, neurovascular coupling, and brain energy metabolism. Cereb. Cortex 2014, 24, 2784–2795. [Google Scholar] [CrossRef] [PubMed]
- Morland, C.; Lauritzen, K.H.; Puchades, M.; Holm-Hansen, S.; Andersson, K.; Gjedde, A.; Attramadal, H.; Storm-Mathisen, J.; Bergersen, L.H. The lactate receptor, G-protein-coupled receptor 81/hydroxycarboxylic acid receptor 1: Expression and action in brain. J. Neurosci. Res. 2015, 93, 1045–1055. [Google Scholar] [CrossRef]
- Magistretti, P.J.; Allaman, I. Lactate in the brain: From metabolic end-product to signalling molecule. Nat. Rev. Neurosci. 2018, 19, 235–249. [Google Scholar] [CrossRef]
- Schurr, A.; Payne, R.S.; Miller, J.J.; Rigor, B.M. Brain lactate is an obligatory aerobic energy substrate for functional recovery after hypoxia: Further in vitro validation. J. Neurochem. 1997, 69, 423–426. [Google Scholar] [CrossRef]
- Schurr, A.; Payne, R.S.; Miller, J.J.; Rigor, B.M. Brain lactate, not glucose, fuels the recovery of synaptic function from hypoxia upon reoxygenation: An in vitro study. Brain Res. 1997, 744, 105–111. [Google Scholar] [CrossRef]
- Herrera-Lopez, G.; Galvan, E.J. Modulation of hippocampal excitability via the hydroxycarboxylic acid receptor 1. Hippocampus 2018, 28, 557–567. [Google Scholar] [CrossRef] [PubMed]
- Buscemi, L.; Blochet, C.; Magistretti, P.J.; Hirt, L. Hydroxycarboxylic Acid Receptor 1 and Neuroprotection in a Mouse Model of Cerebral Ischemia-Reperfusion. Front. Physiol. 2021, 12, 689239. [Google Scholar] [CrossRef] [PubMed]
- Hyacinthe, J.-N.; Buscemi, L.; Lê, T.P.; Lepore, M.; Hirt, L.; Mishkovsky, M. Evaluating the potential of hyperpolarised [1-13C] L-lactate as a neuroprotectant metabolic biosensor for stroke. Sci. Rep. 2020, 10, 5507. [Google Scholar] [CrossRef]
- Schurr, A.; Miller, J.J.; Payne, R.S.; Rigor, B.M. An increase in lactate output by brain tissue serves to meet the energy needs of glutamate-activated neurons. J. Neurosci. Off. J. Soc. Neurosci. 1999, 19, 34–39. [Google Scholar] [CrossRef]
- González-Falcón, A.; Candelario-Jalil, E.; García-Cabrera, M.; León, O.S. Effects of pyruvate administration on infarct volume and neurological deficits following permanent focal cerebral ischemia in rats. Brain Res. 2003, 990, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Tang, F.; Lane, S.; Korsak, A.; Paton, J.F.; Gourine, A.V.; Kasparov, S.; Teschemacher, A.G. Lactate-mediated glia-neuronal signalling in the mammalian brain. Nat. Commun. 2014, 5, 3284. [Google Scholar] [CrossRef] [Green Version]
- Herrera-López, G.; Griego, E.; Galván, E.J. Lactate induces synapse-specific potentiation on CA3 pyramidal cells of rat hippocampus. PLoS ONE 2020, 15, e0242309. [Google Scholar] [CrossRef]
- D’Adamo, P.; Horvat, A.; Gurgone, A.; Mignogna, M.L.; Bianchi, V.; Masetti, M.; Ripamonti, M.; Taverna, S.; Velebit, J.; Malnar, M.; et al. Inhibiting glycolysis rescues memory impairment in an intellectual disability Gdi1-null mouse. Metab. Clin. Exp. 2021, 116, 154463. [Google Scholar] [CrossRef]
- Vardjan, N.; Chowdhury, H.H.; Horvat, A.; Velebit, J.; Malnar, M.; Muhic, M.; Kreft, M.; Krivec, S.G.; Bobnar, S.T.; Mis, K.; et al. Enhancement of Astroglial Aerobic Glycolysis by Extracellular Lactate-Mediated Increase in cAMP. Front. Mol. Neurosci. 2018, 11, 148. [Google Scholar] [CrossRef]
- Manoharan, I.; Prasad, P.D.; Thangaraju, M.; Manicassamy, S. Lactate-Dependent Regulation of Immune Responses by Dendritic Cells and Macrophages. Front. Immunol. 2021, 12, 691134. [Google Scholar] [CrossRef]
- Li, G.; Wang, H.Q.; Wang, L.H.; Chen, R.P.; Liu, J.P. Distinct pathways of ERK1/2 activation by hydroxy-carboxylic acid receptor-1. PLoS ONE 2014, 9, e93041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pötzsch, A.; Zocher, S.; Bernas, S.N.; Leiter, O.; Rünker, A.E.; Kempermann, G. L-lactate exerts a pro-proliferative effect on adult hippocampal precursor cells in vitro. iScience 2021, 24, 102126. [Google Scholar] [CrossRef]
- Wallenius, K.; Thalen, P.; Bjorkman, J.A.; Johannesson, P.; Wiseman, J.; Bottcher, G.; Fjellstrom, O.; Oakes, N.D. Involvement of the metabolic sensor GPR81 in cardiovascular control. JCI Insight 2017, 2, e92564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahmed, K.; Tunaru, S.; Tang, C.; Müller, M.; Gille, A.; Sassmann, A.; Hanson, J.; Offermanns, S. An autocrine lactate loop mediates insulin-dependent inhibition of lipolysis through GPR81. Cell Metab. 2010, 11, 311–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirt, L.; Badaut, J.; Thevenet, J.; Granziera, C.; Regli, L.; Maurer, F.; Bonny, C.; Bogousslavsky, J. D-JNKI1, a cell-penetrating c-Jun-N-terminal kinase inhibitor, protects against cell death in severe cerebral ischemia. Stroke 2004, 35, 1738–1743. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buscemi, L.; Price, M.; Castillo-González, J.; Chatton, J.-Y.; Hirt, L. Lactate Neuroprotection against Transient Ischemic Brain Injury in Mice Appears Independent of HCAR1 Activation. Metabolites 2022, 12, 465. https://doi.org/10.3390/metabo12050465
Buscemi L, Price M, Castillo-González J, Chatton J-Y, Hirt L. Lactate Neuroprotection against Transient Ischemic Brain Injury in Mice Appears Independent of HCAR1 Activation. Metabolites. 2022; 12(5):465. https://doi.org/10.3390/metabo12050465
Chicago/Turabian StyleBuscemi, Lara, Melanie Price, Julia Castillo-González, Jean-Yves Chatton, and Lorenz Hirt. 2022. "Lactate Neuroprotection against Transient Ischemic Brain Injury in Mice Appears Independent of HCAR1 Activation" Metabolites 12, no. 5: 465. https://doi.org/10.3390/metabo12050465
APA StyleBuscemi, L., Price, M., Castillo-González, J., Chatton, J. -Y., & Hirt, L. (2022). Lactate Neuroprotection against Transient Ischemic Brain Injury in Mice Appears Independent of HCAR1 Activation. Metabolites, 12(5), 465. https://doi.org/10.3390/metabo12050465