
Quantitative Profiling of Bile Acids in Feces of Humans and Rodents by Ultra-High-Performance Liquid Chromatography–Quadrupole Time-of-Flight Mass Spectrometry


Abstract
:1. Introduction
2. Result and Discussion
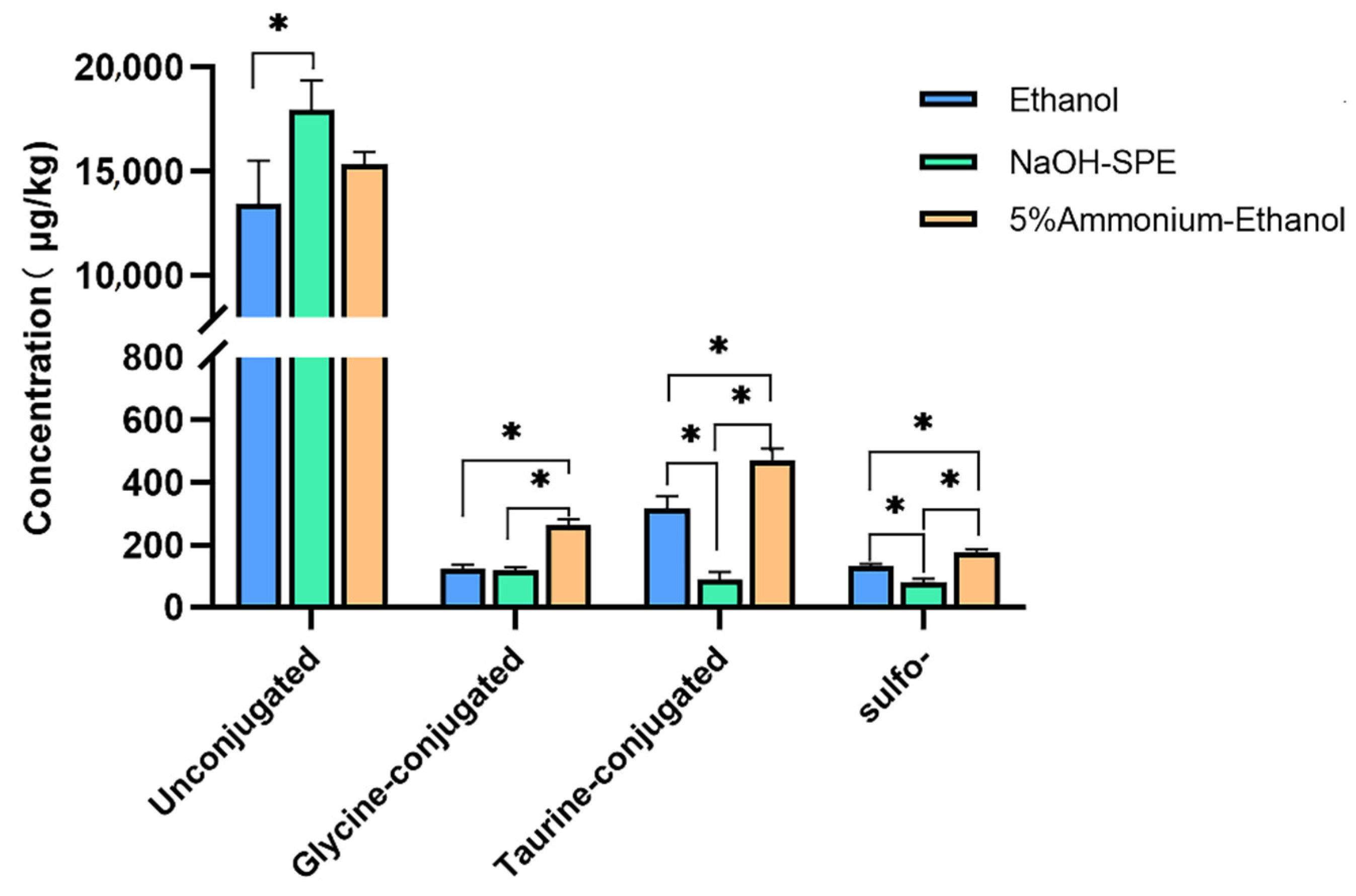
2.1. BA Extraction Methods Comparison
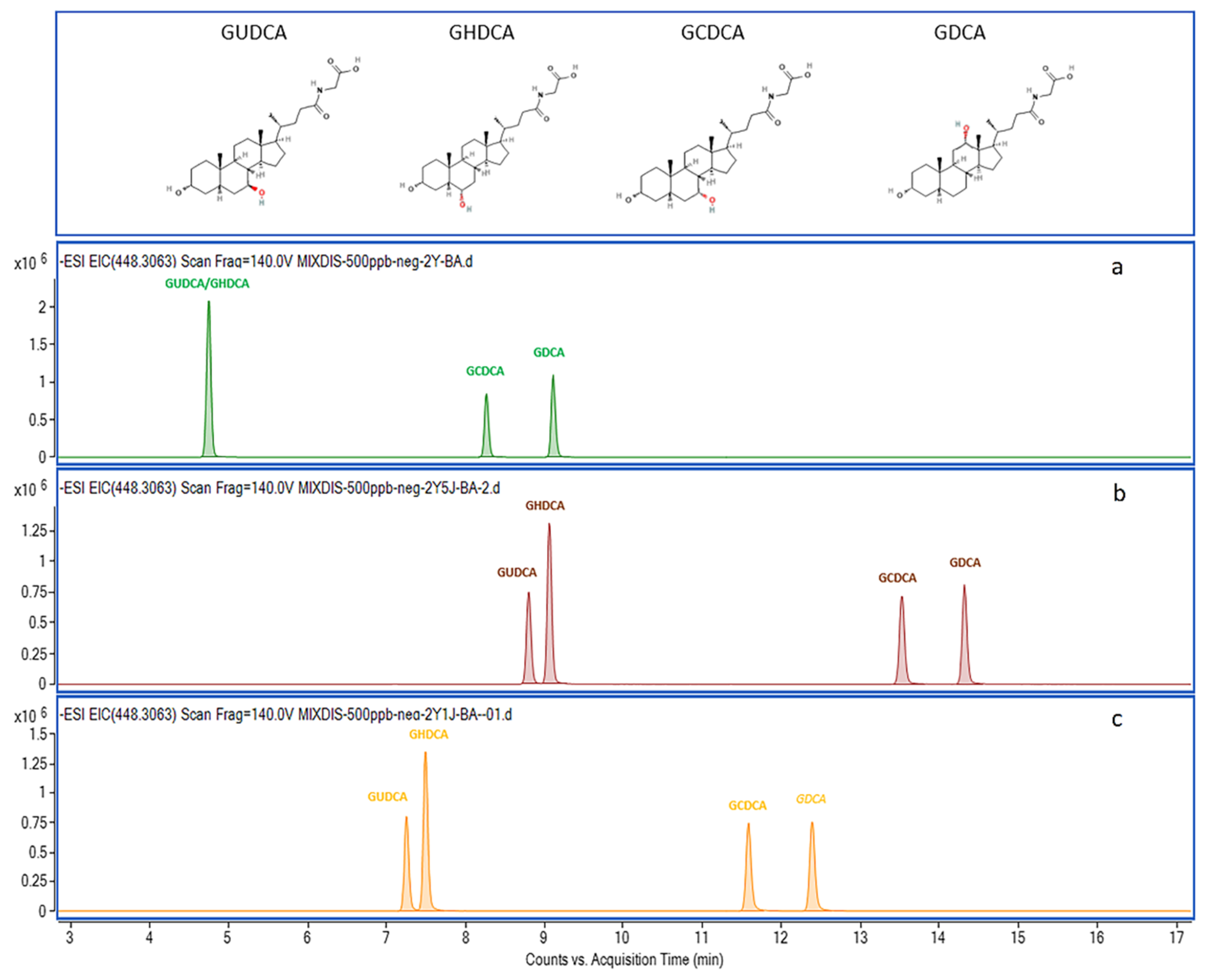
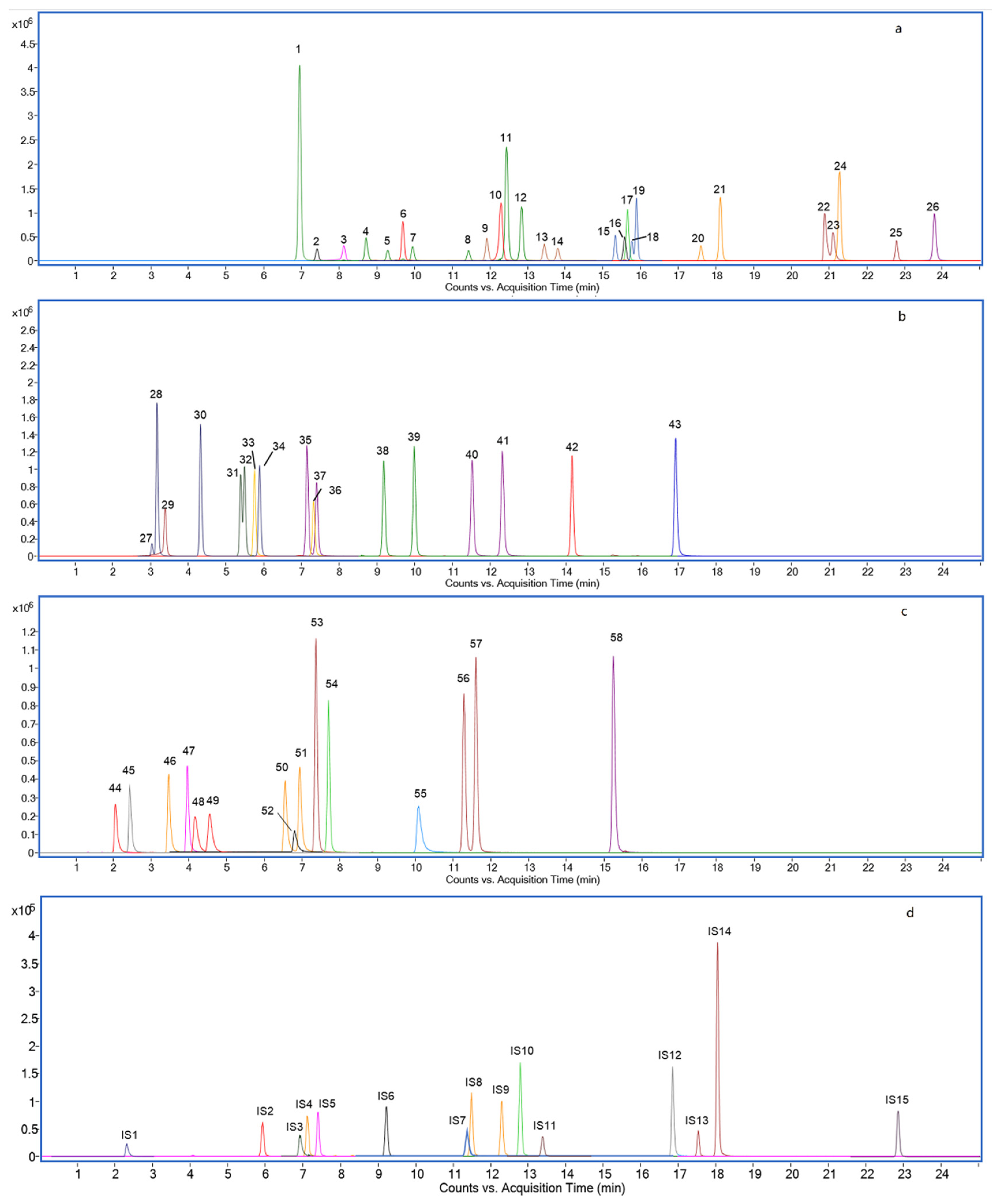
2.2. Chromatography Separation Optimization
2.3. Method Validation
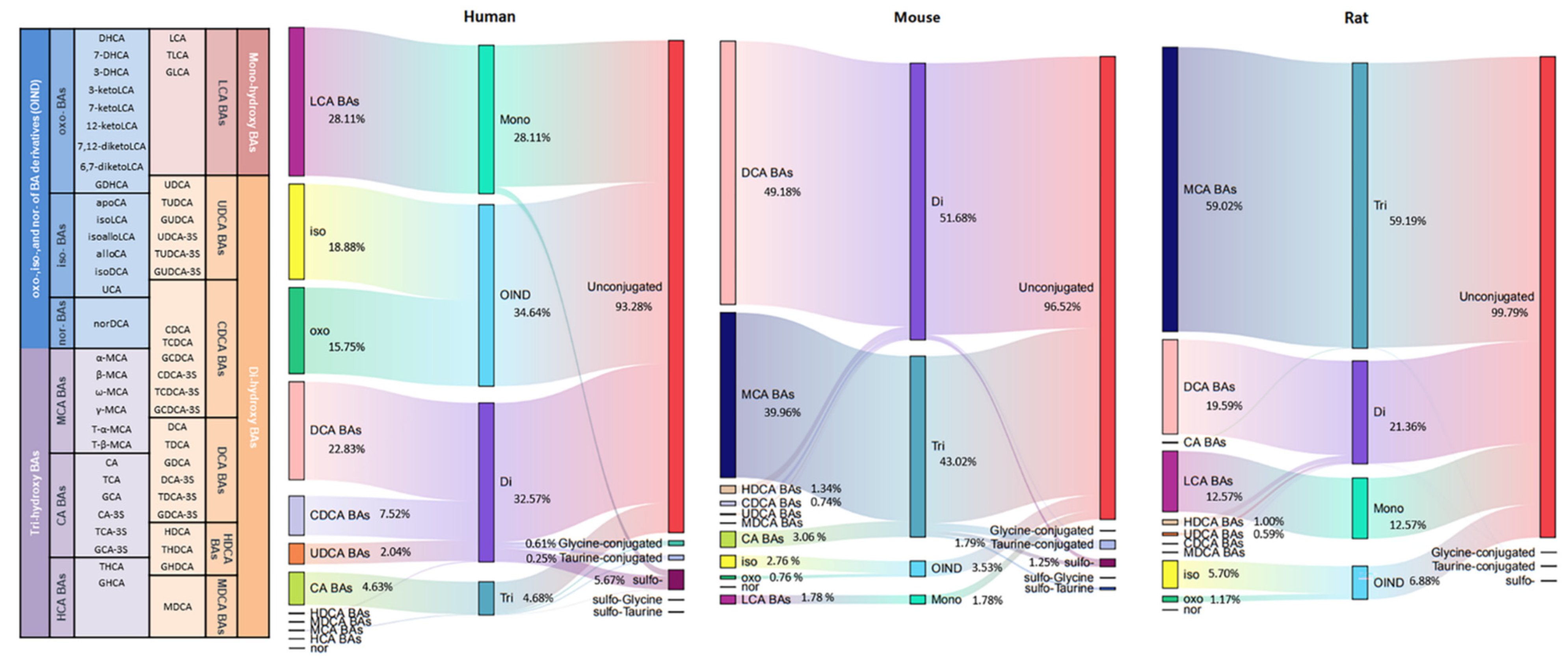
2.4. BA Profiling in Humans, Rats, and Mice
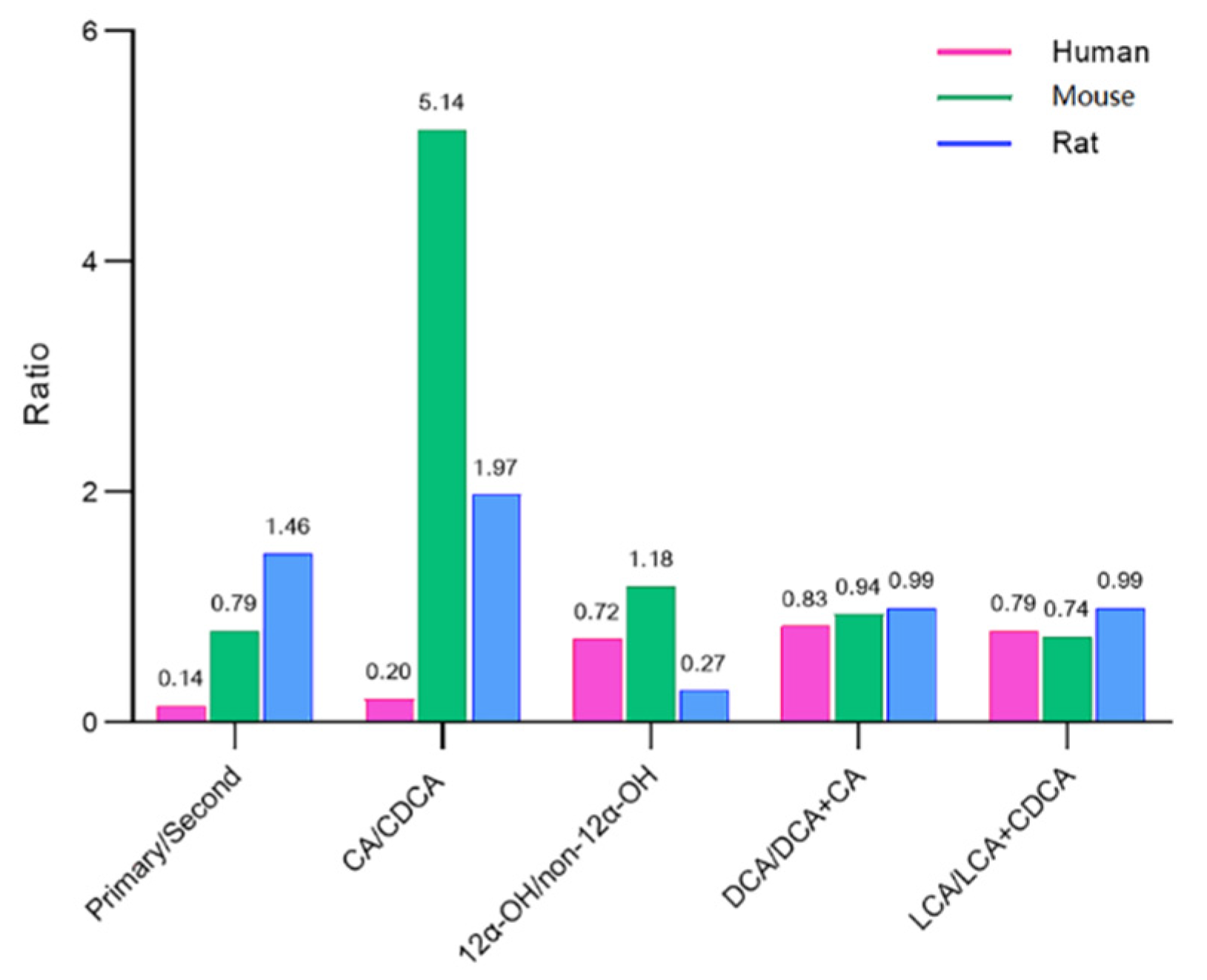
2.5. BA Indices in Humans, Rats, and Mice
3. Materials and Methods
3.1. Materials & Methods
3.2. Standard Solutions and Calibration Curves
3.3. Sample Preparation
3.3.1. S1 Extraction with Ethanol
3.3.2. S2 Extraction with 0.1 mol/L NaOH Followed by SPE
3.3.3. S3 Extraction with 5% Ammonium-Ethanol
- C represents the concentration expressed as μg/kg;
- C0 represents the concentration expressed as μg/L;
- V represents the extraction volume (in L);
- m represents the weight of wet feces (kg) subjected to extraction;
- n represents the dilution ratio;
- MF represents the fecal moisture content (%).
3.4. UPLC–Q-TOF Analysis
3.5. Standard Solutions and Calibration Curves
3.5.1. Linearity, LOD, and LOQ
3.5.2. Recovery and Matrix Effect
3.5.3. Precision and Accuracy
3.5.4. Clinical and Preclinical Samples Collection
3.5.5. Data Processing and Statistical Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.H.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, S4–S14. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.; Wei, M.; Rajani, C.; Zheng, X. Targeting the alternative bile acid synthetic pathway for metabolic diseases. Protein Cell 2021, 12, 411–425. [Google Scholar] [CrossRef] [PubMed]
- Schoeler, M.; Caesar, R. Dietary lipids, gut microbiota and lipid metabolism. Rev. Endocr. Metab. Disord. 2019, 20, 461–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vital, M.; Rud, T.; Rath, S.; Pieper, D.H.; Schlüter, D. Diversity of Bacteria Exhibiting Bile Acid-inducible 7α-dehydroxylation Genes in the Human Gut. Comput. Struct. Biotechnol. J. 2019, 17, 1016–1019. [Google Scholar] [CrossRef]
- Funabashi, M.; Grove, T.L.; Wang, M.; Varma, Y.; McFadden, M.E.; Brown, L.C.; Guo, C.; Higginbottom, S.; Almo, S.C.; Fischbach, M.A. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 2020, 582, 566–570. [Google Scholar] [CrossRef]
- Marion, S.; Desharnais, L.; Studer, N.; Dong, Y.; Notter, M.D.; Poudel, S.; Menin, L.; Janowczyk, A.; Hettich, R.L.; Hapfelmeier, S.; et al. Biogeography of microbial bile acid transformations along the murine gut. J. Lipid Res. 2020, 61, 1450–1463. [Google Scholar] [CrossRef]
- Alnouti, Y. Bile Acid Sulfation: A Pathway of Bile Acid Elimination and Detoxification. Toxicol. Sci. 2009, 108, 225–246. [Google Scholar] [CrossRef]
- Shafaei, A.; Rees, J.; Christophersen, C.T.; Devine, A.; Broadhurst, D.; Boyce, M.C. Extraction and quantitative determination of bile acids in feces. Anal. Chim. Acta 2021, 1150, 338224. [Google Scholar] [CrossRef]
- Fiorucci, S.; Biagioli, M.; Zampella, A.; Distrutti, E. Bile Acids Activated Receptors Regulate Innate Immunity. Front. Immunol. 2018, 9, 1853. [Google Scholar] [CrossRef] [Green Version]
- Thomas, C.; Pellicciari, R.; Pruzanski, M.; Auwerx, J.; Schoonjans, K. Targeting bile-acid signalling for metabolic diseases. Nat. Rev. Drug Discov. 2008, 7, 678–693. [Google Scholar] [CrossRef]
- Chiang, J.Y.L.; Ferrell, J.M. Bile Acids as Metabolic Regulators and Nutrient Sensors. Annu. Rev. Nutr. 2019, 39, 175–200. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Demagny, H.; Velazquez-Villegas, L.; Schoonjans, K. Molecular Physiology of Bile Acid Signaling in Health, Disease, and Aging. Physiol Rev. 2021, 101, 683–731. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Atarashi, K.; Plichta, D.R.; Arai, Y.; Sasajima, S.; Kearney, S.M.; Suda, W.; Takeshita, K.; Sasaki, T.; Okamoto, S.; et al. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature 2021, 599, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Gao, X.; Zhang, X.; Xiao, Y.; Huang, J.; Yu, D.; Li, X.; Hu, H.; Ge, T.; Li, D.; et al. Gut Microbiota Dysbiosis Is Associated with Altered Bile Acid Metabolism in Infantile Cholestasis. mSystems 2019, 4, e00463-19. [Google Scholar] [CrossRef] [Green Version]
- Vasavan, T.; Ferraro, E.; Ibrahim, E.; Dixon, P.; Gorelik, J.; Williamson, C. Heart and bile acids—Clinical consequences of altered bile acid metabolism. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1345–1355. [Google Scholar] [CrossRef]
- Mahmoudian Dehkordi, S.; Arnold, M.; Nho, K.; Ahmad, S.; Jia, W.; Xie, G.; Louie, G.; Kueider-Paisley, A.; Moseley, M.A.; Thompson, J.W.; et al. Altered bile acid profile associates with cognitive impairment in Alzheimer’s disease—An emerging role for gut microbiome. Alzheimer’s Dement. 2018, 15, 76–92. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Sivamaruthi, B.S.; Fern, L.A.; Rashidah Pg Hj Ismail, D.S.N.; Chaiyasut, C. The influence of probiotics on bile acids in diseases and aging. Biomed Pharm. 2020, 128, 110310. [Google Scholar] [CrossRef]
- Gao, T.; Feridooni, H.A.; Howlett, S.E.; Pelis, R.M. Influence of age on intestinal bile acid transport in C57BL/6 mice. Pharm. Res. Perspect. 2017, 5, e00287. [Google Scholar] [CrossRef]
- Han, J.; Liu, Y.; Wang, R.; Yang, J.; Ling, V.; Borchers, C.H. Metabolic Profiling of Bile Acids in Human and Mouse Blood by LC–MS/MS in Combination with Phospholipid-Depletion Solid-Phase Extraction. Anal. Chem. 2015, 87, 1127–1136. [Google Scholar] [CrossRef]
- Sharma, K.R. Review on bile acid analysis. Int. J. Pharm. Biomed. Sci. 2012, 3, 28–34. [Google Scholar]
- Liu, Y.; Rong, Z.; Xiang, D.; Zhang, C.; Liu, D. Detection technologies and metabolic profiling of bile acids: A comprehensive review. Lipids Health Dis. 2018, 17, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Lee, H.; Hong, J.; Lee, S.H.; Jung, B.H. Quantitative profiling of bile acids in rat bile using ultrahigh-performance liquid chromatography–orbitrap mass spectrometry: Alteration of the bile acid composition with aging. J. Chromatogr. B 2016, 1031, 37–49. [Google Scholar] [CrossRef] [PubMed]
- Haag, M.; Hofmann, U.; Mürdter, T.E.; Heinkele, G.; Leuthold, P.; Blank, A.; Haefeli, W.E.; Alexandrov, A.; Urban, S.; Schwab, M. Quantitative bile acid profiling by liquid chromatography quadrupole time-of-flight mass spectrometry: Monitoring hepatitis B therapy by a novel Na(+)-taurocholate cotransporting polypeptide inhibitor. Anal. Bioanal. Chem. 2015, 407, 6815–6825. [Google Scholar] [CrossRef] [PubMed]
- Sangaraju, D.; Shi, Y.; Van Parys, M.; Ray, A.; Walker, A.; Caminiti, R.; Milanowski, D.; Jaochico, A.; Dean, B.; Liang, X. Robust and Comprehensive Targeted Metabolomics Method for Quantification of 50 Different Primary, Secondary, and Sulfated Bile Acids in Multiple Biological Species (Human, Monkey, Rabbit, Dog, and Rat) and Matrices (Plasma and Urine) Using Liquid Chromatography High Resolution Mass Spectrometry (LC-HRMS) Analysis. J. Am. Soc. Mass Spectrom. 2021, 32, 2033–2049. [Google Scholar] [CrossRef] [PubMed]
- Choucair, I.; Nemet, I.; Li, L.; Cole, M.A.; Skye, S.M.; Kirsop, J.D.; Fischbach, M.A.; Gogonea, V.; Brown, J.M.; Tang, W.H.W.; et al. Quantification of bile acids: A mass spectrometry platform for studying gut microbe connection to metabolic diseases. J. Lipid Res. 2020, 61, 159–177. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Zeng, L.; Xie, S.; Chen, J.; Yu, Y.; Zhong, L. Targeted metabolomics study of serum bile acid profile in patients with end-stage renal disease undergoing hemodialysis. PeerJ 2019, 7, e7145. [Google Scholar] [CrossRef] [Green Version]
- Thakare, R.; Alamoudi, J.A.; Gautam, N.; Rodrigues, A.D.; Alnouti, Y. Species differences in bile acids I. Plasma and urine bile acid composition. J. Appl. Toxicol. 2018, 38, 1323–1335. [Google Scholar] [CrossRef]
- Jia, W.; Xie, G.; Jia, W. Bile acid–microbiota crosstalk in gastrointestinal inflammation and carcinogenesis. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 111–128. [Google Scholar] [CrossRef] [Green Version]
- Jones, B.; Begley, M.; Hill, C.; Gahan, C.; Marchesi, J. Functional and comparative metagenomic analysis of bile salt hydrolase activity in the human gut microbiome. Proc. Natl. Acad. Sci. USA 2008, 105, 13580–13585. [Google Scholar] [CrossRef] [Green Version]
- Perwaiz, S.; Mignault, D.; Tuchweber, B.; Yousef, I.M. Rapid and improved method for the determination of bile acids in human feces using MS. Lipids 2002, 37, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Humbert, L.; Maubert, M.; Wolf, C.; Duboc, H.; Mahé, M.; Farabos, D.; Seksik, P.; Mallet, J.-M.; Trugnan, G.; Masliah, J.; et al. Bile acid profiling in human biological samples: Comparison of extraction procedures and application to normal and cholestatic patients. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2012, 899, 135–145. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.; Su, M.; Xie, G.; Li, X.; Wei, R.; Liu, C.; Lan, K.; Jia, W. Factors affecting separation and detection of bile acids by liquid chromatography coupled with mass spectrometry in negative mode. Anal. Bioanal. Chem. 2017, 409, 5533–5545. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, W.J.; Sjovall, J. Bile acids: Analysis in biological fluids and tissues. J. Lipid Res. 2010, 51, 23–41. [Google Scholar] [CrossRef] [Green Version]
- FDA. Guidance for Industry, Bioanalytical Method Validation; U.S. Department of Health and Human Services, Center for Drug Evaluation and Research (CDER), Center for Veterinary Medicine (CV): Silver Spring, MD, USA, 2001. [Google Scholar]
- Thakare, R.; Alamoudi, J.; Gautam, N.; Rodrigues, A.; Alnouti, Y. Species differences in bile acids II. Bile acid metabolism. J. Appl. Toxicol. 2018, 38, 1590–1600. [Google Scholar] [CrossRef] [PubMed]
- García-Cañaveras, J.C.; Donato, M.T.; Castell, J.V.; Lahoz, A. Targeted profiling of circulating and hepatic bile acids in human, mouse, and rat using a UPLC-MRM-MS-validated method. J. Lipid Res. 2012, 53, 2231–2241. [Google Scholar] [CrossRef] [Green Version]
- Haeusler, R.; Pratt-Hyatt, M.; Welch, C.; Klaassen, C.; Accili, D. Impaired Generation of 12-Hydroxylated Bile Acids Links Hepatic Insulin Signaling with Dyslipidemia. Cell Metab. 2011, 15, 65–74. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Formula | RT 1 | Transition | Adduct Ion | IS 2 |
|---|---|---|---|---|---|
| Unconjugated | |||||
| Nordeoxycholic acid (norDCA) | C23H38O4 | 15.63 | 377.2697 | [M − H]− | CA-d4 |
| Dehydrocholic acid (DHCA) | C24H34O5 | 8.10 | 401.2333 | [M − H]− | CA-d4 |
| 7,12-Diketolithocholic acid (7,12-diketoLCA) | C24H36O5 | 7.40 | 449.2545 | [M + COOH]− | CA-d4 |
| 6,7-Diketolithocholic acid (6,7-diketoLCA) | C24H36O5 | 15.56 | 403.2490 | [M − H]− | CA-d4 |
| 3-Ketolithocholic acid (3-ketoLCA) | C24H38O3 | 23.75 | 373.2748 | [M − H]− | CA-d4 |
| 7-Ketolithocholic acid (7-ketoLCA) | C24H38O4 | 15.35 | 435.2752 | [M + COOH]− | CA-d4 |
| 12-Ketolithocholic acid (12-ketoLCA) | C24H38O4 | 9.68 | 405.2646 | [M − H]− | CA-d4 |
| 7-Ketodeoxycholic acid (7-DHCA) | C24H38O5 | 12.29 | 405.2646 | [M − H]− | CA-d4 |
| 3-Ketodeoxycholic acid (3-DHCA) | C24H38O5 | 15.90 | 389.2679 | [M + COOH]− | CA-d4 |
| Apocholic acid (apoCA) | C24H38O4 | 15.75 | 435.2752 | [M − H]− | CA-d4 |
| Isolithocholic acid (isoLCA) | C24H40O3 | 21.23 | 375.2905 | [M − H]− | CA-d4 |
| Allolithocholic acid (isoalloLCA) | C24H40O3 | 21.00 | 375.2905 | [M − H]− | CA-d4 |
| Allocholic acid (alloCA) | C24H40O5 | 12.44 | 407.2803 | [M − H]− | CA-d4 |
| Ursocholic acid (UCA) | C24H40O5 | 6.92 | 407.2803 | [M − H]− | CA-d4 |
| Lithocholic acid (LCA) | C24H40O3 | 22.91 | 421.2959 | [M + COOH]− | LCA-d4 |
| Ursodeoxycholic acid (UDCA) | C24H40O4 | 13.41 | 437.2909 | [M + COOH]− | UDCA-d4 |
| Chenodeoxycholic acid (CDCA) | C24H40O4 | 17.58 | 437.2909 | [M + COOH]− | CDCA-d4 |
| Isodeoxycholic acid (isoDCA) | C24H40O4 | 21.25 | 391.2854 | [M − H]− | DCA-d4 |
| murideoxycholic acid (MDCA) | C24H40O4 | 11.85 | 437.2909 | [M + COOH]− | CA-d4 |
| Deoxycholic acid (DCA) | C24H40O4 | 18.10 | 391.2854 | [M − H]− | CA-d4 |
| Hyodeoxycholic acid (HDCA) | C24H40O4 | 13.78 | 437.2909 | [M + COOH]− | CA-d4 |
| α-Muricholic acid (α-MCA) | C24H40O5 | 9.39 | 453.2860 | [M + COOH]− | CA-d4 |
| β-Muricholic acid (β-MCA) | C24H40O5 | 10.04 | 453.2858 | [M + COOH]− | CA-d4 |
| γ-Muricholic acid (γ-MCA) | C24H40O5 | 11.41 | 453.2858 | [M + COOH]− | CA-d4 |
| ω-muricholic acid (ω-MCA) | C24H40O5 | 8.70 | 453.2858 | [M + COOH]− | CA-d4 |
| Cholic acid (CA) | C24H40O5 | 12.83 | 407.2803 | [M − H]− | CA-d4 |
| Glycine-conjugated | |||||
| Glycodehydrocholic acid (GDHCA) | C26H37NO6 | 3.34 | 458.2548 | [M − H]− | GCA-d4 |
| Glycolithocholic acid (GLCA) | C26H43NO4 | 16.81 | 432.3119 | [M − H]− | GLCA-d4 |
| Glycodeoxycholic acid (GDCA) | C26H43NO5 | 12.25 | 448.3063 | [M − H]− | GDCA-d4 |
| Glycochenodeoxycholic acid (GCDCA) | C26H43NO5 | 11.44 | 448.3063 | [M − H]− | GCDCA-d4 |
| Glycoursodeoxycholic acid (GUDCA) | C26H43NO5 | 7.09 | 448.3068 | [M − H]− | GUDCA-d4 |
| Glycohyocholic acid (GHCA) | C26H43NO6 | 5.78 | 464.3018 | [M − H]− | GCA-d4 |
| Glycohyodeoxycholic acid (GHDCA) | C26H43NO5 | 7.34 | 448.3068 | [M − H]− | GCA-d4 |
| Glycocholic acid (GCA) | C26H43NO6 | 7.36 | 464.3018 | [M − H]− | GCA-d4 |
| Taurine-conjugated | |||||
| Taurolithocholic acid (TLCA) | C26H45NO5S | 14.24 | 482.2946 | [M − H]− | TCA-d4 |
| Taurochenodeoxycholic acid (TCDCA) | C26H45NO6S | 9.23 | 498.2895 | [M − H]− | TCDCA-d5 |
| Taurodeoxycholic acid (TDCA) | C26H45NO6S | 10.05 | 498.2895 | [M − H]− | TCDCA-d5 |
| Tauroursodeoxycholic acid (TUDCA) | C26H45NO6S | 5.48 | 498.2895 | [M − H]− | TCDCA-d5 |
| Taurohyodeoxycholic acid (THDCA) | C26H45NO6S | 5.58 | 498.2895 | [M − H]− | TCDCA-d5 |
| Tauro α-Muricholic acid (T-α-MCA) | C26H45NO7S | 3.19 | 514.2844 | [M − H]− | TCA-d4 |
| Tauro β-Muricholic acid (T-β-MCA) | C26H45NO7S | 4.34 | 514.2844 | [M − H]− | TCA-d4 |
| Taurohyocholic Acid (THCA) | C26H45NO7S | 5.92 | 514.2844 | [M − H]− | TCA-d4 |
| Taurocholic acid (TCA) | C26H45NO7S | 3.04 | 514.2844 | [M − H]− | TCA-d4 |
| Sulfo- | |||||
| Lithocholic Acid-3-Sulfate (LCA-3S) | C24H40O6S | 15.27 | 455.2473 | [M − H]− | CDCA-3S-d4 |
| Ursodeocycholic Acid-3-Sulfate (UDCA-3S) | C24H40O7S | 7.40 | 471.2422 | [M − H]− | CDCA-3S-d4 |
| Chenodeoxycholic Acid-3-Sulfate (CDCA-3S) | C24H40O7S | 11.32 | 471.2422 | [M − H]− | CDCA-3S-d4 |
| Deoxycholic Acid-3-Sulfate (DCA-3S) | C24H40O7S | 11.63 | 471.2422 | [M − H]− | CDCA-3S-d4 |
| Cholic Acid-3-Sulfate (CA-3S) | C24H40O8S | 7.72 | 487.2371 | [M − H]− | CDCA-3S-d4 |
| Glycine-Sulfo- | |||||
| Glycolithocholic Acid-3-Sulfate (GLCA-3S) | C26H43NO7S | 9.98 | 512.2687 | [M − H]− | GDCA-3S-d4 |
| Glycoursodeoxycholic Acid-3-Sulfate (GUDCA-3S) | C26H43NO8S | 3.42 | 528.2637 | [M − H]− | GDCA-3S-d4 |
| Glycochenodeoxychlolic Acid-3-Sulfate (GCDCA-3S) | C26H43NO8S | 6.50 | 528.2637 | [M − H]− | GDCA-3S-d4 |
| Glycodeoxycholic Acid-3-Sulfate (GDCA-3S) | C26H43NO8S | 6.90 | 528.2637 | [M − H]− | GDCA-3S-d4 |
| Glycocholic Acid-3-Sulfate (GCA-3S) | C26H43NO9S | 3.91 | 544.2586 | [M − H]− | GDCA-3S-d4 |
| Taurine-Sulfo- | |||||
| Taurolithocholic Acid-3-Sulfate (TLCA-3S) | C26H45NO8S2 | 6.98 | 562.2514 | [M − H]− | TUDCA-3S-d4 |
| Tauroursodeoxycholic Acid-3-Sulfate (TUDCA-3S) | C26H45NO9S2 | 2.06 | 578.2463 | [M − H]− | TUDCA-3S-d4 |
| Taurochenodeoxycholic Acid-3-Sulfate (TCDCA-3S) | C26H45NO9S2 | 4.19 | 578.2463 | [M − H]− | TUDCA-3S-d4 |
| Taurodeoxycholic Acid-3-Sulfate (TDCA-3S) | C26H45NO9S2 | 4.57 | 578.2463 | [M − H]− | TUDCA-3S-d4 |
| Taurocholic Acid-3-Sulfate (TCA-3S) | C26H45NO10S2 | 2.46 | 594.2412 | [M − H]− | TUDCA-3S-d4 |
| Internal standard | |||||
| Cholic acid-d4 (CA-d4) | C24H36D4O5 | 12.86 | 411.3054 | [M − H]− | |
| Lithocholic Acid-d4 (LCA-d4) | C24H36D4O3 | 22.96 | 379.3156 | [M − H]− | |
| Ursodeoxycholic Acid-d4 (UDCA-d4) | C24H36D4O4 | 13.45 | 395.3105 | [M − H]− | |
| Chenodeoxycholic Acid-d4 (CDCA-d4) | C24H36D4O4 | 17.54 | 395.3105 | [M − H]− | |
| Deoxycholic Acid-d4 (DCA-d4) | C24H36D4O4 | 18.13 | 395.3105 | [M − H]− | |
| Glycolithocholic Acid-d4 (GLCA-d4) | C26H39D4NO4 | 16.86 | 436.3370 | [M − H]− | |
| Glycoursodeoxycholic Acid-d4 (GUDCA-d4) | C26H39D4NO5 | 7.12 | 452.3320 | [M − H]− | |
| Glycodeoxycholic Acid-d4 (GDCA-d4) | C26H39D4NO5 | 6.90 | 452.3320 | [M − H]− | |
| Glycochenodeoxycholic Acid-d4 (GCDCA-d4) | C26H39D4NO5 | 12.26 | 452.3320 | [M − H]− | |
| Glycocholic Acid-d4 (GCA-d4) | C26H39D4NO6 | 7.43 | 468.3269 | [M − H]− | |
| Taurocholic Acid-d4 (TCA-d4) | C26H41D4NO7S | 5.96 | 518.3095 | [M − H]− | |
| Taurochenodeoxycholic Acid-d5 (TCDCA-d5) | C26H40D5NO6S | 9.26 | 503.3209 | [M − H]− | |
| Chenodeoxycholic Acid-3-Suflate-d4 (CDCA-3S-d4) | C24H36D4O7S | 11.38 | 475.2673 | [M − H]− | |
| Glycodeoxycholic Acid-3-Sulfate-d4 (GDCA-3S-d4) | C26H39D4NO8S | 11.42 | 532.2888 | [M − H]− | |
| Tauroursodeoxycholic Acid-3-Sulfate-d4 (TUDCA-3S-d4) | C26H41D4NO9S2 | 2.25 | 582.2714 | [M − H]− |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.; Liu, X.; Yang, J.; Ren, F.; Li, Y. Quantitative Profiling of Bile Acids in Feces of Humans and Rodents by Ultra-High-Performance Liquid Chromatography–Quadrupole Time-of-Flight Mass Spectrometry. Metabolites 2022, 12, 633. https://doi.org/10.3390/metabo12070633
Zhang X, Liu X, Yang J, Ren F, Li Y. Quantitative Profiling of Bile Acids in Feces of Humans and Rodents by Ultra-High-Performance Liquid Chromatography–Quadrupole Time-of-Flight Mass Spectrometry. Metabolites. 2022; 12(7):633. https://doi.org/10.3390/metabo12070633
Chicago/Turabian StyleZhang, Xiaoxu, Xiaoxue Liu, Jiufang Yang, Fazheng Ren, and Yixuan Li. 2022. "Quantitative Profiling of Bile Acids in Feces of Humans and Rodents by Ultra-High-Performance Liquid Chromatography–Quadrupole Time-of-Flight Mass Spectrometry" Metabolites 12, no. 7: 633. https://doi.org/10.3390/metabo12070633
APA StyleZhang, X., Liu, X., Yang, J., Ren, F., & Li, Y. (2022). Quantitative Profiling of Bile Acids in Feces of Humans and Rodents by Ultra-High-Performance Liquid Chromatography–Quadrupole Time-of-Flight Mass Spectrometry. Metabolites, 12(7), 633. https://doi.org/10.3390/metabo12070633